

Abstract
Despite the central role of large multi-protein complexes in many biological processes, it remains challenging to elucidate their structures and particularly problematic to define the structures of native macromolecular assemblies, which are often of low abundance. Here, we present a strategy for isolating such complexes and for extracting distance restraints that allow the determination of their molecular architectures. The method was optimized to allow facile use of the extensive global resources of GFP-tagged transgenic cells and animals.
Where classical structural tools prove insufficient, chemical cross-linking with mass spectrometric readout (CX-MS) is emerging as particularly useful for providing distance restraints between amino acid residues within protein assemblies – data that can be readily combined with other structural information to provide “integrative” structural models1, 2. Because the majority of such studies rely on recombinant protein complexes3–6, the reach and application of CX-MS would be greatly extended were the technique to be optimized for the analysis of native protein complexes isolated directly from their native cellular milieu7, 8. Major challenges to the analysis of such complexes relate to their often low abundance (necessitating ultrahigh affinity reagents for their efficient and pristine capture9), their dynamic nature, and their compositional and structural heterogeneity. Here, we present a pipeline that incorporates ultrahigh affinity reagents tuned to perform optimally with improved CX-MS analyses, in order to provide the requisite sensitivity to analyze native macromolecular assemblies.
Our pipeline (Fig.1) was tuned for affinity capture of native green fluorescence protein (GFP)-tagged protein complexes (with tagged protein expressing from the native genomic loci at close to endogenous levels), to take advantage of this most widely used tag for visualizing proteins in vivo. Indeed the biomedical community has produced an extensive worldwide resource of transgenic organisms and animals that contain GFP-tagged proteins, which should be of great utility in the present application10, 11.
Figure 1.
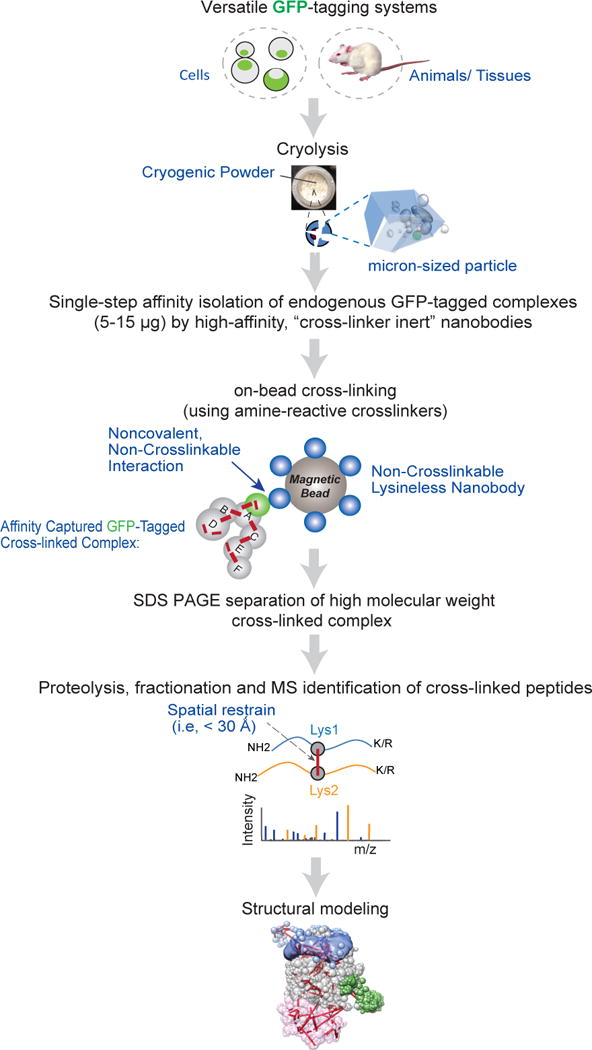
Workflow of nCOGNAC CX-MS. Cells or animal tissues are cryogenically milled into micron-sized particles (powders), which are dissolved in appropriate buffers to efficiently extract the proteins while preserving the architectures of the native protein assemblies. The GFP-tagged protein complex (5–15 μg) is then affinity captured by ultrahigh-affinity, non-cross-linkable nanobody-conjugated magnetic beads. The protein complex is on-bead cross-linked by DSS after which the cross-linked complex is eluted by heated LDS denaturing buffer and separated by SDS-PAGE electrophoresis. The efficiently cross-linked complex is in-gel proteolyzed to generate the cross-linked peptides, which are identified by high-resolution MS. The resulting information can be translated into residue-specific spatial restraints for computing integrative structural models.
The cells or tissues of interest are cryogenically milled into micron-sized particles to maximize the efficiency of solvent extraction of the protein complexes12. The resulting solubilized complexes are affinity captured on magnetic beads, conjugated with anti-GFP VHH nanobodies13, which we have engineered to preclude reaction with amine specific chemical cross-linking reagents such as disuccinimidyl suberate (DSS); they are based on previously described subnanomolar affinity LaG-169 and 3K1K (PDB# 3K1K)14 reagents, but with lysine residues substituted by either arginine or glutamine residues (Supplementary Figs. 1a, 2a). Because these substitutions are distal from the nanobodies’ GFP binding epitopes, they do not alter their binding affinities (Supplementary Figs. 1b, 2b–2d). These nanobodies were also chemically treated to minimize residual reactivity (e.g. at their amino termini) towards amine cross-linkers. The resulting high affinity, “cross-linking inert” reagents allow rapid, efficient affinity capture of complexes followed by on-bead cross-linking, without cross-linking the complexes to the bead-immobilized nanobodies. This property enables efficient removal of the cross-linked complex from the nanobody beads (Fig. 1, Supplementary Fig. 3), circumventing the otherwise formidable challenge of efficiently proteolyzing the highly rigid cross-linked complexes while they are still chemically conjugated to the beads. This “on-bead” cross-linking strategy also simplifies sample handling while providing a means for removal of complexes that are not cleavable from the resin, including most GFP-transgenic systems.
After on-bead cross-linking by DSS, the complexes are eluted from the nanobody beads by denaturing buffer, resolved on SDS-PAGE, and proteolyzed in-gel by trypsin (Methods). The proteolyzed products are fractionated and the cross-link peptides identified by MS. We name this technology nativenativeCOmplex GFP-Nanobody Affinity Capture Cross-linking with MS readout (nnCOGNAC CX-MS). Finally, the resulting distance restraints are used to depict architectures or generate structural models of the protein complexes15.
We illustrate the utility of nnCOGNAC CX-MS to architecturally dissect three exemplary native complexes. First to assess our overall workflow, we examined the Saccharomyces cerevisiae exosome, whose core structure has been elucidated at atomic resolution16, 17. Exosomes are multi-protein assemblies that process and degrade cellular RNA18. An 11-subunit exosome complex (exo11) forms a barrel-like structure composed of two stacked rings – an upper trimeric ring of S1/KH domain proteins (Rrp40, Rrp4, and Csl4) and a lower hexameric ring of PH domain proteins (Rrp45, Rrp46, Rrp42, Rrp43, Mtr3 and Ski6), as well as two catalytic subunits (Rrp6 and Rrp44).
To identify the constituents of the exosome, we affinity captured the complex via a genomically GFP-tagged component of the core (RRP46-GFP). MS analysis identified the eleven components of exo11 as well as two stoichiometric subunits (Lrp1 and Ski7) and a substoichiometric component Mpp6 that were not studied by X-ray crystallography18 (Fig. 2a, Supplementary Table 1).The resulting connectivity map (Fig. 2b) largely recapitulates the previously determined architecture of the complex. For example, the catalytic subunit Rrp6 is located proximal to Rrp43, Csl4, and Rrp4, predominantly via its CTD(residues 421–676), while its NTD (residues 45–379) is extensively cross-linked to Lrp1 to potentially form a heterodimer19 (Figs. 2b, 2c). The other catalytic subunit Rrp44 is crosslinked to the periphery of the hexameric/lower ring via Rrp45 and Ski6. Interestingly, the fact that the majority of the Rrp6 and Ski7 inter-subunit cross-links share identical lysine residues on Rrp4, Csl4 and Rrp43 (Fig. 2c) suggests that Rrp6 and Ski7 are potentially mutually exclusive and exist in two different complexes16. Consistent with this notion, GFP localization and affinity capture coupled to MS (Fig. 2c, Supplementary Fig. 4) show that Rrp6-Lrp1-Mpp6-exo10 (exo11 minus Rrp6) forms a nucleus-localized complex, while the Ski7-exo10 complex is restricted to the cytoplasm (Supplementary Fig. 5, Fig. 2d, Supplementary Tables 2, 3). These results show that nCOGNAC CX-MS can detect and elucidate compositional heterogeneities of native assemblies.
Figure 2.
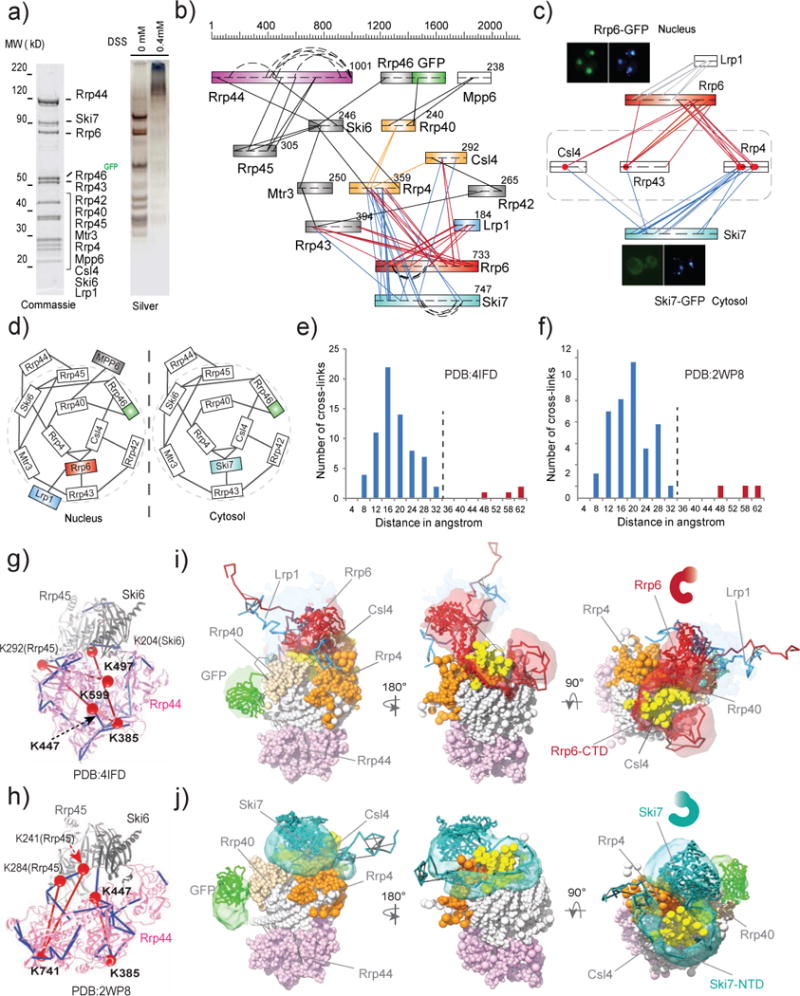
nCOGNAC CX-MS analysis and integrative structural modeling of the eukaryotic exosome complexes. a) SDS-PAGE analyses of affinity-captured and on-bead cross-linked exosome complexes. The protein components were identified by MS. Approximately 30 pmol (~15 μg) of this affinity captured material was used for each CX-MS analysis. b) Cross-link map for exosome complexes. Straight lines represent intersubunit cross-links, while curved lines represent distal intrasubunit cross-links. c) Heterogeneity of the exosome complexes revealed by CX-MS. Red dots represent lysine residues (on Rrp4, Rrp43, and Csl4) that are cross-linked to both Rrp6 and Ski7, indicating the presence of two different complexes in the affinity captured material. Subcellular localizations of Rrp6-GFP (nucleus) and Ski6-GFP (cytosol) were imaged by fluorescence microscopy. d) Cartoon models for the exosome complexes. e) Euclidean Cα-Cα distance distributions of all measured cross-links on the crystal structure of PDB# 4IFD. The Y-axis provides the number of cross-links that are mapped to the crystal structures. Blue bars represent DSS cross-links shorter than 30Å while red bars represent those longer than 44Å. f) Euclidean Cα-Cα distance distributions of all measured cross-links on the crystal structure PDB# 2WP8. g)–h) Observed cross-links displayed on the crystal structures of exosome complexes (PDBs #4IFD and 2WP8). Colors are coded as for e) and f). i)–j) Structural models of nuclear Rrp6-Lrp1-exo10 and cytosolic Ski7-exo10 exosome complexes. Localization density maps of the best 500 scoring models are shown.
To evaluate the cross-link data, we measured the Euclidean Cα-Cα distances between cross-linked lysine residues on the published X-ray structure of exo10 and CTD of Rrp6 (PDB #4IFD) as well as that on the structure of the heterotrimer Rrp45, Ski6 and Rrp4420 (PDB #2WP8). 34% of the cross-linking data could be mapped to the X-ray structures (Supplementary Fig. 6, Supplementary Table 4). Of these, >90% spanned ≤30 Å (the expected maximum reach), while four crosslinks in PDB #4IFD and three in PDB #2WP8 mapped to much longer distances in the X-ray structures (i.e., 44–65 Å) (Figs. 2e, 2f, Supplementary Fig. 7). Significantly, all of these seemingly “violated” cross-links (red) are located on Rrp44 (Figs. 2g, 2h), the essential catalytic subunit that exhibits both 3′–5′ RNA exoribonuclease and endoribonuclease activities, supporting the notion that this enzyme is highly dynamic, forming different conformers in the cell21.
The relatively rich cross-linking information on nuclear Rrp6-Lrp1 and cytosolic Ski7 to the Exo10 core allows us to utilize integrative modeling15 to determine the architectures of these subcellular-specific exosome complexes (Methods). As shown in Fig. 2i, the nuclear Rrp6-Lrp1 heterodimer is localized on top of the trimeric ring, which likely allows recruitment of RNA substrates to the Rrp6 exoribonuclease activity. The position of the corresponding cytosolic component Ski7 with respect to exo10 was resolved with an average residue precision of ~9 Å (RMSF or root-mean-square-fluctuation, Supplementary Fig. 8, Methods), docked on the upper ring with its C-terminal GTPase domain above Csl4 and Rrp4, and close to the central channel (RNA entrance) (Fig. 2j). Intriguingly, its disordered NTD (Fig. 2j) (residues 90–264) curves like a “necklace” that wraps around Csl4 and the N terminus of Rrp4 before it emerges on top of the upper ring where it connects to the folded GTPase domain, reminiscent of the architecture of the nuclear Rrp6 CTD but with the opposite chirality.
To assess nCOGNAC-CX-MS for examining low abundance native complexes (tens to hundreds of copies per cell)22, 23, we studied the S. cerevisiae anaphase-promoting complex (APC/Cyclosome) – a ubiquitin E3 ligase essential for cell cycle progression24. The architecture of the yeast APC/C (13 unique polypeptides) with its co-factor Cdh1 bound to a D-box peptide has been studied by cryoEM and solved using recombinant overexpressed proteins at a resolution of ~10 Å22, and recombinant human APC/C at 7.4 Å, resulting in a characterization of the packing of secondary structure segments for all subunits25.
We employed affinity captured all known subunits of the native complex together with the substoichiometric component Cdh1 (Fig. 3a, Supplementary Table 5). The resulting nCOGNAC connectivity map (Fig. 3b, Supplementary Table 6) is consistent with the architecture revealed by EM22, 25. Thus, for example, the tetratricopeptide repeat (TPR) domain-containing proteins (Cdc27, Cdc16, and Cdc23) are extensively cross-linked, revealing the TPR lobe to which accessory subunits (Apc13, Cdc26 and the yeast specific subunit Apc9) are bound. Cross-links within the base/platform module (Apc1, Apc2, Apc4 and Apc5) are prevalent. The TPR lobe and base modules are bridged by the cross-links Cdc23 - Apc1, Cdc16 - Apc1 and Cdc27 - Apc2, defining a structure that appears conserved from yeast to human. Another yeast-specific subunit, Mnd2, is cross-linked to both the TPR lobe (Cdc23 and Cdc16) and the scaffold (Apc4) via its N terminal region, further connecting the two modules.
Figure 3.
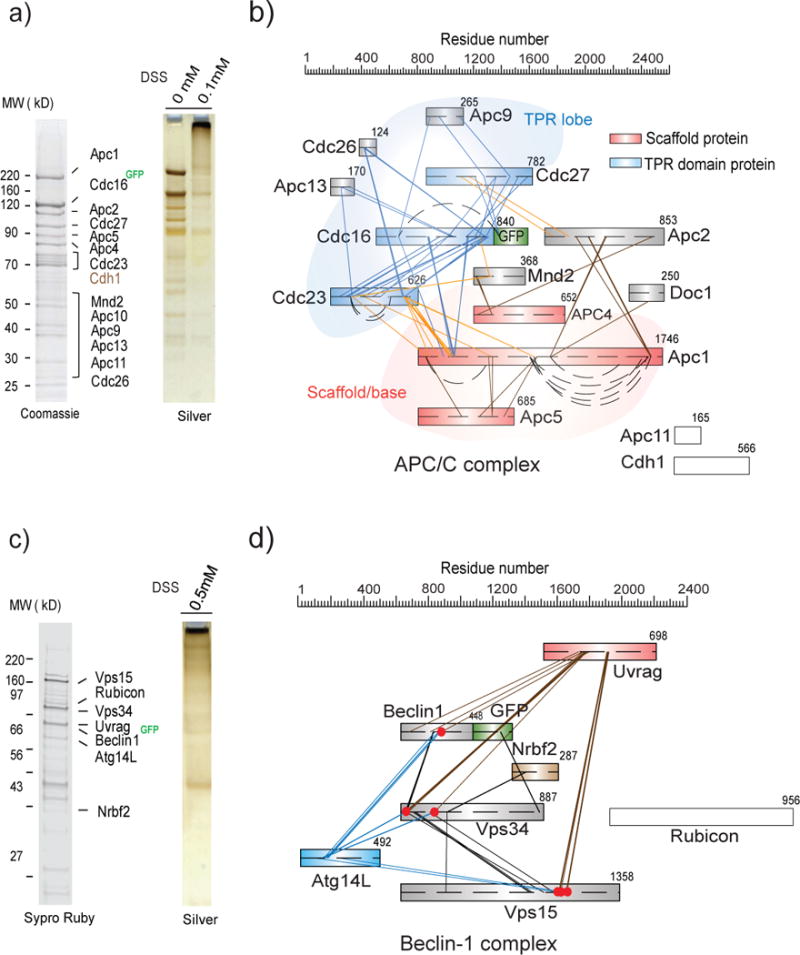
Architectural analyses of eukaryotic APC/C complex and tissue-specific (mouse liver) Beclin1 complexes. a) SDS-PAGE analyses of affinity captured and on-bead cross-linked APC/C complex. Protein subunits were identified by MS. Approximately 15 pmol (15 μg) of the complex was purified from ~6 L of yeast culture for CX-MS analysis. b) Cross-link map for eukaryotic APC/C complex. APC/C Subunits are schematically represented and color coded: red (scaffold subunits of APC1, APC4 and APC5), orange (APC2), blue (TPR lobe subunits of Cdc27, Cdc16, and Cdc23), and gray for all other subunits. Straight lines represent intersubunit cross-links while curved lines represent distal intrasubunit cross-links. c) SDS-PAGE analyses of affinity captured and on-bead cross-linked Beclin1 complexes. Components of the complexes were identified by MS. Affinity captured and on-bead cross-linked complexes were stained by Sypro Ruby and Silver stain, respectively. d) Cross-link map for tissue-specific Beclin1 complexes. Protein subunits are schematically represented and color coded as: red (Uvrag), blue (Atg14L), brown (Nrbf2) and gray (Beclin1, Vps34 and Vps15). Red dots represent lysine residues (on Uvrag, Beclin1, and Atg14L) that are cross-linked to both Uvrag and Atg14L. No intersubunit cross-links were identified on Rubicon.
Although, inter-subunit cross-links provide the most useful distance restraints, certain intra-subunit cross-links may also reveal the architecture of a single subunit – e.g., crosslinks between the C-terminal and mid-domains of Apc1 constrain these regions to be spatially close and proximal to the central region of Apc2 (as in the high-resolution human APC/C structure). We also identified “same-residue” cross-links on Cdc27 and Cdc16 (Supplementary Fig. 9, Supplementary Table 7), consistent with homodimer formation25.
Finally, we explored the possibility of architecturally dissecting native GFP-tagged complexes isolated from a single transgenic mouse (mus musculus) liver, investigating Beclin 1-associated complexes, which are central effectors of autophagy known to regulate aging and neurological disorders26. Despite the biological importance of these complexes, their overall architectures remain to be eludicated27–29. We applied our pipeline to map the arrangement of its seven major subunits30, 31 (Figs. 3c, 3d, Supplementary Tables 7, 8). Multiple cross-links were identified between Beclin 1-EGFP and its binding partner Vps34 through the coiled-coil domain of Beclin 1 (residues 142-267) and the N-terminal C2 domain of Vps34 (residues 1–255). Cross-links also reveal that the C2 domain of Vps34 and Vps15 CTD are in close proximity. Interestingly, Atg14L and Uvrag share many inter-subunit cross-links to the same lysine residues on Beclin 1, Vps34, and Vps15 triad – indicating that Atg14L and Uvrag interact in a mutually exclusive manner with the triad to form two different complexes32 (Figs. 3c, 3d).
nCOGNAC CX-MS yielded useful distance restraints in three exemplary low to medium abundance multi-subunit complexes and elucidated complexes displaying heterogeneity, which is common in native assemblies. Together with integrative modeling, this approach provides an effective means to model structures of proteins that contain extensive disordered or flexible regions, which present a problem for most other structural approaches. Our approach opens the door to large-scale, systematic analyses of native protein complex.
Online Methods
Cloning, purification, and chemical conjugation of anti-GFP VHH nanobodies
Codon optimized DNA sequences of lysineless VHH nanobodies were synthesized (Genscript) and cloned into pET21-pelB-VHH vector using BamHI and XhoI restriction sites. A flexible linker sequence of GGGSGGC was inserted at the carboxyl terminus of the proteins to allow chemical conjugation and proper folding of the proteins. The nanobodies were expressed and were purified by nickel-NTA beads as previously described9. Purified nanobodies were then conjugated to epoxy-activated Dynabeads33 (Invitrogen). For each 1 mg of epoxyl Dynabeads, ~5 μg nanobodies were used for the conjugation. The lysineless nanobody-beads (LaG-16-2K/R or 3K1K-3K/R) were further chemically blocked by 0.5 mM disuccinimidyl suberate for 30–45 minutes at 25 °C with constant agitation on a shaker and subsequently quenched by 100 mM final concentration of Tris-HCl (pH= 8). The reagents were stored at 4 °C and were used within 3 weeks of conjugation.
Kd measurements by Surface Plasmon Resonance (SPR)
SPR measurements were obtained on a Proteon XPR36 Protein Interaction Array System (Bio-Rad). Recombinant GFP was immobilized on a ProteOn GLC sensor chip: the chip surface was first activated with 50 mM sulfo-NHS and 50 mM EDC, run at a flow-rate of 30 μl/min for 300 sec. The ligand was then diluted to 5 μg/ml in 10 mM sodium acetate, pH 5.0, and injected at 25 μl/min for 180 sec. Finally, the surface was deactivated by running 1 M ethanolamine-HCl (pH 8.5) at 30 μl/min for 300 sec. This led to immobilization of approximately 200 response units (RU) of ligand.
The Kd values of recombinant nanobodies were determined by injecting 4 concentrations of each protein, in triplicate, with a running buffer of 20 mM Na-HEPES, pH 8.0, 150 mM NaCl, 0.01% Tween-20. Proteins were injected at 100 μl/min for 90 sec, followed by a dissociation time of 660 sec. Between injections, residual bound protein was eliminated by regeneration with 4.5 M MgCl2 in 10 mM Tris-HCl, pH 7.5, run at 100 μl/min for 90 sec. Binding sensorgrams from these injections were processed and analyzed using the ProteOn Manager software. Binding curves were fit to the data with a Langmuir model, using grouped ka, kd, and Rmax values.
GFP transgenic strains, cell culturing and cryogenic grinding of yeast cells and the mouse liver
The GFP-tagged S. cerevisiae strains of RRP6 (YOR001W), SKI7 (YOR076C), and RRP46 (YGR095C) were purchased from Invitrogen. The GFP-CDC16-3×FLAG-His6 (YKL022C) strain (MATα BY4742) was generated as previously described34. All yeast cells were cultured in YEPD medium (MP Biomedicals) at 30 °C to a density of 3–4 × 107 cells/ml before harvesting. The Becn1−/−; Becn1-EGFP/+ bacterial artificial chromosome (BAC) transgenic mice express Beclin 1-EGFP under endogenous transcriptional control30. One mouse was sacrificed; its liver was collected and immediately frozen in liquid nitrogen for storage. Yeast strains and the mouse liver were cryogenically milled by a planetary ball mill12, 35 (Retsch PM 100) and the cryogenic powders were stored at −80 °C before use.
Fluorescence microscopy
S. cerevisiae strains RRP6-GFP (YOR001W) and SKI7-GFP (YOR076C) were grown to mid-log phase, stained with 2.5 μg/ml DAPI for 20 minutes, washed with PBS and visualized in vivo by a fluorescence microscope (Axioplan 2, Carl Zeiss) equipped with a cooled charge-coupled device camera (ORCA-ER, Hamamatsu). The images were analyzed by Openlab software (Perkin Elmer) and processed using Adobe Photoshop CS5 (Adobe).
Affinity capture and mass spectrometric analysis of endogenous GFP-tagged protein complexes
To identify the compositions of the yeast exosome and APC/C complexes, 200 mg and 500 mg of yeast grindate powder was dissolved in 0.75 ml and 1.5 ml yeast lysis buffer, respectively (100mM Na-HEPES pH=7.9, 0.2% Tween-20, 350 mM sodium citrate, protease inhibitor cocktail (Roche)) and briefly sonicated on ice. The protein extracts were centrifuged at 14,000 rpm for 12 min and the supernatants collected. The protein complexes were affinity isolated by adding ~2 mg of lysineless nanobody-conjugated Dynabeads into the supernatant for 30 min (exosome complex) or 60 min (APC/C complex) at 4°C on a rotating wheel. The beads were then washed four times with 0.5–1ml of yeast pullout buffer and collected using a magnetic stand.
To identify the Bcln1-EGFP complex, ~400 mg of the mouse liver grindate powder was dissolved in 1ml pullout buffer (100 mM Na-HEPES pH=7.9, 0.2% Tween-20, 150 mM sodium chloride, 350 mM sodium citrate, and protease inhibitor cocktail) and was briefly sonicated on ice. The protein extract was centrifuged at 14,000g for 20 min and the supernatant was transferred and incubated with ~ 2 mg of the nanobody-conjugated Dynabeads for ~2 h at 4°C on a rotating wheel. The beads were washed four times and collected using a magnetic stand before elution.
The affinity captured complexes were eluted by heating the beads at 75°C for 5 min in NuPAGE LDS gel sample buffer (Invitrogen) and were subsequently subjected to electrophoresis by SDS-PAGE. The protein-containing gel bands were cut and in-gel digested by trypsin. After peptide extraction and desalting, purified peptides extracted from different gel bands were pooled and analyzed by LC/MS to identify the proteins.
Proteolysis and MS identification of the affinity captured protein complexes
The affinity captured GFP-tagged complexes (from 200mg–500mg of the cell cryogrindate) were separated by electrophoresis on a 4–12% SDS PAGE gel. The protein-containing bands were cut into 3–5 pieces and in-gel digested by trypsin (100–200 ng per gel band) for 6 hours or overnight. After digestion, the resulting tryptic peptides were extracted from the bands, pooled and purified for the subsequent LC/MS analysis.
To characterize the composition of the affinity captured material, a portion of the purified proteolytic peptides (corresponding to 100–200 ng of the purified complex) were loaded onto a self-packed PicoFrit® column (5 cm × 75 μm ID, 3um, New Objective). The column was packed with 5 cm of reverse-phase C18 material (3μm porous silica, 200 Å pore size, Dr. Maisch GmbH). Mobile phase A consisted of 0.5% acetic acid and mobile phase B of 70 % ACN with 0.5 % acetic acid. The peptides were eluted in a linear LC gradient using a HPLC system (Agilent), and analyzed with a LTQ Velos Orbitrap Pro mass spectrometer (Thermo Fisher). The instrument was operated in the data-dependent mode, where the top twenty-most abundant ions were fragmented by CID (normalized energy 35) and analyzed in the linear ion trap. The target resolution for MS1 was 60,000. Other instrumental parameters include: “lock mass” at 371.1012 Da, minimal threshold of 3,000 to trigger an MS/MS event. Ion trap accumulation limits (precursors) were 1 × 105 and 1 × 106 respectively for the linear ion trap and Orbitrap.
For LC/MS analysis of the GFP tagged Ski-exosome complex, ~ 100–200 ng of the complex was affinity isolated using 2K/R nanobody conjugated dynabeads. After reduction and alkylation, the affinity captured proteins were briefly separated on a SDS PAGE by gel electrophoresis (ran ~1/5 of 4–12% Gradient gel) and proteins were in-gel digested by trypsin. After desalting and purification, peptides which correspond to approximately 25–50 ng of the affinity captured complex were loaded on a C18 analytical column (75μm/3μm/200Ǻ/15 cm, Thermo Fisher) and eluted by a 25-min LC gradient and analyzed with an Orbitrap QE plus mass spectrometry (Thermo Fisher).
Raw data on the affinity capture/MS experiments were converted to an mzXML file and were searched online by X! Tandem36 (http://www.thegpm.org/tandem/). Database search parameters include mass accuracies of MS1 < 10 ppm and MS2 < 0.4 Da (or MS2< 20 ppm for the sample analyzed by QE MS), cysteine carbamidomethylation as a fixed modification, methionine oxidation, N-terminal acetylation, and phosphorylation (at serine, threonine, and tyrosine) as variable modifications. Maximum of one trypsin missed-cleavage site was allowed. The false positive rate is estimated about 1%. The search results were provided in the supplementary tables.
Affinity isolation of the yeast exosome complex from whole cell lysates cross-linked with different concentrations of DSS cross-linker
One gram of yeast cryomilled powder (Rrp46-GFP) was resuspended in 3.5ml exosome complex buffer (100mM Na-HEPES pH=7.9/8.0, 0.2% Tween-20, 350 mM sodium citrate, protease inhibitor cocktail, Roche); cell lysate was centrifuged at 13,000 rpm for 15 mins and the supernatant was collected. Equal amount of the soluble, whole protein lysate was then cross-linked for 2 hours at 4°C by different concentrations of DSS crosslinker (0, 0.04, 0.2, 1 and 5 mM (due to the solubility of DSS, we did not intend to further increase its concentration in the whole cell lysate)). After crosslinking of the whole protein lysates, the reaction was quenched by 50mM Tris buffer (final concentration). Cross-linked protein lysates were then centrifuged to remove the precipitated proteins and the supernatants were collected for affinity capture of the exosome complex (using the same protocol as described in the manuscript). Approximately 0.5 mg of 2K/R nanobody-conjugated magnetic beads was used for each affinity capture (from ~200 mg of cryogenic yeast powder).
The affinity isolated proteins were loaded onto a 4–12% Bis-Tris SDS PAGE gel and digested by trypsin. After proteolysis, the purified peptides were analyzed by LC/MS (two analytical replicates, a 25 min LC gradient was used for each analysis) using the QE Plus mass spectrometer (Thermo). The proteins were identified by X! tandem and the relative amounts were quantified by peptide spectra counts using unique peptide identifications (Supplementary Table 9).
On-bead cross-linking, proteolysis and MS analysis cross-linked peptides
Several hundred ng of the complexes were affinity isolated and were cross-linked at various concentrations of DSS cross-linker to empirically decide the optimal reaction conditions – i.e., the minimal concentration of crosslinker so that all component proteins migrated to the high molecular weight regions in the gel (Supplementary Fig. 10). ~40 mg of the nanobody-Dynabeads (3K1K-3K/R or Lag-16-2K/R) were used to isolate the protein complexes for CX-MS analyses from 3–4 g yeast grindate powder of Rrp46-GFP (exosome), ~12–15 g grindate powder of GFP-Cdc16 (APC/C), and the liver from a single transgenic GFP-Bcln1 mouse).
The purified complex (~5–15 μg) was on-bead cross-linked by isotopically labeled DSS (d0:d12=1:1, Creative Molecules) in 100mM K-HEPES, pH=7.9. (Note we found that the isotopic label was not needed in the present experiments and can be omitted). The cross-linked complex was quenched and eluted from the beads by LDS buffer (containing 100mM DTT and 100mM Tris-HCl, pH=8.5) at 85°C for 10 minutes. The sample was cooled at room temperature for cysteine alkylation and separated by electrophoresis in either 4–12% or 3%–8% Bis-Tris SDS PAGE gel (Invitrogen). SDS-PAGE allows (i) separation and enrichment of highly cross-linked materials (which run as high molecular weight products) to reduce the complexity of the digest and (ii) elimination of the detergents needed for solubilization of the complex as well as chemical “bleed” products from the magnetic beads, both of which can greatly reduce MS sensitivity. In addition, the presence of SDS helps denature and disperse the rigid, cross-linked complex, greatly facilitating proteolysis. We have also tested an alternative possibility for obtaining distance restraints by treating whole cell lysate with DSS cross-linker prior to affinity capture of the GFP-tagged protein complexes, but found that this procedure does not provide sufficient inter-subunit crosslinking yield to be useful, while producing a high level of non-specific protein background. (Supplementary Fig. 11 and Supplementary Table 9).
A central feature of the nCOGNAC CX-MS strategy are specially engineered, optimized affinity reagents that are available at minimal cost in virtually unlimited supply to any scientist in the world. Our pre-treatment with crosslinking reagent to render the affinity reagent inert to crosslinking may also allow the more general use of standard antibodies for on-bead crosslinking provided that they have sufficiently high affinity and that the pre-treatment does not affect the antigen binding site (Supplementary Fig. 3). Although our strategy significantly expands the utility of CX-MS for studies of native complexes, further improvements (e.g., more efficient sample handling and more sensitive MS readout) are still desired to increase the sensitivity of crosslink detections, and the richness of the resulting restraint information, which is especially important for modeling of complexes8. In the present work, we have explored protein assemblies with molecular masses ranging to ~1 MDa, which represents the great majority of stable and soluble complexes in cells37, 38. Additional work will be needed to ascertain how best to deal with even larger systems.
The cross-linked complex was digested in-gel with trypsin to generate cross-linked peptides. The gel region above 220 kDa was sliced, crushed into small pieces and incubated overnight with trypsin at 1:20 (w/w) ratio in 50mM ammonium bicarbonate buffer containing 0.1% Rapigest (Waters) at 37°C. The cross-linked complex was then re-digested by a second bolus of the same amount of trypsin for four hours. The digestion mixture was acidified by formic acid and peptides were extracted twice by adding a stepwise gradient of acetonitrile and shaking vigorously at room temperature. The peptide extracts were pooled, and centrifuged at 13,000 rpm for 10min. The supernatant was collected, flash frozen and lyophilized to ~30 μl. The peptide mixture was then reconstituted in 5% MeOH, 0.2% FA and desalted by a C18 SPE column (Waters). After lyophilization (to ~5 μl), the resulting peptides were reconstituted in 20 μl of a solution containing 30% acetonitrile (ACN) and 0.2% formic acid (FA) and fractionated by peptide SEC39 (Superdex Peptide PC 3.2/30, GE Healthcare) using offline HPLC separation and autosampler (Agilent Technologies). Two or three SEC fractions covering the molecular mass range of ~2.5 kD to ~8 kD were collected and analyzed by LC/MS.
For cross-link identifications, the purified peptides were dissolved in the sample loading buffer (5% MeOH, 0.2% FA) and analyzed by a LTQ Velos Orbitrap Pro mass spectrometer (for most analyses) or an Oribtrap Q Exactive (QE) Plus mass spectrometer (Thermo Fisher). For the analysis by the Velos Orbitrap mass spectrometer, the dissolved peptides were pressure loaded onto a self-packed PicoFrit® column with integrated electrospray ionization emitter tip (360 O.D, 75 I.D with 15 μm tip, New Objective). The column was packed with 8 cm of reverse-phase C18 material (3μm porous silica, 200 Å pore size, Dr. Maisch GmbH). Mobile phase A consisted of 0.5% acetic acid and mobile phase B of 70 % ACN with 0.5 % acetic acid. The peptides were eluted in a 120 minute LC gradient (8% B to 50% B, 0–93 minutes, followed by 50% B to 100% B, 93–110 minutes and equilibrated with 100% A until 120 minutes) using a HPLC system (Agilent), and analyzed with a LTQ Velos Orbitrap Pro mass spectrometer. The flow rate was ~200 nL/min. The spray voltage was set at 1.9–2.3 kV. The capillary temperature was 275°C and ion transmission on Velos S lenses was set at 35%. The instrument was operated in the data-dependent mode, where the top eight-most abundant ions were fragmented by higher energy collisional dissociation40(HCD) (HCD energy 27–29, 0.1 ms activation time) and analyzed in the Orbitrap mass analyzer. The target resolution for MS1 was 60,000 and 7,500 for MS2. Ions (370–1700 m/z) with charge state of >3 were selected for fragmentation. A dynamic exclusion of (15 s / 2 / 55 s) was used. Other instrumental parameters include: “lock mass” at 371.1012 Da, a mass exclusion window of 1.5 Th, and a minimal threshold of 5,000 to trigger an MS/MS event. Ion trap accumulation limits (precursors) were 1 × 105 and 1 × 106 respectively for the linear ion trap and Orbitrap. For MS2, the Orbitrap ion accumulation limit was 5 × 105. The maximum ion injection time for Orbitrap was 500–700 milliseconds. The QE plus instrument was directly coupled to an EasyLC system (Thermo Fisher) and experimental parameters were similar to those of the Velos Orbitrap. The cross-linked peptides were loaded onto an Easy-Spray column heated at 35 °C (C18, 3μm particle size, 200 Å pore size, and 50 μm × 15cm, Thermo fisher) and eluted using a 120-min LC gradient (2% B to 10% B, 0–6 minutes, 10% B – 35% B, 6–102 minutes, 35% B–100% B, 102–113 minutes followed by equilibration, where mobile phase A consisted 0.1% formic acid and mobile phase B consisted 0.1% formic acid in acetonitrile). The flow rate was ~300 nl/min. The spray voltage was 2.0 kV and the top 10 most abundant ions (with charge stage of 3–7) were selected and fragmented by HCD.
The raw data were transformed to MGF (mascot generic format) by pXtract 1.0 and searched by pLink41 using a FASTA database containing protein sequences of the identified subunits of each complex. An initial MS1 search window of 5 Da was allowed to cover all isotopic peaks of the cross-linked peptides. The data were automatically filtered using a mass accuracy of MS1 ≤ 10 ppm (parts per million) and MS2 ≤ 20 ppm of the theoretical monoisotopic (A0) and other isotopic masses (A+1, A+2, A+3, and A+4) as specified in the software. Other search parameters include cysteine carboxymethylation as a fixed modification, and methionine oxidation as a variable modification. A maximum of two trypsin missed-cleavage sites was allowed. The initial search results were obtained using the default 5% false discovery rate (FDR) – estimated using a target-decoy search strategy. In our analysis, we treated the 5% FDR as a rough initial filter of the raw data (albeit quite permissive). Next, we manually applied additional filters to remove potential false positive identifications from our dataset8. For positive identifications, both peptide chains should contain four or more amino acids. In addition for both peptide chains, the major MS/MS fragmentation peaks should be assigned and follow a pattern that contains a continuous stretch of fragmentations. The cross-link maps were generated using AUTOCAD (Autodesk, Inc., educational version).
Proteins used for cross-linking database search
For the exosome cross-linking database search, the budding yeast protein sequences of 14 known exosome subunits of YOL021C/ Rrp44, YOL142W/ Rrp40, YGR095C/ Rrp46, YHR069C/ Rrp4, YGR158C/ Mtr3, YNL232W/ Csl4, YOR001W/ Rrp6, YDR280W/ Rrp45, YOR076C/ Ski7, YCR035C/ Rrp43, YDL111C/ Rrp42, YHR081W/LRP1, YGR195W/Ski6, YNR024W/Mpp6, together with GFP and two other relatively abundant proteins (YNL178W/ RPS3, YGR178C/PBP1, which were identified with more than 10 unique peptides by affinity capture/MS in Supplementary table 1) were included. For the APC/C cross-linking database search, the budding yeast protein sequences of 11 known APC/C subunits of YDL008W/Apc11, YKL022C/Cdc16, YHR166C/Cdc23, YBL084C/Cdc27, YDR260C/Swm1/, YNL172W/Apc1, YLR127C/Apc2, YDR118W/Apc4, YOR249C/Apc5, YGL240W/Doc1, YFR036W/Cd26, YIR025W/Mnd2, YGL003C/Cdh1, YLR102C/Apc9, and GFP were included. For the mouse (Mus musculus) Beclin1 complex cross-linking database search, seven subunits (Beclin1, Uvrag, Atg14L, Vps34, Vps15, Rubicon, and Nrbf2) of and the GFP protein were used.
Determining the structures of the subcellular specific exosome complexes by Integrative Modeling Platform (IMP)
Our integrative approach to determine the exosome complex structure proceeds through four stages: (1) gathering of data, (2) representation of subunits and translation of the data into spatial restraints, (3) configurational sampling to produce an ensemble of models that satisfies the restraints as well as possible, and (4) analysis and assessment of the ensemble. The modeling protocol was scripted using the Python Modeling Interface, a library to model macromolecular complexes based on the open-source Integrative Modeling Platform15 (IMP) package release 2.3.0. Briefly, we represented the protein subunits of the exosome by beads of varying sizes, arranged into rigid or flexible strings, based on the available exosome crystallographic structures and a comparative model for Ski7(259–747) (Supplementary Fig. 12). The relative stoichiometries of the key components were determined to be in 1:1 ratios within both the nuclear and cytosolic complexes16, 42 (Supplementary Fig. 13). The cross-linking data were then encoded into a Bayesian scoring function that restrained the distances spanned by the cross-linked residues3, 8, 43. Cytosolic Ski7-exo10 and nuclear Rrp6-Lrp1-exo10 complexes were computed separately. The 500 best scoring models for each subcellular complex were clustered, yielding the localization density maps (solutions) shown in Figs. 2i – 2j. The resulting solutions satisfy 95% of the input cross-link restraints (Supplementary Fig. 14). The precisions of Ski7 and Rrp6 solutions (average RMSD (root-mean-square deviation) with respect to the cluster center) are respectively 22.7 Å and 40.0 Å, even though these two proteins contain long disordered regions (Supplementary Fig. 12).
Specifically, The integrative approach to determine the Ski7-exo10 and Rrp6-Lrp16-ex10 complexes proceeds through four stages7, 44–47: (1) gathering of data, (2) representation of subunits and translation of the cross-linking data and the prior knowledge into a Bayesian scoring function, (3) configurational sampling to produce an ensemble of models that minimize the Bayesian scoring function, and (4) analysis of the ensemble.
Gathering of data
We used the crystallographic structures of the exosome complex (PDB# 4IFD and # 2HBJ) as well as the GFP (PDB #1GFL)48 and a comparative model of Ski7 259–747 structured domain generated using Phyre249 (Supplementary Fig. 12). 216 cross-links identified by mass spectrometry were used for modeling.
Representation of subunits
The domains of the two exosome complexes were represented by beads, arranged into either a rigid body or a flexible string, based on the available crystallographic structures and comparative models. To balance the thoroughness of configurational sampling and precision of model representation, we represented the structures in a multi-scale fashion. Sequence segments missing in the crystal structures were substituted by a single or multiple beads of the corresponding size. For each domain and interface with an atomic model, the beads representing a structured region were kept rigid with respect to each other during configurational sampling (i.e., rigid bodies). The rigid bodies include: the exo10 complex, GFP which is fused to the C terminus of Rrp46, Rrp6 (residues 127–516), and Ski7 (residues 259–747). Segments without a crystallographic structure or comparative model (i.e., with an unknown structure) were represented by a flexible string of beads corresponding to a maximum of 5 residues each (Supplementary Fig. 12).
Bayesian scoring function
The Bayesian approach estimates the probability of a model, given information available about the system, including both prior knowledge and newly acquired experimental data. The model includes the structure coordinates X and additional parameters . Using Bayes’ theorem, the posterior probability , given data D and prior knowledge I, is , where the likelihood function is the probability of observing data D, given I and M, and the prior is the probability of model M, given I. To define the likelihood function, one needs a forward model that predicts the data point (i.e., the presence of a cross-link between two given residues) given any model M, and a noise model that specifies the distribution of the deviation between the observed and predicted data points. The Bayesian scoring function is the negative logarithm of , which ranks the models identically to the posterior probability.
Briefly, the forward model fn is computed as the probability of randomly picking two points and within the spheres centered on the Cα atoms of the cross-linked residues, with coordinates ri and rj, with unknown radii σi and σj, such that the distance between them is lower than the maximum cross-linker length lXL; the radii σi and σj are proxies for the uncertainty of forming a cross-link, given structural model X. To reduce the number of parameters in the model, we utilized a single uncertainty parameter σ for all residues. We imposed lXL = 21 Å for the DSS cross-linker.
The likelihood function for a cross-link dn is , where is the uncertainty of observing a cross-link, and is approximately equal to the expected fraction cross-links that are inconsistent with the structure X. We set Ψ to 5%. The joint likelihood function for a dataset of NXL independently observed cross-links is the product of likelihood functions for each data point.
The model prior is defined as a product of the priors p(x) and p(σ) on the structural coordinates X and uncertainty σ, respectively. The prior p(X) includes the excluded volume restraints, the sequence connectivity restraints, and a weak restraint whose score depends linearly on the distance between cross-linked residues, with a slope of 0.01 Å-1. p(σ) is a uniform distribution over the interval [0,100].
Sampling model configurations
Structural models of the Rrp6-Lrp1-exo10 and Ski7-exo10 complexes were computed by Replica Exchange Gibbs sampling, based on Metropolis Monte Carlo sampling43. This sampling was used to generate configurations of the system as well as values for the uncertainty parameters. The Monte Carlo moves included random translation and rotation of rigid bodies (1 Å and 0.025 radian maximum, respectively), random translation of individual beads in the flexible segments (1 Å maximum), as well as a Gaussian perturbation of the uncertainty parameters. The sampling was run on 64 replicas, with temperatures ranging between 1.0 and 2.5. 2 independent sampling calculations were run for each complex, each one starting with a random initial configuration, for a total of 100,000 models per complex. This set of models was divided into two ensembles of the same size to assess sampling convergence (data not shown).
Analysis of the model ensemble
For each ensemble, the 500 best-scoring models (i.e., the solutions) were grouped by k-means clustering50 based on the root-mean-square deviation (RMSD) between the Rrp6-Lrp1 and Ski7 subunits, after the superposition of the exo10 subunits. Two dominant clusters of similar structures were identified, with one cluster scoring better than the other. The precision of a cluster was calculated as the average RMSD with respect to the cluster center (i.e., the solution that had the minimal RMSD distance with respect to the others). The per-residue precision of a cluster (the root-mean-square fluctuation or RMSF, Supplementary Fig. 9) is calculated as the average RMSD distance of a residue in a solution with respect to the cluster center. The solutions of a cluster, superposed on the exo10 structure, were converted into the probability of any volume element being occupied by Rrp6, Ski7, Lrp1, and the GFP tag (localization density)44, 46.
Relative stoichiometry quantification of affinity captured exosome complexes by SYPRO Ruby stain
To quantify the relative stoichiometries of the complexes, SDS-PAGE protein gels were stained with SYPRO Ruby according to the manufacturer instructions (Life Technologies) and were visualized using a LAS-3000 system (linear detection range; Fujifilm). The intensities of the different protein bands on the gels were measured using ImageJ software (National Institutes of Health) and were normalized according to the predicted molecular weights of the quantifiable subunit proteins. The relative stoichiometries of the exosome components were calculated as relative to Rrp6-GFP or Ski7-GFP. Note that Mpp6 has the exact same molecular weight as Lrp1 (21 kD) but is much less abundant in the affinity capture complex (the ratio of normalized ion intensities -iBAQ ratio51, 52 of Mpp6/ Lrp1 is ~1/10, data not shown). We therefore did not consider Mpp6 in the stoichiometry determination.
Comparison of efficacies of three different anti-GFP affinity reagents for on-bead cross-linking
Equal amounts of Ilama polyclonal antibody (Poly, generated in-house), wild-type LaG-16, and lysine-less LaG-16-2K/R (2K/R) nanobodies were conjugated in parallel to epoxy magnetic beads. The antibody-conjugated magnetic beads were pre-treated with DSS cross-linker to block residual reactive amines and were then used for the affinity isolation of the APC/C complex. The isolated complex was on-bead cross-linked by 0.1 mM DSS for 25 mins and eluted by hot LDS buffer. The complex was subjected to SDS PAGE electrophoresis; gel regions corresponding to the GFP-tagged Cdc16 (~70–160 kD for non cross-linked Cdc16 and >350 kD for cross-linked Cdc16) were excised from the gel and digested by trypsin.
The purified peptides were analyzed by an Orbitrap Fusion mass spectrometer coupled online to the easy LC system (Thermo). For each analysis, peptides corresponding to ~20–40 ng of the purified complex were loaded onto an Easy-Spray column heated at 35 °C (C18, 3μm particle size, 200 Å pore size, and 50 μm × 15cm, Thermo Fisher) and eluted using a 25 min LC gradient (2% B to 7% B, 0–3 minutes, 7% B – 42% B, 3–21 minutes, 42% B–100% B, 21–25 minutes, where mobile phase A consisted 0.1% formic acid and mobile phase B consisted of 0.1% formic acid in acetonitrile). The flow rate was ~325 nl/min. The instrument was operated in the data-dependent mode, where the top eight-most abundant ions were fragmented by higher energy collisional dissociation (HCD) (normalized HCD energy 30) and analyzed in the Orbitrap mass analyzer. The target resolution for MS1 was 60,000 and 15,000 for MS2. Peptides (300–1500 m/z) with charge state of 2–5 were isolated by the ion trap (isolation window 1.8 Th) and detected by the orbitrap analyzer. The experiments were repeated three times and each repeated sample was measured three times to minimize the LC-MS analytical variations.
For label free quantification (LFQ), the raw data were searched against the yeast ORF database (01/05/2010) and quantified by MaxQuant (version 1.5)52. The search parameters include: initial MS accuracy <20 ppm, the main search MS accuracy tolerance < 4.5 ppm, cysteine carbamidomethyl as a fixed modification, protein N terminal acetylation and methionine oxidation as variable modifications. A maximum of two trypsin missed-cleavage sites was allowed. More than 30 unique peptides from Cdc16 were identified and used for the quantification. For LFQ, ion intensities of Cdc16 (which in essence represents the relative signals of the complex) from the cross-linked APC/C complex (isolated by three different anti-GFP affinity reagents) were then normalized by those of input signals; the resulting ratios were normalized to 2K/R and plotted in Supplementary Fig 3.
Structure PDB
PDBs# 4IFD, 2WP8 and 3Q8T are used in this study. Figs. 2, Supplementary Figs. 6, 15 (cross-links mapping to the crystal structure of the Beclin1 coil-coil domain) are prepared using UCSF Chimera53.
Annotated high-resolution crosslink MS/MS spectra are provided in the zip file (Supplementary Figs.16–18)
Supplementary Material
Acknowledgments
We are grateful to A.N. Krutchinsky for providing the yeast GFP-CDC16 strain, S. Obado for technical assistance, E. Jacobs, J. Lacava, SJ. Kim and B. Webb for discussions and assistance; Z. Yue (Icahn School of Medicine at Mount Sinai) for sharing Becn1-EGFP/+ and Becn1+/− mice. Y.S acknowledges M. Chen (R.G. Roeder lab, The Rockefeller University) for generating a reagent that was not used for this study. This work was funded by US NIH grants P41 GM103314 (B.T.C), R01 GM083960 (A.S.), U54 GM103511 (A.S., M.P.R., and B.T.C.) and Ellison Medical Foundation (Q.J.W.).
Footnotes
Code availability
The scripts and models are available at http://salilab.org/exosome. The modeling results are provided in the zip file (exosome_models).
Contributions
Y.S and B.T.C conceived the research, with input from M.P.R, J.F-M, R.P, Q.J.W, A.S and P.F. Y.S, J.F-M, P.F, and M.T cloned and purified the nanobodies. P.F performed the SPR measurements. Y.S and J.F-M carried out the microscopic imaging. Y.S performed biochemical and mass spectrometric analyses. R.P performed the modeling analyses. Q.W contributed reagents and discussed the results. P.F and Y.Y.L discovered the LaG-16 nanobody. Y.S and B.T.C wrote the paper with input from M.P.R, J.F-M, R.P, Q.J.W, A.S and P.F. All authors reviewed the manuscript.
References
- 1.Ward AB, Sali A, Wilson IA. Biochemistry. Integrative structural biology. Science. 2013;339:913–915. doi: 10.1126/science.1228565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walzthoeni T, Leitner A, Stengel F, Aebersold R. Mass spectrometry supported determination of protein complex structure. Curr Opin Struct Biol. 2013;23:252–260. doi: 10.1016/j.sbi.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 3.Erzberger JP, et al. Molecular architecture of the 40SeIF1eIF3 translation initiation complex. Cell. 2014;158:1123–1135. doi: 10.1016/j.cell.2014.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knutson BA, Luo J, Ranish J, Hahn S. Architecture of the Saccharomyces cerevisiae RNA polymerase I Core Factor complex. Nat Struct Mol Biol. 2014;21:810–816. doi: 10.1038/nsmb.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordiyenko Y, et al. eIF2B is a decameric guanine nucleotide exchange factor with a gamma2epsilon2 tetrameric core. Nat Commun. 2014;5:3902. doi: 10.1038/ncomms4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cevher MA, et al. Reconstitution of active human core Mediator complex reveals a critical role of the MED14 subunit. Nat Struct Mol Biol. 2014 doi: 10.1038/nsmb.2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lasker K, et al. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci U S A. 2012;109:1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi Y, et al. Structural Characterization by Cross-linking Reveals the Detailed Architecture of a Coatomer-related Heptameric Module from the Nuclear Pore Complex. Molecular & cellular proteomics: MCP. 2014;13:2927–2943. doi: 10.1074/mcp.M114.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fridy PC, et al. A robust pipeline for rapid production of versatile nanobody repertoires. Nature methods. 2014;11:1253–1260. doi: 10.1038/nmeth.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsien RY. The green fluorescent protein. Annual review of biochemistry. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 11.Huh WK, et al. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 12.Oeffinger M, et al. Comprehensive analysis of diverse ribonucleoprotein complexes. Nature methods. 2007;4:951–956. doi: 10.1038/nmeth1101. [DOI] [PubMed] [Google Scholar]
- 13.Muyldermans S. Nanobodies: natural single-domain antibodies. Annual review of biochemistry. 2013;82:775–797. doi: 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- 14.Kirchhofer A, et al. Modulation of protein properties in living cells using nanobodies. Nat Struct Mol Biol. 2010;17:133–138. doi: 10.1038/nsmb.1727. [DOI] [PubMed] [Google Scholar]
- 15.Russel D, et al. Putting the pieces together: integrative modeling platform software for structure determination of macromolecular assemblies. PLoS biology. 2012;10:e1001244. doi: 10.1371/journal.pbio.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makino DL, Baumgartner M, Conti E. Crystal structure of an RNA-bound 11-subunit eukaryotic exosome complex. Nature. 2013;495:70–75. doi: 10.1038/nature11870. [DOI] [PubMed] [Google Scholar]
- 17.Wasmuth EV, Januszyk K, Lima CD. Structure of an Rrp6-RNA exosome complex bound to poly(A) RNA. Nature. 2014;511:435–439. doi: 10.1038/nature13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Houseley J, LaCava J, Tollervey D. RNA-quality control by the exosome. Nat Rev Mol Cell Biol. 2006;7:529–539. doi: 10.1038/nrm1964. [DOI] [PubMed] [Google Scholar]
- 19.Feigenbutz M, Jones R, Besong TM, Harding SE, Mitchell P. Assembly of the yeast exoribonuclease Rrp6 with its associated cofactor Rrp47 occurs in the nucleus and is critical for the controlled expression of Rrp47. The Journal of biological chemistry. 2013;288:15959–15970. doi: 10.1074/jbc.M112.445759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonneau F, Basquin J, Ebert J, Lorentzen E, Conti E. The yeast exosome functions as a macromolecular cage to channel RNA substrates for degradation. Cell. 2009;139:547–559. doi: 10.1016/j.cell.2009.08.042. [DOI] [PubMed] [Google Scholar]
- 21.Liu JJ, et al. Visualization of distinct substrate-recruitment pathways in the yeast exosome by EM. Nat Struct Mol Biol. 2014;21:95–102. doi: 10.1038/nsmb.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schreiber A, et al. Structural basis for the subunit assembly of the anaphase-promoting complex. Nature. 2011;470:227–232. doi: 10.1038/nature09756. [DOI] [PubMed] [Google Scholar]
- 23.Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nature methods. 2014;11:319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- 24.Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644–656. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- 25.Chang L, Zhang Z, Yang J, McLaughlin SH, Barford D. Molecular architecture and mechanism of the anaphase-promoting complex. Nature. 2014;513:388–393. doi: 10.1038/nature13543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vidal RL, Matus S, Bargsted L, Hetz C. Targeting autophagy in neurodegenerative diseases. Trends in pharmacological sciences. 2014 doi: 10.1016/j.tips.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Li X, et al. Imperfect interface of Beclin1 coiled-coil domain regulates homodimer and heterodimer formation with Atg14L and UVRAG. Nat Commun. 2012;3:662. doi: 10.1038/ncomms1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller S, et al. Shaping development of autophagy inhibitors with the structure of the lipid kinase Vps34. Science. 2010;327:1638–1642. doi: 10.1126/science.1184429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baskaran S, et al. Architecture and dynamics of the autophagic phosphatidylinositol 3-kinase complex. eLife. 2014;3 doi: 10.7554/eLife.05115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong Y, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong Y, et al. Nrbf2 protein suppresses autophagy by modulating Atg14L protein-containing Beclin 1-Vps34 complex architecture and reducing intracellular phosphatidylinositol-3 phosphate levels. The Journal of biological chemistry. 2014;289:26021–26037. doi: 10.1074/jbc.M114.561134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–5372. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cristea IM, Chait BT. Conjugation of magnetic beads for immunopurification of protein complexes. Cold Spring Harbor protocols. 2011;2011:pdb prot5610. doi: 10.1101/pdb.prot5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng C, Krutchinsky AN. See & Catch method for studying protein complexes in yeast cells: a technique unifying fluorescence microscopy and mass spectrometry. Methods in molecular biology. 2014;1163:75–95. doi: 10.1007/978-1-4939-0799-1_6. [DOI] [PubMed] [Google Scholar]
- 35.Hakhverdyan Z, et al. Rapid, optimized interactomic screening. Nature methods. 2015;12:553–+. doi: 10.1038/nmeth.3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 37.Krogan NJ, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 38.Havugimana PC, et al. A census of human soluble protein complexes. Cell. 2012;150:1068–1081. doi: 10.1016/j.cell.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leitner A, et al. Expanding the chemical cross-linking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Molecular & cellular proteomics: MCP. 2012;11:M111 014126. doi: 10.1074/mcp.M111.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olsen JV, et al. Higher-energy C-trap dissociation for peptide modification analysis. Nature methods. 2007;4:709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 41.Yang B, et al. Identification of cross-linked peptides from complex samples. Nature methods. 2012;9:904–906. doi: 10.1038/nmeth.2099. [DOI] [PubMed] [Google Scholar]
- 42.Synowsky SA, van den Heuvel RH, Mohammed S, Pijnappel PW, Heck AJ. Probing genuine strong interactions and post-translational modifications in the heterogeneous yeast exosome protein complex. Molecular & cellular proteomics: MCP. 2006;5:1581–1592. doi: 10.1074/mcp.M600043-MCP200. [DOI] [PubMed] [Google Scholar]
- 43.Rieping W, Habeck M, Nilges M. Inferential structure determination. Science. 2005;309:303–306. doi: 10.1126/science.1110428. [DOI] [PubMed] [Google Scholar]
- 44.Alber F, et al. Determining the architectures of macromolecular assemblies. Nature. 2007;450:683–694. doi: 10.1038/nature06404. [DOI] [PubMed] [Google Scholar]
- 45.Alber F, et al. The molecular architecture of the nuclear pore complex. Nature. 2007;450:695–701. doi: 10.1038/nature06405. [DOI] [PubMed] [Google Scholar]
- 46.Fernandez-Martinez J, et al. Structure-function mapping of a heptameric module in the nuclear pore complex. The Journal of cell biology. 2012;196:419–434. doi: 10.1083/jcb.201109008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Algret R, et al. Molecular architecture and function of the SEA complex, a modulator of the TORC1 pathway. Molecular & cellular proteomics: MCP. 2014 doi: 10.1074/mcp.M114.039388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang F, Moss LG, Phillips GN., Jr The molecular structure of green fluorescent protein. Nature biotechnology. 1996;14:1246–1251. doi: 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]
- 49.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nature protocols. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MacQueen JB. Some Methods for Classification and Analysis of Multivariate Observations. Proceeding of the Fifth Berkeley Symposium on Mathematical Statistics and Probability. 1965:281–297. [Google Scholar]
- 51.Schwanhausser B, et al. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 52.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature biotechnology. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 53.Pettersen EF, et al. UCSF Chimera–a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.