

Abstract
Lung cancer is one of the most lethal forms of cancer and current chemotherapeutic strategies lack broad specificity and efficacy. Recently, β-lapachone (β-lap) was shown to be highly efficacious in killing non-small cell lung cancer (NSCLC) cells regardless of their p53, cell cycle and caspase status. Pre-clinical and clinical use of β-lap (clinical form, ARQ501 or 761) is hampered by poor pharmacokinetics and toxicity due to hemolytic anemia. Here, we report the development and preclinical evaluation of β-lap prodrug nanotherapeutics consisting of diester derivatives of β-lap encapsulated in biocompatible and biodegradable poly(ethylene glycol)-b-poly(d,l-lactic acid) (PEG-b-PLA) micelles. Compared to the parent drug, diester derivatives of β-lap showed higher drug loading densities inside PEG-b-PLA micelles. After esterase treatment, micelle-delivered β-lap-dC3 and -dC6 prodrugs were converted to β-lap. Cytotoxicity assays using A549 and H596 lung cancer cells showed that both micelle formulations maintained NAD(P)H:quinone oxidoreductase 1 (NQO1)-dependent cytotoxicity. However, antitumor efficacy study of β-lap-dC3 micelles against orthotopic A549 NSCLC xenograft-bearing mice showed significantly greater long-term survival over β-lap-dC6 micelles or β-lap-HPβCD complexes. Improved therapeutic efficacy of β-lap-dC3 micelles correlated with higher area under the concentration-time curves of β-lap in tumors, and enhanced pharmacodynamic endpoints (e.g., PARP1 hyperactivation, γH2AX, and ATP depletion). β-Lap-dC3 prodrug micelles provide a promising strategy for NQO1-targeted therapy of lung cancer with improved safety and antitumor efficacy.
Keywords: β-Lapachone, Prodrug therapy, Polymeric micelles, Non-small cell lung cancer, Cancer nanomedicine
1. Introduction
Lung cancer has the highest rate of mortality in both male and female populations in the US. Non-small cell lung cancer (NSCLC) accounts for 85% of lung cancer patients with a low survival rate of 15% after 5 years [1]. Conventional cytotoxic chemotherapy (e.g. Carbo-Taxol, a front line therapy using a combination of carboplatin and paclitaxel) causes significant patient morbidity and limited response in lung cancer patients. Novel NSCLC treatments that focus on identification of cancer-selective targets and development of target-specific therapies are needed. Successful examples include gefitinib, a small molecular kinase inhibitor that targets the cytosolic portion of epidermal growth factor receptor (EGFR) on the cancer cell surface. Despite reported clinical success, gefitinib is only effective in ∼30% of NSCLC patients. Furthermore, prolonged gefitinib treatment leads to drug resistant mutations in EGFR (e.g., T790M) and relapse. New therapeutic strategies that attack specific cancer targets with efficacy against a broad range of cancers, and that have a broad range of effects downstream from their targets to prevent resistance, are greatly needed.
β-Lapachone (β-lap) is a novel therapeutic agent that kills a broad spectrum of cancer cells through p53-, cell cycle-, and caspase-independent mechanisms. Its mechanism of action is dependent on expression of NAD(P)H:quinone oxidoreductase 1 (NQO1, a.k.a. DT-diaphorase, xip3, E.C.1.6.5.2), a two-electron oxidoreductase that typically detoxifies quinones after environmental exposures [2,3]. NQO1 is a homodimeric protein (MW: ∼60 kDa) whose expression is regulated by antioxidant and xenobiotic response elements [4,5]. In multiple tumor types with higher levels of reactive oxygen species (ROS), NQO1 is constitutively over-expressed at levels 5- to 100-fold greater than in associated normal tissues [6–8]. Research by our group and others have demonstrated up to 100-fold over-expression of NQO1 in ∼90% NSCLC and pancreatic cancers, and up to 10-fold over-expression in ∼60% of prostate [8] and breast cancers [9]. Attempts to exploit NQO1 in the past included using mitomycin C, E09 or streptonigrin, where NQO1 converts these agents to DNA alkylating agents in a one-step, two-electron reduction reaction. Their efficacies are, thereby, restricted to DNA alkylation-mediated damage, killing in cell cycle-dependent mechanisms [10,11]. In addition, they are less effective against cancer cells that have lost tumor suppressor (e.g., p53) function, and are subject to major resistance mechanisms (e.g. loss of caspase) [12].
Unlike all other quinone drugs, β-lap undergoes a futile redox cycle resulting in rapid production of reactive oxygen species (ROS), specifically catalyzed by NQO1 [13]. For every mole of β-lap, >60 mol of NAD(P)H is consumed and >120 mol of H2O2 is generated in ∼2 min [13,14]. Elevated cytoplasmic H2O2 causes DNA single strand breaks (SSBs), hyperactivation of poly(ADP-ribose) polymerase-1 (PARP-1), loss of NAD+ and ATP pools, and ultimately a unique pattern of cell death referred to as “programmed necrosis” (Fig. 1) [15]. Cell death occurs specifically in cancer cells overexpressing NQO1, while normal cells and tissue with low endogenous levels of the enzyme are spared. While β-lap is a promising agent from a mechanistic standpoint, its clinical use is hampered by low water solubility (0.038 mg/mL), poor pharmacokinetics and methemoglobinemia [16,17]. The use of hydroxypropyl β-cyclodextrin (HPβCD) to formulate β-lap (ARQ501) increased the drug's solubility by ∼400-fold [18]. However, rapid drug clearance from the blood (t1/2, β = 24 min), hemolysis due to the HPβCD carrier and β-lap-induced methemoglobinemia [19] were noted, limiting its success as a therapeutic agent in clinical trials.
Fig. 1.
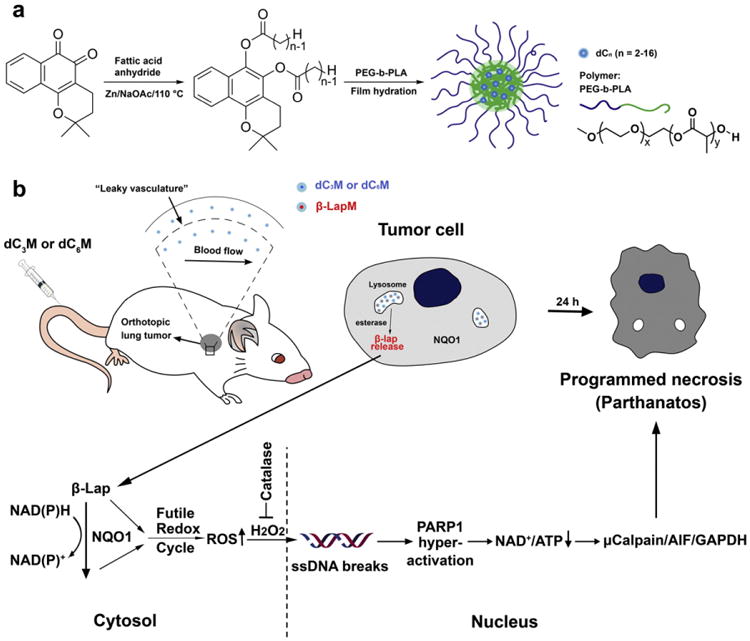
β-Lap prodrug micelles for lung cancer-targeted therapy. (a) Syntheses of diester derivatives (β-lap-dCn, n = 2–16) of β-lap and production of β-lap prodrug micelles by a film hydration method. (b) β-Lap-dC3 and β-lap-dC6 micelles were selected for NSCLC therapy, wherein micelle carriers provide prolonged circulation and enhanced accumulation in tumor tissue. Esterase converts the prodrugs into parent drug (i.e., β-lap), which induces programmed necrosis. In the cytosol of cancer cells,1 equivalent (e.q.)of released β-lap undergoes a futile redox cycle to produce ∼120 e.q. H2O2, in 2–5 min (depending on cancer cell), which results in DNA damage, PARP1 hyperactivation, NAD+/ATP depletion and ultimately programmed necrosis.
We previously reported the development of polymeric micelles for the delivery of β-lap [20], however, low drug loading efficiencies and problems with scale-up prevented their clinical development. Polymeric micelles are nanosized (∼10–200 nm) supramolecular constructs composed of amphiphilic block-copolymers. Hydrophobic cores of the micelles provide a natural carrier environment for hydrophobic drugs, and the hydrophilic outer shell prevents particle aggregation and opsonization [21,22]. Prior studies showed that poly(ethylene glycol)-b-poly(d,l-lactic acid) (PEG-b-PLA) micelles exhibited prolonged blood circulation times of β-lap, increased tumor accumulation due to the enhanced permeability and retention (EPR) effect [23,24], and improved safety and antitumor efficacy over ARQ501 (i.e., β-lap-HPβCD) [19]. However, low drug loading density (2.2 wt.%) caused by crystallization of β-lap (yellow needle crystals appearing in attempts to scale-up) presented a major limitation in achieving higher drug loading content and stable β-lap nanotherapeutics. To overcome this challenge, we generated a novel prodrug micelle strategy using diester derivatives of β-lap: β-lap-dC3 and β-lap-dC6 (hereafter referred to as dC3 and dC6, respectively). We demonstrate that dC3 and dC6 micelles (dC3M and dC6M, respectively) have greatly improved drug loading content (∼10%) and efficiency (>95%) in PEG-b-PLA micelles. This strategy allows easy scale-up formulations with high apparent drug solubility (>7 mg/mL), physical stability, and the ability for reconstitution after lyophilization [25].
In this study, we report the preclinical evaluation of two β-lap prodrug micelle formulations (i.e., dC3M and dC6M) in an orthotopic lung cancer model in comparison to the current clinical form of β-lap, β-lap-HPβCD complex. While cell culture studies in vitro showed similar drug potency (i.e., LD50) and NQO1 specificity for both micelle formulations, studies using tumor-bearing mice in vivo demonstrated a significantly improved long-term survival for dC3M over dC6M or β-lap-HPβCD. The improved antitumor response was supported by drug pharmacokinetics in tumor tissues and pharmacodynamic end point assays that strongly suggest that the antitumor responses noted were NQO1-specific causing NAD+-keresis cell death in vivo [26].
2. Materials and methods
2.1. Materials
All chemicals were purchased from Sigma-Aldrich or TCI America and used as received. Organic solvents (analytical grade), phosphate buffered saline (PBS, pH 7.4) and normal saline were purchased from Fisher Scientific Inc. H596 and A549 non-small cell lung carcinoma (NSCLC) cells were grown in DMEM with 10% fetal bovine serum, 2 mM l-glutamine, 100 units/mL penicillin, and 100 mg/mL streptomycin at 37 °C in a humidified incubator with a 5% CO2–95% air atmosphere. A549 cells were infected with a lentivirus construct that contained the luciferase gene with a cytomegalovirus promoter. All cells were routinely found free of mycoplasma infection. All animal procedures adhered to NIH guidelines, following approved protocols by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center at Dallas.
2.2. Synthesis of β-lap prodrugs and PEG-b-PLA block copolymer
β-Lap prodrugs and poly(ethylene glycol)-block-poly(d,l-lactic acid) (PEG-b-PLA) block copolymer (Mn = 10 kD, the PEG and PLA segments were 5 kD) were synthesized following previously reported procedures [25,27]. Briefly for dC3, β-Lap (242 mg, 1 mmol), zinc powder (320 mg, 4.9 mmol), 40 mg sodium acetate (0.49 mmol), and 1 mL anhydrous propionic anhydride were mixed and stirred at 110 °C for 1 h. After reaction, the mixture was cooled to room temperature, filtered, and washed with 10 mL ethyl acetate. The filtrate was distilled under reduced pressure to remove propionic anhydride and ethyl acetate. The residue was dissolved in 20 mL CH2Cl2 and washed with water. The organic extract was dried over sodium sulfate and concentrated. The residue was recrystallized from isopropanol. PEG-b-PLA was synthesized by ring opening polymerization of d,l-lactide at 110 °C. Poly(ethylene glycol) monoethyl ether (Mn = 5 kD) was used as a macro-initiator. d,l-lactide was added as a monomer and Stannous (II) octoate (Sn(Oct)2) was added as a catalyst. After reacting for 4 h, the mixture was allowed to cool down to room temperature. PEG-b-PLA was purified by redissolving in THF and precipitating in hexane 3 times. After synthesis, the copolymer was characterized by 1H and 13C NMR and gel permeation chromatography using THF as an eluent.
2.3. Production of β-lap prodrug micelles by film hydration
β-Lap prodrug (dC3 and dC6) micelles were prepared by a film hydration method following a published protocol [25]. Briefly, dC3 or dC6 (10 mg) and PEG-b-PLA (90 mg) were first dissolved in acetonitrile (1 mL) and the solvent evaporated in vacuum using a rotary evaporator to form a solid thin film. Normal saline was added to the film at 60 °C and vortexed for 5 min. The resulting micelle solution was stored at 4 °C for 1 h and filtered through a 0.45 μm nylon filter to remove non-encapsulated drug aggregates. Micelle solutions were then lyophilized and the resulting freeze-dried powder was accurately weighed, dissolved in a mixture of methanol and deionized water (v/v = 9/1), and analyzed using a Shimadzu UV-1800 UV–Vis spectrophotometer (λ = 240 nm, extinction coefficient = 2.0 × 104 M−1 cm−1) to calculate the total amount of micelle encapsulated prodrug. Nanoparticles were also characterized by dynamic light scattering (DLS) and transmission electron microscopy (TEM) to examine particle size and morphology.
2.4. Conversion of β-lap prodrug micelles in vitro
Based on different absorption properties of β-lap and β-lap prodrugs, we used UV–Vis spectroscopy to measure prodrug conversions. In a typical procedure, a stock solution of prodrug micelles in water (10 μg/mL) was added in 1 mL PBS buffer (pH 7.4) in a quartz cuvette. Porcine liver esterase (PLE) was added at a concentration of 1 unit/mL (1 U/mL). Solutions were then incubated at 37 °C and absorbance spectra measured using UV–Vis spectrophotometer over time. Eqs. (1)–(3) were used to calculate the percentage of prodrug conversion to β-lap:
| (1) |
| (2) |
| (3) |
where A1 and A2 were absorbance at 240 and 257 nm, respectively; ε1 and ε2 are extinction coefficients of dC3 and β-lap at 240 nm ε1 = 2.0 × 104 M−1 cm−1, ε2 = 9.0 × 103 M−1 cm−1, respectively); ε3 and ε4 are extinction coefficients of dC3 and β-lap at 257 nm (ε3 = 1.1 × 103 M−1 cm−1, ε4 = 1.9 × 104 M −1 cm−1, respectively); L was the path length (1 cm); c1 and c2 were concentrations of dC3 and β-lap, respectively. c0 is the initial molar concentration of dC3
2.5. In vitro drug release of β-lap from prodrug micelles
β-Lap release from dC3M and dC6M was measured using a centrifugation method. In a typical procedure, dC3M solution (50 mL, 10 μg/mL) and porcine liver esterase (PLE, 10 U/mL) was incubated at 37 °C under shaking. At different time points, 1 mL solution was removed and placed inside a centrifuge tube and centrifuged at 12,000 rpm for 2 min. The released β-lap was determined by measuring the UV–Vis absorbance of the supernatant based on the standard curve of β-Lap. Percentage of drug release was plotted as a function of time to show the drug release kinetics.
2.6. NQO1 enzyme assay of converted β-lap
β-Lap, β-lap prodrug micelles with or without PLE were examined using an NADH (400 mM) recycling assay as catalyzed by recombinant NQO1 (1.5 μg). NADH oxidation to NAD+ was monitored by absorbance (λmax = 340 nm) and data recorded at 2 s intervals for 20 min. NADH oxidation rates of prodrugs with or without PLE were compared with β-lap.
2.7. Cytotoxicity evaluation of β-lap prodrug micelles in vitro
DNA assays were performed in NQO1 + A549, as well as in genetically matched NQO1 — vs NQO1 + H596 NSCLC cells as described [28]. Briefly, NSCLC cells were seeded (10,000 cells/well) into each lane of 48-well plates, and 12 h later cells were exposed to various doses of free β-lap (dissolved in DMSO) or prodrug micelles with or without PLE (10 units) for 2 h. For A549 cells, dicoumarol (50 μM, a fairly specific inhibitor of NQO1) was added to block NQO1 activity. After 2 h exposures, media were replaced with fresh growth media and cells were allowed to grow for an additional 7 days [29]. DNA content was determined by Hoescht dye 33258, using a modified method of Labarca and Paigen [30] and assessed in a Perkin-Elmer HTS 7000 BioAssay Reader (Waltham, MA). Data were expressed as means ± SE for treated/control (T/C) values from six wells per treatment.
2.8. Alkaline comet assays
DNA lesions, including DNA single and double strand breaks (SSBs, DSBs, respectively), as well as DNA base damage, were assessed in single cells treated with dC3M and dC6M (6 μM) with or without PLE using alkaline comet assays as previously described [28,31]. Slides were stained with SYBR-green and visualized using a Nikon Eclipse TE2000-E fluorescence microscope (Melville, NY). Digital photomicrographs were taken and comet tail lengths quantified using the NIH Image J software.
2.9. Hemolytic assays
Red blood cells (RBCs) from female NOD-SCID mice (∼20 g) were extracted, separated from mouse plasma and incubated 1:20 (v:v) with varying concentrations of either β-lap-HPβCD, dC3 micelles, or dC6 micelles. Samples were then incubated (37 °C, 1 h), centrifuged (10,000 rpm, 1 h), and supernatants analyzed for hemoglobin (Hb) released via UV-Vis (λmax: 408 nm). For the micelle samples, 10 U/mL PLE (comparable to esterase level in mouse plasma) was added. The percentage of hemolysis was calculated based on Hb present in 0.2% Triton X-100 (complete hemolysis) samples. A sample consisting of RBCs incubated with normal saline was included as a negative control. All experiments were conducted in triplicate. Percent (%) hemolysis was plotted as a function of β-lap or prodrug (converted to β-lap) concentrations. Formation of methemoglobin was assessed by the peak at 628 nm that is characteristic of the oxidized hemoglobin.
2.10. Examination of β-lap-HPβCD and prodrug micelle toxicity in NOD-SCID mice
In attempts to evaluate the safety of dC3M and dC6M in comparison to a previously established β-lap-HPβCD formulation, we performed dose escalation studies as previously described [19,32]. Morbidity and mortality responses were recorded in mice (Supplemental Table 1) injected with five i.v. administrations of varying doses of β-lap-HPβCD or prodrug micelles every other day, and responses examined immediately following injection.
2.11. Evaluation of antitumor efficacies of dC3M or dC6M
Antitumor efficacy studies of dC3M and dC6M compared to β-lap-HPβCD were performed in 6- to 8-week-old female tumor-bearing NOD-SCID mice (18–20 g each). Firefly luciferase gene-transfected A549 NSCLC cells (1.2 × 106) were injected by tail vein into mice that were randomized into groups (n = 10/group), monitored fortumor formation in the lungs using bioluminescence (BLI) following treatment with dC3 or dC6 micelles, β-lap-HPβCD or control blank micelles. An average of 1.0 × 107 total photons was used before any therapies were given as described [32]. Animals were treated with dC3 micelle (30, 50 and 70 mg/kg e.q. dose of β-lap), dC6M(70 mg/kg e.q. dose of β-lap), blank micelles or β-lap-HPβCD (ARQ501, 25 mg/kg), administered via tail vein and repeated five times every other day over a 10 day period. Tumor-bearing mice were imaged using BLI to estimate relative tumor volumes as described [19,32] using d-luciferin (2.5 mg, subcutaneously injected). Relative tumor volume estimates were derived from BLI images of mice captured using a Xenogen Vivovision IVIS Lumina Imager (60 s exposure time). Animals were monitored daily for survival and morbidity. Mortality was recorded over time as a result of cancer-related deaths or drug-related toxicities (≥20% body weight loss relative to the start of therapy). Overall toxicities in separate organs were assessed by tissue immunohistology as previously described [19,32].
2.12. Pharmacokinetic (PK) analyses of dC3M and dC6M
Pharmacokinetic studies were performed in 6 to 8-week-old female tumor-bearing NOD-SCID mice (15–20 g each). The A549 orthotopic lung cancer model was also used in this study. The dC3M (50 mg/kg e.q. dose of β-lap), dC6M (50 mg/kg e.q. dose of β-lap), or β-lap (25 mg/kg) were first injected into mice via the tail vein. Animals were sacrificed at different time points (from 1 min to 24 h). Organs were harvested, weighed and snap frozen. Tumor tissues were homogenized in acetonitrile. PBS was added to each sample which was then spun for 5 min at 16,100 × g at 4 °C. The supernatant was processed by passage over a solid phase extraction column primed by the addition of 2 mL acetonitrile followed by 2 mL of water. The column was washed twice with 2 mL of 20% acetonitrile/80% H2O, and compound was eluted by addition of 2 mL of acetonitrile. The eluent was concentrated and all liquid evaporated in a speed-vac. The sample was resuspended in 200 μL 100% acetonitrile with 0.1% formic acid and 10 ng internal standard (tolbutamide) and then analyzed by LC–MS/MS using an Applied Biosystems/MDS Sciex 4000-QTRAP coupled to a Shimadzu Prominence LC. For standards, 100 μL of tumor tissue homogenate was mixed with 300 μL of acetonitrile, vortexed, and then spiked with 2 μL of varying concentrations of compound. β-Lap was detected by following the precursor to fragment ion 243.1 to 187.2. An Agilent Zorbax XDB-C18 column (50 × 4.6 mm, 5 μM packing) was used for chromatography. Areas under the concentration time curve (AUCs) were calculated for 0–2 h using the noncompartmental analysis tool of WinNonLin (Pharsight).
2.13. Pharmacodynamic (PD) analyses
Samples for PD analyses were obtained from the same animals for the PK study. Normal lung and tumor tissues were collected and immediately snap-frozen in liquid nitrogen. Protein extracts of combined tissue from 3 mice were separated by SDS-PAGE, transferred onto PVDF membranes, and probed with antibodies against γH2AX (05-636, 1:1000, Millipore), PARP-PAR (4335-mc-100, 1:1000, Trevigen) or α-tubulin (T6199,1:5000, Sigma). Proteins of interest were detected with HRP-conjugated goat α-mouse IgG antibody (sc-2031, 1: 5000, Santa Cruz Biotechnology) and visualized by ECL Western immunoblot-ting (Pierce, Thermo Scientific, Rockford, IL).
2.14. Statistical analysis
Student's t-tests were used to determine statistical significance from experiments repeated at least 3 independent times. P values were reported by asterisks as indicated and considered significant when P< 0.05.
3. Results
3.1. Physical properties of dC3 and dC6 micelles
Diester derivatives of β-lap were synthesized with varying alkyl chain lengths as reported [25]. The dC2 prodrug was still easily crystallized from micelles in aqueous solution and was not used in this study. We chose dC3 and dC6 for further development to investigate the effect of chain length on micelle formulation and antitumor efficacy. The dC3M and dC6M nanoparticles were formed by a film hydration method [25] using a biocompatible and biodegradable PEG-b-PLA polymer at a theoretical loading density of 10 wt.%. Experimental loading contents were 9.7 ± 0.4 and 10.0 ± 0.3 wt.% for dC3 and dC6 prodrugs, respectively, demonstrating high drug loading efficiency (>95%, Fig. 2a, b). These values were significantly higher than β-lap micelles (β-lapM, 2.2 wt.% loading content) as previously reported [19], which indicates improved compatibility of the prodrug molecules with the PEG-b-PLA copolymer (or carrier). After micelle production, dynamic light scattering (DLS) was used to measure the hydrodynamic diameters (Dh) of dC3M and dC6M. The dC3M had a Dh value of 35 ± 4.0 nm, while dC6M was 42 ± 13 nm (Fig. 2c). Transmission electron microscopy (TEM) showed that both micelles hada spherical morphology and furthermore, dC6M had a larger size and size variation over dC3M (Fig. 2d and e). The zeta potentials of dC3M and dC6M were −0.40 and −0.76 mV, respectively. Both prodrug micelles achieved high apparent drug solubility (7 mg/mL, > 180-fold increase over free β-lap in water), were stable at 4 °C for over two days, and can be reconstituted after lyophilization using polyethylene glycol (Mn = 2000) as a lyoprotectant. Compared to β-lapM, the prodrug micelles are superior in meeting the formulation criteria for subsequent biological studies.
Fig. 2.
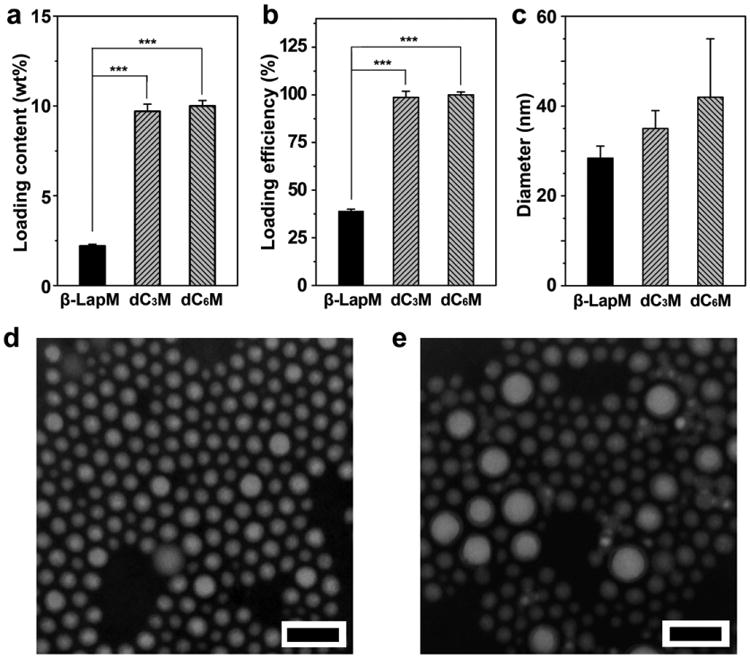
Characterization of dC3 and dC6 micelles. (a) Drugloading content and (b) drug loading efficiency of dC3M and dC6M were compared to β-LapM. PEG-b-PLA copolymer was used for drug encapsulation. (c) Hydrodynamic diameters of dC3M and dC6M were measured by DLS. TEM images of dC3M (d) or dC6M (e) show spherical morphology of both prodrug micelles (scale bars = 100 nm in both images). ***, p ≤ 0.001.
3.2. Esterase conversion of prodrugs and release of β-lap from micelles
Many forms of esterase are present in the body and play important roles in the metabolism of lipids and breakdown of organic molecules [33]. Several reports indicated tumors of the lung, colon and liver had high esterase expressions and could be exploited for selective drug conversion using an ester prodrug strategy [34,35]. For our in vitro studies, we chose porcine liver esterase (PLE) as a model enzyme to test the feasibility of conversion of micelle-delivered prodrugs [36]. At 1 U/mL PLE, the rate of prodrug conversion was much faster for micelle-delivered dC3 than dC6 (t1/2, c = 30 ± 1 vs 135 ± 10 min, respectively, Fig. 3a). Without PLE, both micelles were stable without detectable prodrug conversion in the PBS buffer (pH = 7.4) over 7 days (data not shown). After prodrug conversion, the release of parent drug (i.e. β-lap) was measured, where 50% of β-lap was released (t1/2,r) from dC3M and dC6M at 7.9 ± 0.5 and >48 h, respectively. The slower release kinetics of β-lap from dC6M may be a result of slower prodrug conversion, as well as the more hydrophobic core environment created by the unconverted dC6 prodrugs.
Fig. 3.
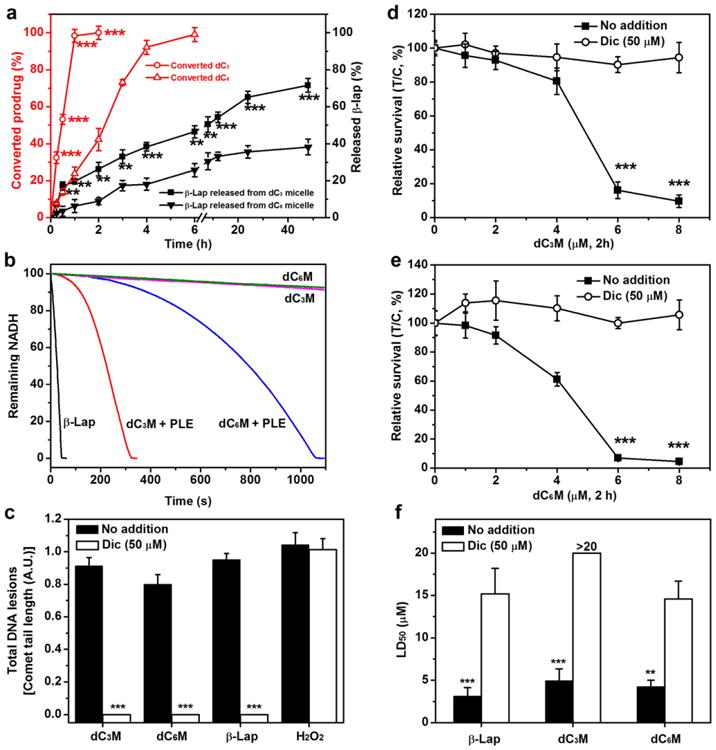
Prodrug conversion and NQO1-dependent DNA damage and lethality. (a) Comparison of dC3M and dC6M conversion and release kinetics versus β-lap (n =3) from polymeric micelles in the presence of PLE (1U/mL). Prodrug conversion occurred in the first 4h before release from micelles. dC3M or dC6M concentrations were at 0.1 mg/mL. (b) The NADH consumption rates of dC3M and dC6M catalyzed by NQO1 using recycling assays. (c) DNA lesion assessments in endogenously overexpressed NQO1+A549 NSCLC cells. A549 cells were exposed to dC3M (6 μM, 2 h), dC6M (6 μM, 2 h), β-lap-HPβCD (ARQ501, 6 μM,2 h) or H2O2 (500 μM, 15 min). After 2 h,DNA lesions were assessed by alkaline comet assays as described (35). (d–e) Relative survival assays of A549 NSCLC cells exposed to dC3M or dC6M at the indicated doses for 2 h (p ≤ 0.001). (f) LD50 values of dC3M and dC6M in A549 NSCLC cancer cells in the presence of PLE with or without dicoumarol. Dic, dicoumarol (50 μM, 2 h).
3.3. Micelle-delivered prodrugs kill in an NQO1-dependent manner
We next examined the NQO1 target specificity of prodrug micelles, with or without esterase activation. Fig. 3b shows the results from NQO1 recycling assays, where NADH consumption was recorded after prodrug micelles were added to a solution containing 1.5 μg recombinant NQO1 and 400 mM NADH as described [13]. In the presence of 10 U/mL PLE, it took 300 and 1000 s to consume 90% of NADH for dC3M and dC6M, respectively. In contrast, in the absence of PLE, <5% of NADH was consumed within the same time-span with either prodrug micelles. Cells exposed to dC3M or dC6M in the presence of PLE also induced DNA damage (DNA single strand breaks) in an NQO1-dependent manner, as measured by alkaline comet assays (Supplementary Fig. S1). A549 cells that endogenously overexpress NQO1, showed extensive comet tail formation in response to treatment with dC3M or dC6M in the presence of 10 U/ml PLE. Co-incubation with dicoumarol, a specific NQO1 inhibitor, blocked the DNA damage (Fig. 3c).
To investigate whether dC3 or dC6 retained NQO1-specific cytotoxicityin NSCLC cells in vitro, we measured survival of A549 and NQO1+ vs NQO1– H596 lung cancer cells using a 7-day DNA survival assay. Original H596 cells contain a homozygous *2 NQO1 polymorphism and lack NQO1 expression. Genetically matched NQO1+ H596 cells with unaltered growth rates were previously generated to test NQO1 specific lethality responses to β-lap [13]. In these experiments, prodrugs alone were directly dissolved in DMSO and DMSO content in the medium was less than 0.1%. Data show dC3 prodrug has NQO1-dependent toxicity to both A549 and H596 cancer cells and the cytotoxicity was considerably increased in the presence of 10 U/mL PLE (Supplementary Figs. S2–3). For dC6 prodrug, we did not observe any NQO1 dependent toxicity in the two cell lines even co-administered with 10 U/mL PLE. The lack of cytotoxicity from dC6 prodrug only may be due to its low water solubility and precipitation from medium as a result of the two long hydrophobic hexyl chains.
Fig. 3d, e shows the relative survival (%T/C) of A549 cells treated with dC3M and dC6M at different drug doses, respectively. After 2 h incubation with dC3M in the absence of PLE, little cytotoxicity was noted in A549 cells at 6 μM (> 80% survival), with or without dicoumarol (Supplementary Fig. S4). In contrast, when 10 U/mL PLE was added to the cell culture medium, a significant increase in NQO1-dependent cytotoxicity was detected. Over 90% A549 cells were killed by dC3M at 6 μM, and this toxicity was blocked by 50 μM dicoumarol (>90% survival). Similarly, dC3M treatments resulted in PLE-dependent, NQO1-mediated dose–response lethality in genetically matched NQO1+ but not NQO1 — H596 cells (Supplementary Fig. S5b), further confirming that dC3M can be activated by PLE and converted to free β-lap, causing cell death by the NQO1-specific mechanism as β-lap. For dC6M, the same pattern of NQO1-dependent cytotoxicity was also observed for NQO1-overexpressing NSCLC cells (Fig. 3e and Supplementary Fig. S5c). A comparison of LD50 values showed that both dC3M and dC6M in the presence of PLE had similar LD50 values as β-lapM for either NQO1+ A549 or H596 lung cancer cells (Fig. 3f and Supplementary Fig. 5d), and that NQO1 — H596 cells were not responsive to any of these drugs.
3.4. Safety and antitumor efficacy of dC3M and dC6M
Prior clinical trial data suggested that improved β-lap formulations are necessary to reduce hemolytic anemia, the dose-limiting toxicity of β-lap-HPβCD [17,37]. We first investigated the percentage of hemolysis of dC3M or dC6M in comparison with β-lap-HPβCD. In samples co-incubated with 0.2% Triton X-100 treatment, two hemoglobin (Hb) peaks were noted at 541 and 576 nm (Supplementary Fig. S6a). The characteristic peak at 576 nm was also apparent in samples with β-lap-HPβCD-induced hemolysis. Data show that β-lap-HPβCD indeed caused hemolysis, with concentrations at 1.5 and 2.0 mg/mL β-lap resulting in 51 ± 3% and 55 ± 1% hemolysis, respectively (Supplementary Fig. S6b). In contrast, dC3M or dC6M samples did not show significant hemolysis (5.5 ± 0.4 and 5.2 ± 1.1%, respectively) at 2.0 mg/mL. In samples treated with β-lap-HPβCD, a new peak was observed at 628 nm that was absent in samples treated with Triton X-100, dC3M and dC6M. The peak at 628 nm is indicative of conversion of the ferrous (Fe2+) ions of Hb to ferric (Fe3+) ions of methemoglobin [19].
To investigate the safety of dC3M or dC6M, we first analyzed the morbidity and mortality responses in healthy NOD-SCID mice at different doses (Supplementary Table S1). Five injections were administered intravenously every other day. For β-lap-HPβCD, a dose of 25 mg/kg caused severe muscle contractions, labored breathing and an irregular gait in mice, symptoms consistent with methemoglobinemia [19]. No weight loss was noted in the following week for the recovered mice. In contrast, the exposure of mice to dC3M at doses ranging from 30 to 70 mg/kg (converted to β-lap equivalence) did not show severe side-effects, with no significant weight loss and no lethality noted. At 100 mg/kg of dC3M, we observed significant loss of animals (80% death). In contrast, for dC6M, altered breathing and mild morbidity were observed at 100 mg/kg, with no ensuing animal deaths or weight loss. Based on these data, we estimated the maximum tolerated doses (MTDs) for β-lap-HPβCD, dC3M and dC6M to be 20, 70 and 100 mg/kg, respectively.
The antitumor efficacy studies of dC3M or dC6M were performed in female NOD-SCID mice bearing orthotopic fire-fly luciferase-transfected A549 tumors. Six days after i.v. inoculation of A549-luc cells (1.2 × 106), mice were randomly divided into different groups and treated every other day for 5 injections with dC3M or dC6M (30, 50, and 70 mg/kg e.q. dose to β-lap), β-lap-HPβCD (25 mg/kg) or blank micelles. Changes in tumor volumes (monitored by BLI intensity) in the control group showed rapid tumor growth at day 35 (Fig. 4a and Supplementary Fig. S7). Quantitative analyses of BLI intensities (Fig. 4b) showed that treatment with dC3M resulted in obvious tumor suppression, especially at higher doses of dC3M (50 and 70 mg/kg). In contrast, dC6M showed only minor antitumor effects even at 70 mg/kg (Fig. 4c). To compare the antitumor efficacies of the two different prodrug micelles, we performed long-term survival assessments at the same dose of 70 mg/kg and graphed all data on Kaplan–Meier survival curves with appropriate statistical comparison. Data show that 50% of control animals treated with blank micelles died at day 88, and treatment of mice with dC6M did not show any significantly increased survival benefit (Fig. 4d). In contrast, treatment of orthotopic A549 tumor-bearing mice with dC3M showed significantly increased antitumor efficacy (Fig. 4e) at all doses tested. At 30 mg/kg dC3M, the average 50% survival time was 108 days, statistically similar to the average survival of mice exposed to β-lap-HPβCD (ARQ501, 25mg/kg) at 104 days. The medium survival times for tumor-bearing mice treated with 50 or 70 mg/kg dC3M were significantly longer at 115 and 139 days, respectively. It is worth noting that 20% of the 70 mg/kg dC3M-treated mice were still alive (‘apparently cured’) even after 250 days (not graphed) and remained disease free at the time of reporting these data. Kaplan-Meier survival curves indicated a statistically significant survival advantage of 70 mg/kg dC3M over blank micelle carrier alone (p ≤ 0.0004) or β-lap-HPβCD (p ≤ 0.008) (Fig. 4e).
Fig. 4.
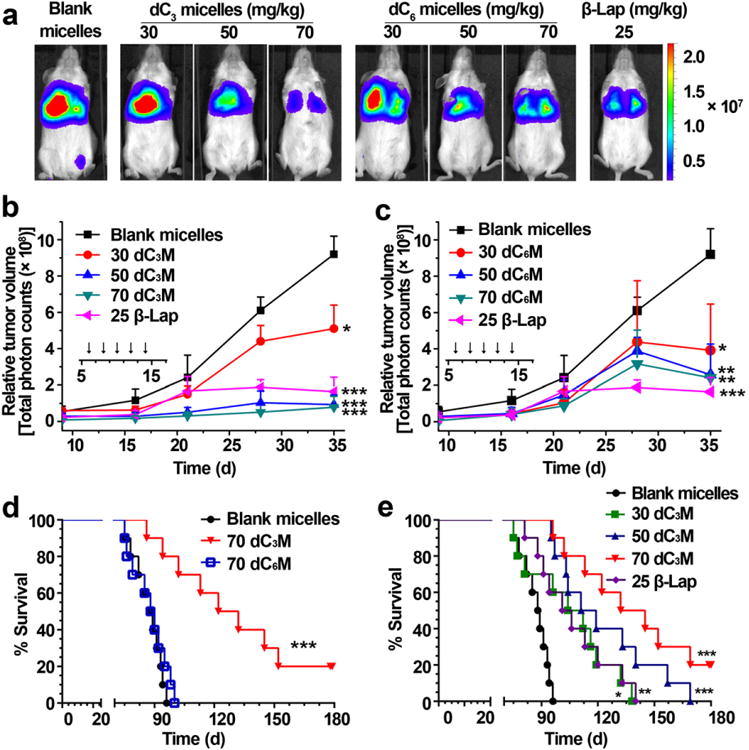
Antitumor efficacy of dC3M and dC6M against orthotopic A549 tumors in female NOD-SCID mice. (a) BLIs of anesthetized mice after treatment with either blank micelles (vehicle alone), β-lap-HPβCD, dC3M or dC6M at day 35. Day 0 was designated as the day after injection of A549 cancer cells. (b, c) Quantitation of luciferase levels in mice after dC3M (b) or dC6M (c) treatment as a function of time. Time of treatment is indicated by a solid bar in the insets. (d) Kaplan–Meier survival curve of female NOD-SCID mice (n = 10) bearing orthotopic A549 NSCLC xenografts after blank micelle, dC3M (70 mg/kg) or dC6M (70 mg/kg) treatments. (e) Kaplan–Meier survival curve of female NOD-SCID mice (n = 10) bearing orthotropic A549 NSCLC xenografts after blank micelles, β-lap-HPβCD (25 mg/kg), or dC3M treatment (30, 50 and 70 mg/kg). *, p ≤ 0.05; **, p ≤ 0.01; *** p ≤ 0.001.
3.5. PK and PD analyses of tumor and associated normal lung tissues
The pharmacokinetics (PK) of β-lap in tumors and normal tissues from β-lap-HPβCD and β-lap prodrug micelles (dC3M or dC6M) were examined in mice bearing orthotopic A549 NSCLC tumors. β-Lap prodrug and converted β-lap concentration-time curves in tumor tissues are shown in Supplementary Fig. S8. At 2 min after a single dose of dC3M injection (50 mg/kg of β-lap), β-lap concentration reached the maximum (1.25 ± 0.04 × 103 μM) and dC3 prodrug (0.21 ±0.12 μM) was almost completely converted to β-lap. In comparison, β-lap from dC6M converted slowly and the maximum concentration was only 201 ± 65 μM at 2 min after administration, while dC6 prodrug concentration was high (443 ±131 μM) at this time. Calculation of areas under the concentration time curves (AUCs) showed that significantly more β-lap accumulated in the tumor tissues of mice treated with dC3M compared to β-lap-HPβCD or dC6M in the first 2 h after a single dose of drug administration (7.3 ± 0.61 vs 1.4 ± 0.16 vs 0.85 ± 0.10 × 106 ng*min/g, respectively, Fig. 5a). For direct comparison purpose, dC3M and dC6M were given as the same 50 mg/kg e.q. dose of β-lap while β-lap-HPβCD was at 25 mg/kg (MTD).
Fig.5.
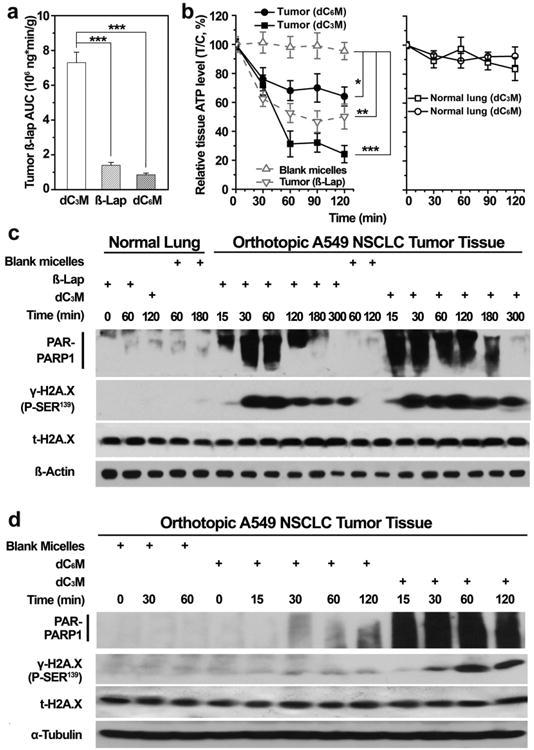
In vivo PK and PD study of dC3M and dC6M. (a) β-Lap AUC in tumors of mice treated with 1 dose of dC3M (50 mg/kg), dC6M(50mg/kg) and β-lap-HPβCD (25 mg/kg). β-Lap concentrations were measured by LC/MS/MS. (b) PAR-PARP1 formation, a consequence of PARP1 hyperactivation, results in NAD+ loss and consequentially, ATP depletion at the indicated times (min) in dC3M-treated mice bearing A549 tumor and surrounding normal lung tissues. (c, d) PAR formation confirmed PARP1 hyperactivation and the subsequent programmed necrosis mechanism of cell death in the lung tumor tissue of dC3M- or dC6M-treated NQO1+ A549 NSCLC tumors. Note the lack of PAR formation, ATP loss and DNA damage (γH2AX formation) responses in associated normal lung tissue from tumor-bearing mice. Mice treated with dC6M (70 mg/kg) showed significantly less antitumor responses. *, p ≤ 0.05; **, p ≤ 0.01. ***, p ≤ 0.001.
Consistent with AUC values, higher antitumor efficacy was noted (e.g., tumor regression as monitored by BLI) in mice treated with dC3M compared to dC6M (Fig. 4b, c). Blank micelle injections did not alter the growth of orthotopic tumors. Moreover, we evaluated downstream pharmacodynamic (PD) biomarkers of tumor tissues compared to associated normal lung as early indicators for drug response. Clear evidence of PARP-1 hyperactivation by dC3M or β-lap-HPβCD in tumor tissue was noted by the formation of PAR-formed PARP1 (Fig. 5c). The dC3M treatment induced significant PARP-1 hyperactivation as early as 15 min and lasted for 180 min. β-Lap-HPβCD caused less PARP-1 hyperactivation and stopped at 120 min. As a negative control, blank micelle in tumors and β-Lap in normal tissue didn't induce any PARP-1 formation. In contrast, dC6M stimulated negligible PARP-1 formation in 120 min (Fig. 5d) and adjacent normal tissue showed no PARP-1 hyperactivation (Fig. 5c and d). The ATP loss of tumor tissue treated with dC3M or β-lap-HPβCD were coincident with their PARP1 formation. The ATP level of tumor tissue treated with β-lap-HPβCD reached a minimum at 90 min and showed some recovery at 120 min, while dC3M was still effective at this time (Fig. 5b).
4. Discussion
A major limitation of current formulations of β-lap in Phase I clinical trials (i.e., ARQ761) is methemoglobinemia (MH), limiting the antitumor efficacy of this otherwise NQO1-targeted antitumor agent. Both ARQ501 and ARQ761, HPβCD formulations of β-lap or reduced β-lap, respectively, showed adequate drug solubility that enabled clinical testing, however, dose-limiting hemolytic anemia hampered their clinical potential [38]. Micellar delivery of β-lap effectively reduced hemolysis and methemoglobinemia [19]. Unfortunately, the low drug loading content (∼2.2 wt.%) prevented further development of a β-lap micelle as a clinically translatable formulation. Based on this prior research, we proposed a prodrug micelle strategy to improve micelle loading efficiency and safety of β-lap, with the objective of increasing the therapeutic window. Prior research from our lab [39] reported that modification of the β-lap's α-keto group with aryl imines led to pH-sensitive prodrugs. Although these prodrugs can be adequately loaded inside polymeric micelles (>50% loading efficiency), they are not stable at physiological pH due to hydrolytic degradation (data not shown) and therefore were not pursued in the current study.
Prodrugs have been widely used in pharmaceutical industry to improve the physicochemical and biopharmaceutical properties of parent drugs. They can undergo enzymatic or chemical transformations to convert to the active parent drug. Among these, ester groups are most commonly used to improve lipophilicity and membrane permeability of drugs containing carboxylate or phosphate groups [40,41]. Ester groups are readily hydrolyzed via many types of esterases to convert inactive prodrugs into active drugs in the body. To reduce crystallization and increase compatibility with micelles, β-lap prodrugs with carbonic ester side-chains for the 1,2-ortho keto positions of β-lap were synthesized and successfully loaded into PEG-b-PLA micelles [25]. Among these prodrug micelles, we choose dC3 and dC6 micelles for in vitro and in vivo correlation studies. Compared with the β-lap micelles, dC3 and dC6 micelles had higher drug loading content (∼10 wt.%, 5-fold over β-lap micelles) and higher loading efficiencies (>95%, p ≤ 0.001) (Fig. 2b).
Our data show that dC3 and dC6 prodrugs were readily converted to β-lap inside the micelles by PLE [42], however, the dC6 micelles had slower release kinetics of β-lap from micelles than dC3 micelles (Fig. 3a). In cell culture experiments, both dC3 and dC6 micelles induced programmed necrosis similar to β-lap in the presence of PLE. NQO1+ NSCLC cells were selectively killed by PLE-treated dC3 and dC6 micelles, whereas genetically matched NQO1 — H596, or dicoumarol-exposed A549 cells, were resistant. Comet assays highlighted the NQO1-dependent DNA damage and cytotoxicity of prodrug micelles, and confirmed NQO1-induced redox-mediated futile cycling of prodrug micelles (Fig. 3c and Supplementary Fig. S1).
While the MTD results showed that dC3 and dC6 micelles were 3.5-to >5-fold safer in vivo than β-lap (i.e., 70 and >100 mg/kg, respectively, vs 20 mg/kg β-lap-HPβCD), respectively, only dC3 prodrug micelles showed efficacious antitumor activity with appropriate pharmacodynamic endpoints. At MTD doses of prodrug micelles, mice showed less severe reactions than what are typically noted with β-lap-HPβCD, such as labored breathing, irregular gait or muscle contractions noted as a result of hemolysis and MH (Supplementary Table 1). Instead, mice exhibited minor side-effects 30 min after intravenous injections of dC3 or dC6 prodrug micelles. In comparison, mice exposed to β-lap-HPβCD had severe reactions immediately after treatment that dissipated 30-45 min after injection and were not associated with weight loss or toxicities to normal tissues [9,19,32]. UV–Vis spectroscopy analyses of mouse red blood cells found only negligible hemolysis or MH by dC3 micelles compared to treatment with β-lap-HPβCD at the same concentration of 2 mg/mL β-lap (Supplementary Fig. S6b, <5% vs >50%, respectively). In addition to hemolysis, β-lap-HPβCD also caused MH, where it reacts with hemoglobin, causing iron oxidation (Fe2+ to Fe3+, 628 nm, Supplementary Fig. S6a) [43]. In contrast, the exposure of mouse red blood cells to dC3 or dC6 micelles did not show observable MH. Despite comparable NQO1-dependent cytotoxicities to NSCLC cell lines in vitro, dC3M and dC6M showed different antitumor efficacies in vivo at 70 mg/kg (Fig. 4d). To achieve optimal antitumor efficacy, the prodrugs must be converted to β-lap effectively in tumor tissue to kill NQO1+ cancer cells. PD endpoint assay showed only dC3 micelles persistently stimulated ROS-induced PARP1 hyperactivation (Fig. 5c). In contrast, treatment of animals with dC6 micelles failed to cause high level of PARP1 hyperactivation as monitored by PAR-PARP1 formation and NAD+/ATP depletion. Tumor pharmacokinetic studies show that micelle-delivered dC3 prodrugs were efficiently converted to β-lap in the tumors as early as the first 2 min after administration, leading to high β-lap tumor AUC (7.3 ± 0.61 × 106 ng*min/g, Fig. 5a, Supplementary Fig. S8). In contrast, a majority of micelle-delivered dC6 prodrugs were still present in the first 30 min, leading to significantly smaller β-lap tumor AUC value (0.85 ± 0.10 × 106 ng*min/g). As our PK analyses cannot distinguish between micelle encapsulated β-lap and free β-lap, the AUC value for bioavailable β-lap from dC6 micelles may be further decreased if β-lap release is retarded as shown by in vitro drug release studies from the dC6 micelles (Fig. 3a). Overall, the current study found excellent agreement between tumor pharmacokinetic data (i.e., AUC) with pharmacodynamic endpoints for NAD+-keresis (e.g., PARP1 hyperactivation) and long-term survival outcome (i.e., Kaplan-Meier curves) for different drug formulations. These results also illustrate the value of pharmacodynamic endpoints as early biomarkers to predict antitumor response in drug delivery systems where pharmacokinetic analysis may not be able to differentiate bio-available drugs from encapsulated drugs.
5. Conclusions
In summary, we report the preclinical evaluation of β-lap prodrug nanotherapeutics for the treatment of NSCLC cancers that over-express NQO1. Both dC3 and dC6 prodrugs achieved high drug loading densities and efficiencies (>95%) with significantly reduced hemolysis and methemoglobinemia that currently limits ARQ761 formulations. The dC3 prodrug micelles showed excellent antitumor efficacy in treating orthotopic NSCLC tumors that overexpress NQO1, with target validation in pharmacodynamic endpoints. The advantages of high loading content, ease of scale-up, low toxicity and broader therapeutic window, suggest that dC3 prodrug micelles are feasible for clinical evaluation. Synergistic approaches with ionizing radiation [44], paclitaxel [45,46] and gemcitabine [47] are also under development in our labs.
Supplementary Material
Acknowledgments
This work is supported by grants from the Cancer Prevention Research Institute of Texas(RP120897) to JG and DAB, and the National Institutes of Health (5 R01 CA102792) to DAB and JG.
Footnotes
Appendix A. Supplementary data: Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jconrel.2014.12.027.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013, CA Cancer. J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Ross D, Siegel D. NAD(P)H:quinone oxidoreductase 1 (NQO1, DT-diaphorase), functions and pharmacogenetics. Methods Enzymol. 2004;382:115–144. doi: 10.1016/S0076-6879(04)82008-1. [DOI] [PubMed] [Google Scholar]
- 3.Jaiswal AK. Human NAD(P)H:quinone oxidoreductase (NQO1) gene structure and induction by dioxin. Biochemistry (Washington) 1991;30:10647–10653. doi: 10.1021/bi00108a007. [DOI] [PubMed] [Google Scholar]
- 4.Boothman DA, Meyers M, Fukunaga N, Lee SW. Isolation of X-ray-inducible transcripts from radioresistant human melanoma cells. Proc Natl Acad Sci U S A. 1993;90:7200–7204. doi: 10.1073/pnas.90.15.7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ross D, Kepa JK, Winski SL, Beall HD, Anwar A, Siegel D. NAD(P)H:quinone oxidoreductase 1 (NQO1): chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem Biol Interact. 2000;129:77–97. doi: 10.1016/s0009-2797(00)00199-x. [DOI] [PubMed] [Google Scholar]
- 6.Marin A, Lopez de Cerain A, Hamilton E, Lewis AD, Martinez-Penuela JM, Idoate MA, Bello J. DT-diaphorase and cytochrome B5 reductase in human lung and breast tumours. Br J Cancer. 1997;76:923–929. doi: 10.1038/bjc.1997.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belinsky M, Jaiswal AK. NAD(P)H:quinone oxidoreductase1 (DT-diaphorase) expression in normal and tumor tissues. Cancer Metastasis Rev. 1993;12:103–117. doi: 10.1007/BF00689804. [DOI] [PubMed] [Google Scholar]
- 8.Ough M, Lewis A, Bey EA, Gao J, Ritchie JM, Bornmann W, Boothman DA, Oberley LW, Cullen JJ. Efficacy of beta-lapachone in pancreatic cancer treatment: exploiting the novel, therapeutic target NQO1. Cancer Biol Ther. 2005;4:102–109. doi: 10.4161/cbt.4.1.1382. [DOI] [PubMed] [Google Scholar]
- 9.Dong Y, Bey EA, Li LS, Kabbani W, Yan J, Xie XJ, Hsieh JT, Gao J, Boothman DA. Prostate cancer radiosensitization through poly(ADP-Ribose) polymerase-1 hyperactivation. Cancer Res. 2010;70:8088–8096. doi: 10.1158/0008-5472.CAN-10-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prakash AS, Beall H, Ross D, Gibson NW. Sequence-selective alkylation and cross-linking induced by mitomycin C upon activation by DT-diaphorase. Biochemistry. 1993;32:5518–5525. doi: 10.1021/bi00072a005. [DOI] [PubMed] [Google Scholar]
- 11.Siegel D, Gibson NW, Preusch PC, Ross D. Metabolism of mitomycin C by DT-diaphorase: role in mitomycin C-induced DNA damage and cytotoxicity in human colon carcinoma cells. Cancer Res. 1990;50:7483–7489. [PubMed] [Google Scholar]
- 12.Rekha GK, Sladek NE. Multienzyme-mediated stable and transient multidrug resistance and collateral sensitivity induced by xenobiotics. Cancer Chemother Pharmacol. 1997;40:215–224. doi: 10.1007/s002800050649. [DOI] [PubMed] [Google Scholar]
- 13.Pink JJ, Planchon SM, Tagliarino C, Varnes ME, Siegel D, Boothman DA. NAD(P) H:quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J Biol Chem. 2000;275:5416–5424. doi: 10.1074/jbc.275.8.5416. [DOI] [PubMed] [Google Scholar]
- 14.Bey EA, Reinicke KE, Srougi MC, Varnes M, Anderson VE, Pink JJ, Li LS, Patel M, Cao L, Moore Z, Rommel A, Boatman M, Lewis C, Euhus DM, Bornmann WG, Buchsbaum DJ, Spitz DR, Gao J, Boothman DA. Catalase abrogates β-lapachone-induced PARP1 hyperactivation-directed programmed necrosis in NQO1-positive breast cancers. Mol Cancer Ther. 2013;12:2110–2120. doi: 10.1158/1535-7163.MCT-12-0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bey EA, Bentle MS, Reinicke KE, Dong Y, Yang CR, Girard L, Minna JD, Bornmann WG, Gao J, Boothman DA. An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proc Natl Acad Sci U S A. 2007;104:11832–11837. doi: 10.1073/pnas.0702176104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khong HT, Dreisbach L, Kindler HL, Trent DF, Jeziorski KG, Bonderenko I, Popiela T, Yagovane DM, Dombal G. A phase 2 study of ARQ 501 in combination with gemcitabine in adult patients with treatment naive, unresectable pancreatic adenocarcinoma. J Clin Oncol (Meeting Abstracts) 2007;25:15017. [Google Scholar]
- 17.Hartner L, Rosen L, Hensley M, Mendelson D, Staddon A, Chow W, Kovalyov O, Ruka W, Skladowski K, Jagiello-Gruszfeld A, Byakhov M. Phase 2 dose multi-center, open-label study of ARQ 501, a checkpoint activator, in adult patients with persistent, recurrent or metastatic leiomyosarcoma (LMS) J Clin Oncol (Meeting Abstracts) 2007;25:20521. [Google Scholar]
- 18.Nasongkla N, Wiedmann A, Bruening A, Beman M, Ray D, Bornmann W, Boothman D, Gao J. Enhancement of solubility and bioavailability of β-lapachone using cyclodextrin inclusion complexes. Pharm Res. 2003;20:1626–1633. doi: 10.1023/a:1026143519395. [DOI] [PubMed] [Google Scholar]
- 19.Blanco E, Bey EA, Khemtong C, Yang SG, Setti-Guthi J, Chen H, Kessinger CW, Carnevale KA, Bornmann WG, Boothman DA, Gao J. β-Lapachone micellar nanotherapeutics for non-small cell lung cancer therapy. Cancer Res. 2010;70:3896–3904. doi: 10.1158/0008-5472.CAN-09-3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanco E, Bey EA, Dong Y, Weinberg BD, Sutton DM, Boothman DA, Gao J. β-Lapachone-containing PEG–PLA polymer micelles as novel nanotherapeutics against NQO1-overexpressing tumor cells. J Control Release. 2007;122:365–374. doi: 10.1016/j.jconrel.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Torchilin VP. Structure and design of polymeric surfactant-based drug delivery systems. J Control Release. 2001;73:137–172. doi: 10.1016/s0168-3659(01)00299-1. [DOI] [PubMed] [Google Scholar]
- 22.Gref R, Minamitake Y, Peracchia M, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263:1600–1603. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 23.Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156:1363–1380. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzym Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 25.Ma X, Huang X, Huang G, Li L, Wang Y, Luo X, Boothman DA, Gao J. Prodrug strategy to achieve lyophilizable, high drug loading micelle formulations through diester derivatives of β-lapachone. Adv Healthc Mater. 2014;3:1210–1216. doi: 10.1002/adhm.201300590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore Z, Chakrabarti G, Lou X, Ali A, Deberadinis R, Brekken R, Boothman DA. NAMPT inhibition sensitizes pancreatic adenocarcinoma cells to tumor-selective, PAR-independent metabolic catastrophe and cell death induced by β-lapachone. Cell Death Dis. 2015 doi: 10.1038/cddis.2014.564. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Planchon SM, Wuerzberger S, Frydman B, Witiak DT, Hutson P, Church DR, Wilding G, Boothman DA. β-Lapachone-mediated apoptosis inhuman promyelocytic leukemia (HL-60) and human prostate cancer cells: A p53-independent response. Cancer Res. 1995;55:3706–3711. [PMC free article] [PubMed] [Google Scholar]
- 28.Bentle MS, Reinicke KE, Bey EA, Spitz DR, Boothman DA. Calcium-dependent modulation of poly(ADP-ribose) polymerase-1 alters cellular metabolism and DNA repair. J Biol Chem. 2006;281:33684–33696. doi: 10.1074/jbc.M603678200. [DOI] [PubMed] [Google Scholar]
- 29.Pink JJ, Planchon SM, Tagliarino C, Wuerzberger-Davis SM, Varnes ME, Siegel D, Boothman DA. NAD(P)H:quinone oxidoreductase (NQO1) activity is the principal determinant of β-lapachone cytotoxicity. J Biol Chem. 2000;275:5416–5424. doi: 10.1074/jbc.275.8.5416. [DOI] [PubMed] [Google Scholar]
- 30.Labarca C, Paigen K. A simple, rapid, and sensitive DNA assay procedure. Anal Biochem. 1980;102:344–352. doi: 10.1016/0003-2697(80)90165-7. [DOI] [PubMed] [Google Scholar]
- 31.Olive PL, Banáth JP, Durand RE. Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the “comet” assay. Radiat Res. 1990;122:86–94. [PubMed] [Google Scholar]
- 32.Huang X, Dong Y, Bey EA, Kilgore JA, Bair JS, Li LS, Patel M, Parkinson EI, Wang Y, Williams NS, Gao J, Hergenrother PJ, Boothman DA. An NQO1 substrate with potent antitumor activity that selectively kills by PARP1-induced programmednecrosis. Cancer Res. 2012;72:3038–3047. doi: 10.1158/0008-5472.CAN-11-3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ross MK, Crow JA. Human carboxylesterases and their role in xenobiotic and endobiotic metabolism. J Biochem Mol Toxicol. 2007;21:187–196. doi: 10.1002/jbt.20178. [DOI] [PubMed] [Google Scholar]
- 34.Xu G, Zhang W, Ma MK, McLeod HL. Human carboxylesterase 2 is commonly expressed in tumor tissue and is correlated with activation of irinotecan. Clin Cancer Res. 2002;8:2605–2611. [PubMed] [Google Scholar]
- 35.Lu X, Howard MD, Talbert DR, Rinehart JJ, Potter PM, Jay M, Leggas M. Nano- particles containing anti-inflammatory agents as chemotherapy adjuvants II: role of plasma esterases in drug release. AAPS J. 2009;11:120–122. doi: 10.1208/s12248-009-9086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen Y, Jin E, Zhang B, Murphy CJ, Sui M, Zhao J, Wang J, Tang J, Fan M, Van Kirk E, Murdoch WJ. Prodrugs forming high drug loading multifunctional nanocapsules for intracellular cancer drug delivery. J Am Chem Soc. 2010;132:4259–4265. doi: 10.1021/ja909475m. [DOI] [PubMed] [Google Scholar]
- 37.Kawecki A, Adkins DR, Cunningham CC, Vokes E, Yagovane DM, Dombal G, Koralewski P, Hotko Y, Vladimirov V. A phase II study of ARQ501 in patients with advanced squamous cell carcinoma of the head and neck. J Clin Oncol (Meeting Abstracts) 2007;25:16509. [Google Scholar]
- 38.Hartner PL RL, Hensley M, Mendelson D, Staddon AP, Chow W, Kovalyov O, Ruka W, Skladowski K, Jagiello-Gruszfeld A, Byakhov M. Phase 2 dose multi-center, open-label study of ARQ 501, a checkpoint activator, in adult patients with persistent, recurrent or metastatic leiomyosarcoma (LMS) J Clin Oncol. 2007;25 [Google Scholar]
- 39.Reinicke KE, Bey EA, Bentle MS, Pink JJ, Ingalls ST, Hoppel CL, Misico RI, Arzac GM, Burton G, Bornmann WG, Sutton D, Gao J, Boothman DA. Development of β-lapachone prodrugs for therapy against human cancer cells with elevated NAD(P)H:quinone oxidoreductase 1 levels. Clin Cancer Res. 2005;11:3055–3064. doi: 10.1158/1078-0432.CCR-04-2185. [DOI] [PubMed] [Google Scholar]
- 40.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T, Savolainen J. Prodrugs: design and clinical applications. Nat Rev Drug Discov. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 41.Müller CE. Prodrug approaches for enhancing the bioavailability of drugs with low solubility. Chem Biodivers. 2009;6:2071–2083. doi: 10.1002/cbdv.200900114. [DOI] [PubMed] [Google Scholar]
- 42.Samarajeewa S, Shrestha R, Li Y, Wooley KL. Degradability of poly(lactic acid)-containing nanoparticles: enzymatic access through a cross-linked shell barrier. J Am Chem Soc. 2012;134:1235–1242. doi: 10.1021/ja2095602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cenas N, Ollinger K. Redox conversions of methemoglobin during redox cycling of quinones and aromatic nitrocompounds. Arch Biochem Biophys. 1994;315:170–176. doi: 10.1006/abbi.1994.1486. [DOI] [PubMed] [Google Scholar]
- 44.Boothman DA, Trask DK, Pardee AB. Inhibition of potentially lethal DNA damage repair in human tumor cells by β-lapachone, an activator of topoisomerase I. Cancer Res. 1989;49:605–612. [PubMed] [Google Scholar]
- 45.Li CJ, Li YZ, Pinto AV, Pardee AB. Potent inhibition of tumor survival in vivo by β-lapachone plus taxol: combining drugs imposes different artificial checkpoints. Proc Natl Acad Sci U S A. 1999;96:13369–13374. doi: 10.1073/pnas.96.23.13369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.D'Anneo A, Augello G, Santulli A, Giuliano M, di Fiore R, Messina C, Tesoriere G, Vento R. Paclitaxel and beta-lapachone synergistically induce apoptosis in human retinoblastoma Y79 cells by downregulating the levels of phospho-Akt. J Cell Physiol. 2010;222:433–443. doi: 10.1002/jcp.21983. [DOI] [PubMed] [Google Scholar]
- 47.Kim H, Rigell C, Zhai G, Lee SK, Samuel S, Martin A, Umphrey H, Stockard C, Beasley TM, Buchsbaum D, Li L, Boothman D, Zinn K. Antagonistic effects of anti-EMMPRIN antibody when combined with chemotherapy against hypovascular pancreatic cancers. Mol Imaging Biol. 2014;16:85–94. doi: 10.1007/s11307-013-0665-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.