

High-resolution profiling of the genomic and transcriptomic landscapes of visceral cancers using the DNA and RNA of tumor shed exosomes has the potential to be used as a clinical tool for cancer diagnosis, therapeutic stratification and treatment monitoring, precluding the need for direct tumor sampling.
Keywords: liquid biopsy, exosomes, pancreatic cancer, next-generation sequencing
Abstract
Background
The ability to perform comprehensive profiling of cancers at high resolution is essential for precision medicine. Liquid biopsies using shed exosomes provide high-quality nucleic acids to obtain molecular characterization, which may be especially useful for visceral cancers that are not amenable to routine biopsies.
Patients and methods
We isolated shed exosomes in biofluids from three patients with pancreaticobiliary cancers (two pancreatic, one ampullary). We performed comprehensive profiling of exoDNA and exoRNA by whole genome, exome and transcriptome sequencing using the Illumina HiSeq 2500 sequencer. We assessed the feasibility of calling copy number events, detecting mutational signatures and identifying potentially actionable mutations in exoDNA sequencing data, as well as expressed point mutations and gene fusions in exoRNA sequencing data.
Results
Whole-exome sequencing resulted in 95%–99% of the target regions covered at a mean depth of 133–490×. Genome-wide copy number profiles, and high estimates of tumor fractions (ranging from 56% to 82%), suggest robust representation of the tumor DNA within the shed exosomal compartment. Multiple actionable mutations, including alterations in NOTCH1 and BRCA2, were found in patient exoDNA samples. Further, RNA sequencing of shed exosomes identified the presence of expressed fusion genes, representing an avenue for elucidation of tumor neoantigens.
Conclusions
We have demonstrated high-resolution profiling of the genomic and transcriptomic landscapes of visceral cancers. A wide range of cancer-derived biomarkers could be detected within the nucleic acid cargo of shed exosomes, including copy number profiles, point mutations, insertions, deletions, gene fusions and mutational signatures. Liquid biopsies using shed exosomes has the potential to be used as a clinical tool for cancer diagnosis, therapeutic stratification and treatment monitoring, precluding the need for direct tumor sampling.
introduction
For many visceral cancers, such as pancreatic ductal adenocarcinoma (PDAC), the availability of tissue-based companion diagnostics may be limited or precluded secondary to clinical factors such as tumor location, amount of tumor tissue-sampled or procedure-associated risk, hindering the progress of precision medicine [1]. Relatively noninvasive liquid biopsies offer a promising alternative for tumor characterization and disease monitoring. To this end, several investigators have identified tumor-specific genetic mutations in patient plasma-derived circulating cell-free DNA (cfDNA) including activating mutations in KRAS, BRAF, epidermal growth factor receptor (EGFR) and other cancer genes using highly sensitive targeted approaches such as digital PCR and targeted amplicon sequencing on cfDNA [2,3,4]. Recently, whole-genome and exome sequencing have been performed using the cfDNA of plasma samples in an effort to estimate tumor copy number profiles and identify actionable mutations in a more agnostic manner [5–7]. However, the extensively fragmented nature of cfDNA in circulation makes it difficult for this format to become generalizable in the context of genomic characterization of tumors through current next-generation sequencing (NGS) platforms [8]. This limitation is even more profound in the context of circulating RNAs, where profiling is essentially restricted to small microRNAs, due to extensive fragmentation of coding transcripts [9–11].
Exosomes are 40–150 nanometer-sized membrane-bound extracellular vesicles that arise by specific endosomal biogenesis pathways [12]. Functionally, exosomes have been shown to influence the tumor microenvironment as vehicles for cell–cell communication in cancer, harboring a diverse repertoire of molecular cargo that are shielded from degradation in circulation and that are representative of their originating cells [12–14]. Therefore, the quality, diversity and tumor-specific nature of exosomal DNA (exoDNA), and exoRNA provide a potentially favorable alternative compared with cell-free nucleic acids for comprehensive tumor profiling at high resolution. Indeed, recent publications have shown that exosomes contain genomic representations of high molecular weight (HMW; >10 kb), double-stranded fragments of DNA [15, 16].
We sought to assess the feasibility and potential clinical utility of characterizing the entire genomic and transcriptomic profiles of visceral cancers using the nucleic acid cargo within shed exosomes obtained from a single specimen of patient biofluid. We show, for the first time, that it is possible to perform integrative profiling of tumors from shed exosomes by analyzing the DNA and RNA cargo using standard NGS platforms, and that this approach has the potential to circumvent the need for direct tumor sampling in visceral cancers.
materials and methods
Three patients with pancreaticobiliary cancers were included in our study (supplementary Table S1, available at Annals of Oncology online), and each was consented following institutional review board approval (PA15-014). Case LBx01 is a 57-year-old man who initially presented with stage IIA PDAC. Fifteen months after surgical metastatectomy, the patient developed evidence of pleural effusion, and therapeutic thoracentesis yielded 800 ml of pleural fluid from which exosomes were isolated using a serial ultracentrifugation protocol (supplementary Figure S1, available at Annals of Oncology online) and downstream whole genome, exome and RNA sequencing using an Illumina HiSeq 2500 were performed for tumor profiling. Case LBx02 is a 68-year-old woman with PDAC primary and hepatic metastases. Thirty milliliter of whole blood was collected via blood draw before initiation of chemotherapy and exosomes were isolated for tumor profiling. Case LBx03 is a 74-year-old man who underwent an upfront pancreaticoduodenectomy for an ampullary mass. Final pathology confirmed a stage IIB pancreatobiliary type adenocarcinoma of the ampulla. Thirty milliliter of peripheral whole blood was collected and plasma exosomes were isolated for tumor profiling. See supplementary Methods, available at Annals of Oncology online, for our liquid biopsy workflow and sequencing analysis details.
results
plasma and pleural effusion exosome isolations are enriched with high molecular weight double-stranded genomic DNA
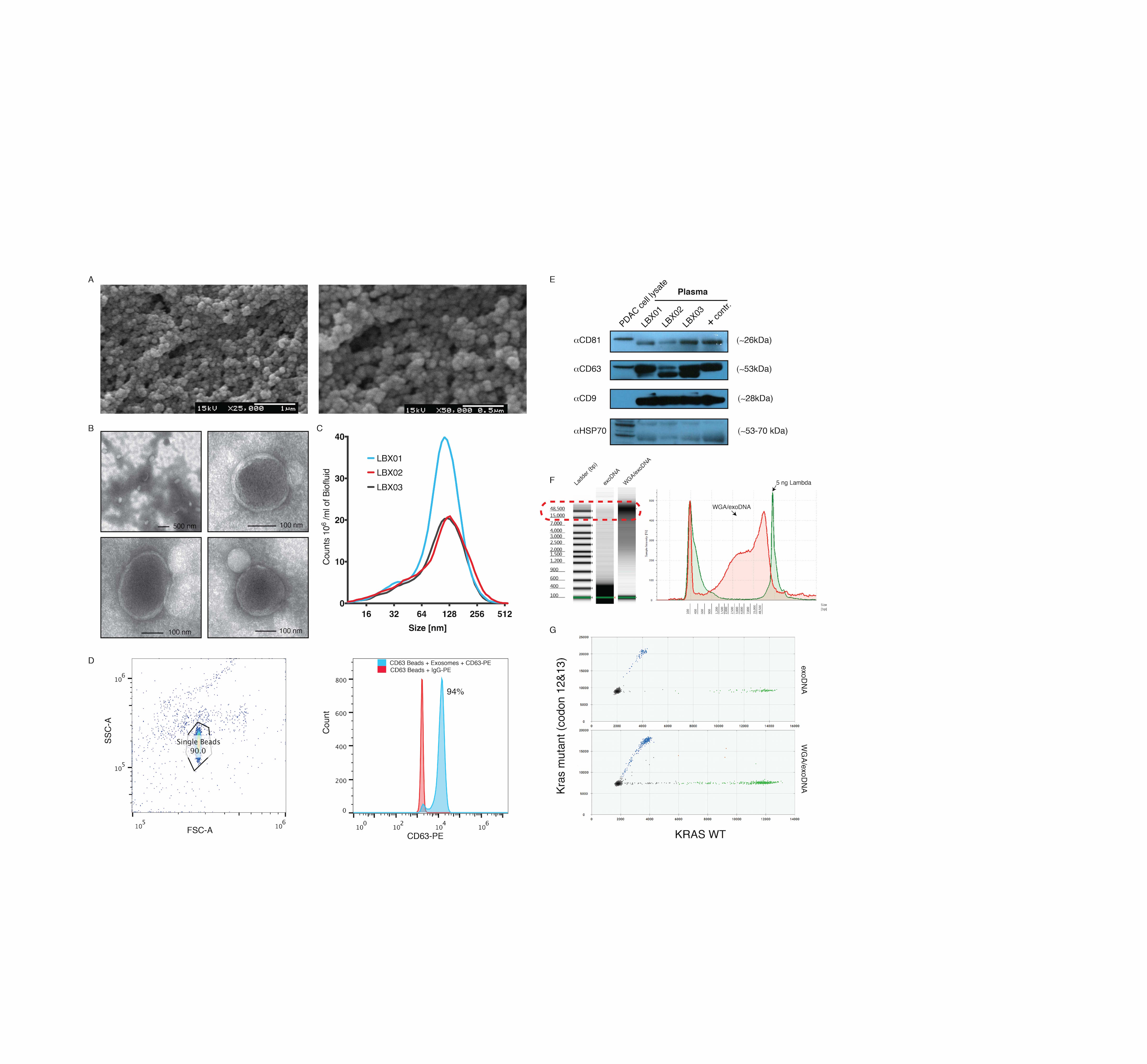
Shed exosome populations were confirmed by scanning electron microscopy and transmission electron microscopy (supplementary Figure S3A and B, available at Annals of Oncology online). Nanoparticle-tracking analysis (Particle Metrix, Inc.) confirmed the presence of exosome-sized vesicles in the liquid biopsy of all three patients (supplementary Figure S2C, available at Annals of Oncology online). Expression of canonical exosome surface markers, including the tetraspanin CD63 by flow cytometry and CD9, CD63, CD81 and HSP70 by western blots (supplementary Figure S2D and E, available at Annals of Oncology online), also established the presence of exosomes in our isolations. Extraction of exoDNA revealed quantifiable HMW double-stranded DNA (>10 kb in size) as seen in supplementary Figure S2F, available at Annals of Oncology online.
exosomes contain a large fraction of tumor DNA
KRAS mutant allele frequency (MAF) was determined using the KRAS multiplex screening assay and droplet digital PCR platform (ddPCR, BioRad Technologies, see supplementary Methods, available at Annals of Oncology online) demonstrating tumor presence in our exosome isolations (supplementary Figure S2G, available at Annals of Oncology online). PCR-based analysis of mutant KRAS and BRCA2 pre- and post-whole-genome amplification demonstrated conserved MAFs (supplementary Figure S3, available at Annals of Oncology online). In addition, genome-wide copy number profiling identified somatic copy number changes across the genomes of each patient. High estimates of tumor fractions ranging from 56% to 82% for each liquid biopsy sample suggest stout representation of the tumor within the circulating exosomes of each patient.
ExoDNA is representative of the entire human genome
Whole-genome sequencing covered 65%–91% of the human genome at a mean depth of 12–35× at high-quality sequencing with 88.2%–92.5% of bases having greater than or equal to sequencing quality scores of Q30 (supplementary Table S2, available at Annals of Oncology online). Exome sequencing covered 95%–99% of the targeted genome (54 megabases) with at least one read at a mean depth of 133–490× with 73%–96% being covered by at least 10 reads. Ninety percent to 94.6% of bases represented high-quality sequence suggesting that exoDNA in our samples is representative of the entire human genome.
comprehensive profiling of tumors using exoDNA and mRNA
LBx01: Tumor profiling using pleural effusion exosomes from a patient with pancreatic ductal adenocarcinoma with previously resected lung metastasis
LBx01 is a patient PDAC, who underwent a thoracoscopic resection of a suspicious pulmonary nodule, subsequently confirmed to be metastasis. Fifteen months later, he developed a pleural effusion, which contained <1% malignant cells on cytospin, per final cytopathology report. A deep NGS assay performed on the pleural fluid cytospin failed to detect any evidence of tumor DNA (data not shown). In contrast, abundant cancer-derived exosomes were present in the pleural fluid even with the marked paucity of cancer cells. The pleural effusion exoDNA had a computationally estimated tumor fraction of 82% [95% confidence region (CR) of 81% to 83%] and a mean genome copy number of 2.57 (see supplementary Methods, available at Annals of Oncology online). The exoDNA tumor fraction estimate was higher than that compared with the previously resected metastatic lung tissue (23%, 95% CR 22% to 24%). The exoDNA mutation rate was estimated at 341 mutations/Mb compared with 2.06 mutations/Mb in the metastatic lung tissue DNA 15 months before the liquid biopsy sampling. This substantially higher mutational load is not surprising given the time between metastectomy and manifestation of pleural effusion, and the multidrug cytotoxic chemotherapy regimen administered to the patient.
Potentially actionable mutations are listed in supplementary Table S3, available at Annals of Oncology online. We considered mutations to be potentially actionable if they are either putative drivers (recurrently mutated in COSMIC [17]) that could be monitored over the course of patient management, or COSMIC mutations that reside in genes associated with a clinical trial or cancer drug (see supplementary Methods, available at Annals of Oncology online). Mutations in KRAS and TP53 were identified in both the lung metastatic tissue and the subsequent pleural effusion. Mutations likely representative of progression include those in adenomatous polyposis coli and CHEK2. The metastatic lung tissue harbored a mutation signature with peaks at C-to-T base substitutions that are consistent with Signature 1 of the COSMIC mutational signatures (Figure 1C), a common cancer signature proposed to be involved in spontaneous deamination of 5-methylcytosine [17, 18]. The exoDNA mutational signature deviates from this, which suggests that additional mutational processes may have contributed to tumor progression, possibly driven by cytotoxic chemotherapy.
Figure 1.

LBx01 tumor profiling. (A) Copy number profile comparison between the metastatic lung tissue (top) sampled 15 months before the pleural effusion exoDNA (bottom). The cancer-related genes on the light-red vertical bars have copy number gains and those on the light-blue vertical bars have copy number losses, where the numbered labels represent the estimated copy numbers. The yellow vertical bar annotates putatively actionable copy number variations (CNVs) (e.g. ERBB2). The arrow to the left depicts the progression of cancer-associated CNVs between the 2 time points. These happen to all be amplifications, which were also confirmed to be upregulated in the exoRNA compared with that in the metastatic lung tissue RNA-seq. (B) Mutant KRAS was identified in the mRNA (RNA sequencing) as well as DNA (exome and genome sequencing) of the pleural effusion exosomes. (C) Mutational signature of the plasma exosomes derived from exome sequencing (top) and genome sequencing (middle) compared with the mutational signature of the metastatic lung tissue (bottom). (D) Circos plot illustrating putative gene fusions (blue), lung metastatic copy number profile (inner-most ring), pleural effusion exosomes copy number profile (second inner-most ring) and gene aberrations. Mutations seen in the pleural effusion are black and those seen in both the metastatic lung tissue and pleural effusion are in bolded black.
ExoDNA copy number profiling showed that 27% of the genome exhibited copy number variation (Figure 1A). This included amplification of MYC [copy number (CN) = 3; P = 1.3e−72], KRAS (CN = 6; P = 2.7e−11), EGFR (CN = 3; P = 1.3e−138) and ERBB2 (CN = 5; P = 6.1e−10) (supplementary Table S4, available at Annals of Oncology online). ERBB2 amplification, in particular, was also identified in the previously resected lung metastasis, albeit at a lower copy number. We confirmed the amplification and overexpression, respectively, ERBB2 in both exoDNA and exoRNA, where the estimated copy number was 5 and ERBB2 was overexpressed at 85.13 transcripts per million (TPM) which represents a 3.62 times higher expression compared with normal pancreas tissue [23.52 TPM—the median ERBB2 expression in normal pancreas tissue identified from the Genotype-Tissue Expression (GTEx) project [19]].
A key advantage of exoRNA (in contrast to cell-free nucleic acids) is the preserved quality of transcripts that allows for assessment of an aberrant transcriptome in the same liquid biosample from which the genomic landscape is derived. As exemplified with ERBB2, cross-comparison of exoDNA and exoRNA data permits precise delineation of the oncogenic targets of genomic copy number aberrations. Another potential clinical benefit of this approach is identification of expressed neoantigens from the tumor, be it missense mutations, or unique cancer-derived fusion transcripts, which can serve as the basis for identification of neoantigen-targeted humoral or cellular immune responses [20, 21]. For example, the exoRNA confirmed the presence of a KRAS G12D mutation in the transcriptome (Figure 1B and Supplementary Table S3, available at Annals of Oncology online). Furthermore, 87.8% of protein-coding transcripts were expressed (having greater than or equal to 2 TPM; supplementary Table S5, available at Annals of Oncology online) and 40 putative expressed gene fusions were identified (supplementary Table S6, available at Annals of Oncology online; Figure 1D), though no delineated cancer signaling pathways were overrepresented in the exoRNA data (supplementary Table S7, available at Annals of Oncology online).
LBx02: Tumor profiling using blood-derived exosomes of a pancreatic ductal adenocarcinoma patient
LBx02 is a treatment-naïve patient with PDAC and hepatic metastases. The plasma exoDNA estimated tumor fraction was 56% (95% CR 54% to 57%) with a mean genome copy number of 2.12. Copy number profiling showed that 9% of the genome exhibited copy number variation (Figure 2A). This included amplification of MYC (CN = 13; P = 4.7e−08), KRAS (CN = 3; P = 6.5e−24) and loss of TP53 (CN = 1; P = 3.6e−39) (supplementary Table S4, available at Annals of Oncology online). Potential actionable mutations (supplementary Table S8, available at Annals of Oncology online) include mutations in ERBB2, KRAS, NRAS and NOTCH1 (in practice, KRAS or NRAS mutations are not strictly actionable, although many commercially available or academic center-initiated sequencing panels list them as such). The exoDNA exhibited a mutation rate of 77 mutations/Mb and a profile resembling Signature 1 of the COSMIC mutational signatures (Figure 2B) [17, 18].
Figure 2.
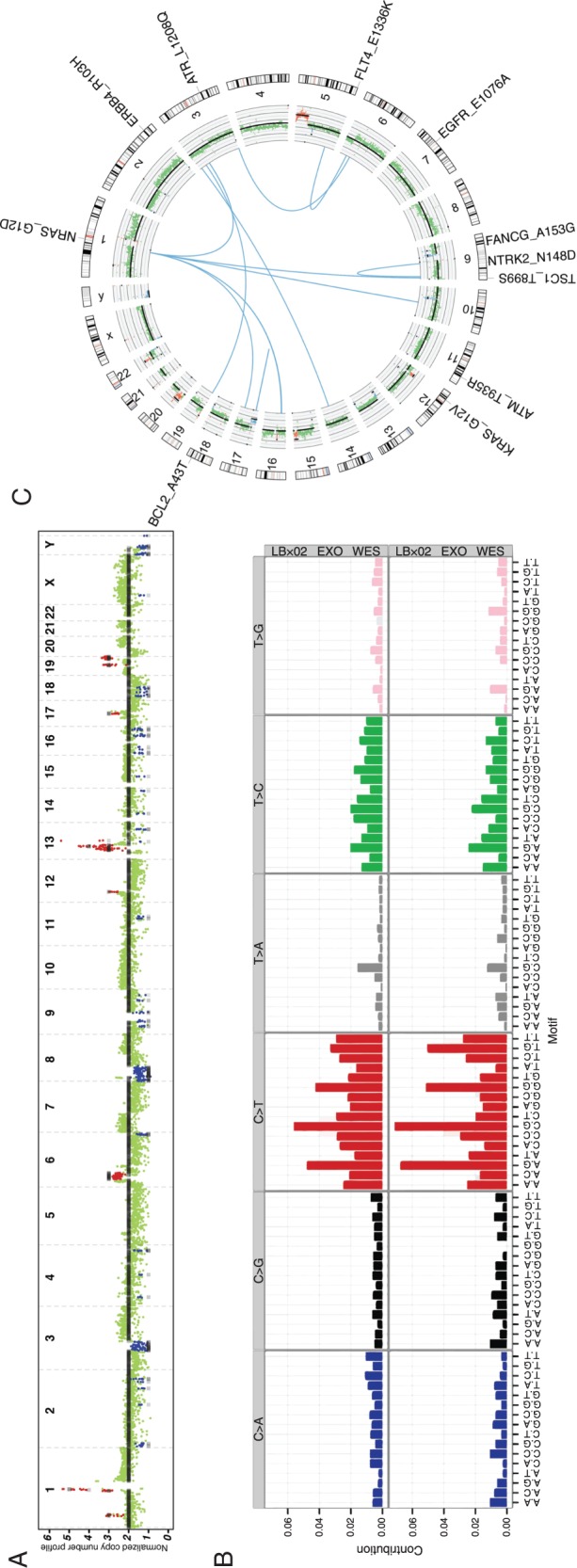
LBx02 tumor profiling. (A) Copy number profile of the plasma exoDNA. (B) Mutational signature of the pleural effusion exosomes derived from exome sequencing (top) and genome sequencing (bottom). (C) Circos plot illustrating putative gene fusions (blue), plasma exoDNA copy number profile (inner-most ring) and potential actionable genes (blue, deletions; red, amplifications; black, somatic point mutations).
Approximately 9% of protein-coding transcripts were expressed (supplementary Table S9, available at Annals of Oncology online) in the exoRNA and 16 putative expressed fusions were identified (supplementary Table S10, available at Annals of Oncology online; Figure 2C). The mechanistic target of rapamycin signaling pathway was overrepresented in the exoRNA transcripts (Benjamini-Hochberg, P = 0.027; supplementary Table S11, available at Annals of Oncology online).
LBx03: Tumor profiling using blood-derived exosomes of an ampullary carcinoma patient identifies an unexpected therapeutic vulnerability
LBx03 plasma exoDNA had an estimated tumor fraction of 82% (95% CR 81% to 84%) and a mean genome copy number of 2.5. Copy number profiling showed that 53% of the genome exhibited copy number variation (Figure 3A), which suggests the presence of an ‘unstable’ genome phenotype [22]. Copy number aberrations include amplification of MYC (CN = 4; P = 6.7e−129) and KRAS (CN = 3; P = 3.8e−69) (supplementary Table S4, available at Annals of Oncology online). The exoDNA mutation rate is estimated at 125 mutation/Mb exhibiting a relatively large proportion of C-to-A and C-to-T base substitutions (Figure 3B). Several potentially actionable mutations were identified (supplementary Table S12, available at Annals of Oncology online) including an unexpected somatic mutation of BRCA2, which was not present in the germline DNA. Specifically, the BRCA2 V3091I mutation has previously been reported as conferring a homologous recombination defect in cancer cells [23]. Three lines of evidence suggest that this BRCA2 mutation is indeed pathogenic: first, the high MAF in exoDNA, underscoring its ‘driver’ status; second, the ‘unstable’ genome phenotype on genome-wide copy number assessment [22]; and third, the exceptional response to a platinum-containing adjuvant regimen that this patient has had to date (although the overall follow-up period remains limited).
Figure 3.
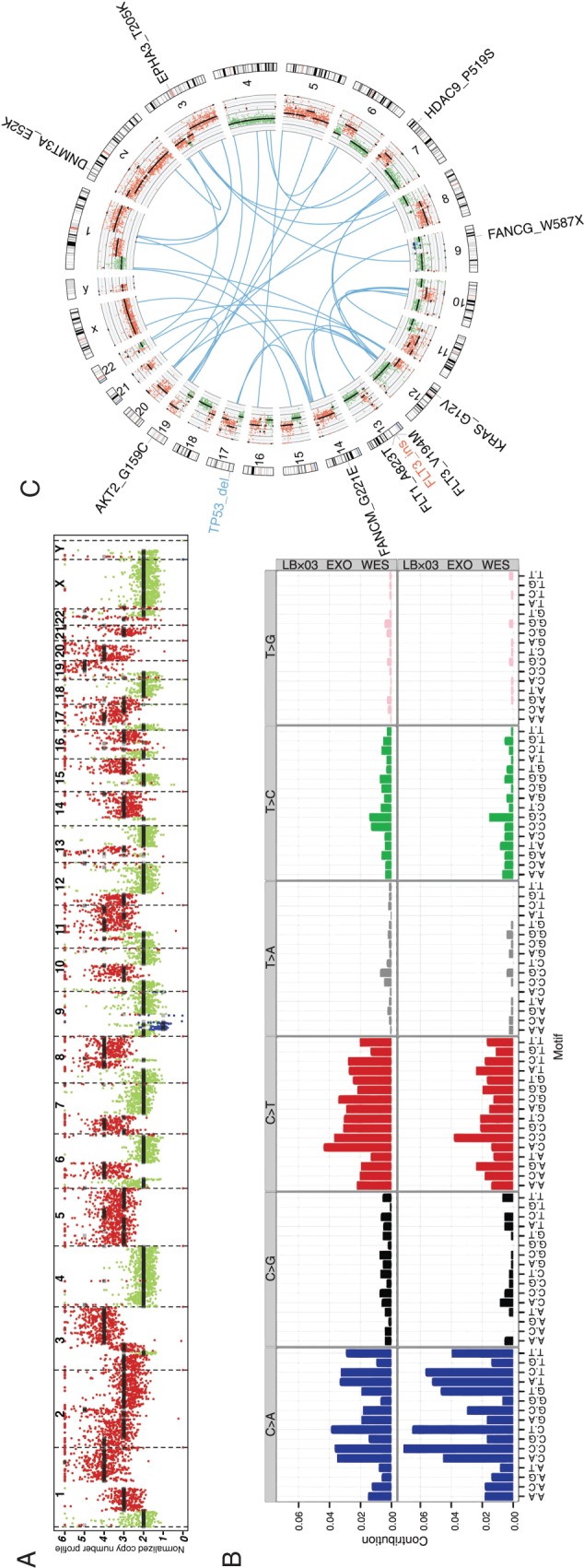
LBx03 tumor profiling. (A) Copy number profile of the plasma exoDNA. (B) Mutational signature of the plasma exosomes derived from exome sequencing (top) and genome sequencing (bottom). (C) Circos plot illustrating putative gene fusions (blue), plasma exoDNA copy number profile (inner-most ring) and potential actionable genes.
In the exoRNA, 16.6% of protein-coding transcripts were expressed (supplementary Table S13, available at Annals of Oncology online), including 40 putative expressed gene fusions (supplementary Table S14, available at Annals of Oncology online; Figure 3C). No cancer signaling pathways were overrepresented in the exoRNA data (supplementary Table S15, available at Annals of Oncology online).
discussion
We have demonstrated the feasibility of using peripheral blood and pleural effusion-based liquid biopsies to comprehensively profile the genomes and transcriptomes of deeply located visceral cancers for which traditional tissue biopsies may be difficult, risky or unachievable in less-specialized centers. In addition, exosome-based liquid biopsy results demonstrate the potential for identifying unexpected therapeutic vulnerabilities. Of particular importance regarding patient LBx03 is the presence of a BRCA2 mutation, which has been shown to predict responsiveness to platinum-based chemotherapies. Currently, several clinical trials are ongoing that incorporate platinum-based regimens of poly ADP ribose polymerase inhibitors in PDAC patients with such DNA damage repair defects [24]. In addition to identifying actionable mutations at the time of presentation, exosome-based liquid biopsies provide an opportunity to identify new therapeutic vulnerabilities that emerge over the course of treatment, or elucidate potential mechanisms of resistance to administered targeted therapies. For example, at the time of metastectomy of the lung metastasis from LBx01, the patient was found to have evidence of ERBB2 amplification in the pulmonary nodule, leading to subsequent attempt of targeted therapy with Trastuzumab. However, no meaningful response was found to the agent. Two months following completion of this therapy, subsequent liquid biopsy from this patient confirmed the ERBB2 amplification, as well as the emergence of an EGFR amplification, which might represent a clonal selection in response to the trial of a targeted agent [25]. Liquid biopsy in this patient far exceeded standard-of-care laboratory metrics where <1% tumor cells were detected in the pleural effusion, precluding further analysis. Cancer-derived exosomes were able to enrich for the genetic makeup of the local tumor tissue, recapitulating the molecular identity of the diseased lung. It is important to note that such ‘serial’ sampling of the tumor genomic landscape, while possible in superficial cancers like melanomas, is almost unheard of in visceral malignancies, due to logistical or reimbursement limitations.
While cfDNA platforms can certainly elucidate limited panels of genomic abnormalities and even map the emergence of resistance mechanisms during the course of targeted therapies, exosome-based liquid biopsy approaches have the additional benefit of being able to comprehensively profile the cancer transcriptome from the same biosample. In particular, the ability to identify expressed neoantigens (point mutations or fusion transcripts) represents an avenue to interrogate the humoral or cellular responses to such neoantigens in visceral cancers [20, 23]. For example, emerging ‘personalized’ adoptive T-cell therapies require elucidation of cancer-specific neoantigens that are expressed and processed in an human leukocyte antigen context [26]. Typically, this requires a tissue biopsy and RNA profiling of the tumor. Exosome-based liquid biopsy can identify such expressed neoantigens without the need for tissue sampling, and moreover, map the response to immunotherapy through quantitative estimates of neoantigen load in circulation. In addition, since the peripheral blood is a sampling of all body tissues, this genetic analysis presumably has the power to characterize the patient's entire tumor burden: primary tumor and any metastatic disease. This is of particular importance when considering that primary tumors and associated metastases are of a heterogeneous genetic makeup with compounded temporal heterogeneity [27].
Our study is not without limitation. Conceptually, many will desire to see liquid biopsy-detected mutations validated in primary tissue. For visceral cancers, the acquisition of such tissues may be limited and localized, thus detection of mutations for validation may not be ideal. Of note, our mutation rates of 341, 77 and 125 mutations/Mb are substantially higher than the average of 2.64 mutations/Mb (range 0.65–28.2) estimated by Waddell et al. [22] from PDAC tissue whole-genome sequencing. We suspect that a large degree of this discrepancy is due to exoDNA representing tumor heterogeneity at a level that is not attainable through tissue sequencing. A potential strategy to confirm these liquid biopsy findings is to compare serial samples in the same patient, to validate over time the identification of mutations at varying allelic frequency. Such serial profiling is the subject of further study. Nonetheless, our proof of concept results demonstrate that seamless coordination between clinical and research efforts can produce a workflow from blood draw to sequencing results in a period of 14 days, acquiring results in a clinically relevant timeframe for patients with visceral cancers.
funding
This work was supported by the MD Anderson Moonshot Program in Pancreatic Cancer and the Sheikh Ahmed Pancreatic Cancer Research Center; the National Institute of Health (5T32CA009599-27 to KA, R01CA113669 to AM, U01CA196403 to AM, U01DK108328 to AM, P30CA016672 for the MD Anderson High-Resolution Electron Microscopy Facility); the Translational Molecular Pathology Fellowship at MD Anderson Cancer Center to FS; the Cancer Prevention Research Institute of Texas (RP140106 to VB); and the MD Anderson Institutional Cancer Center Support Grant CA16672 for the High-Resolution Electron Microscopy Facility.
disclosure
The authors have declared no conflicts of interest.
Supplementary Material
acknowledgements
We are grateful to Arnold Levine, Institute for Advanced Study, Princeton University for his insightful guidance, and to the Precision Medicine Research Associates (PMRA)/Fox Family Foundation for partially supporting the studies described in this manuscript. We thank Arul Chinnaiyan at the University of Michigan for generously sharing exome-sequencing data on the surgically resected LBx01 metastatic lung tissue.
references
- 1.Chantrill LA, Nagrial AM, Watson C et al. Precision medicine for advanced pancreas cancer: the Individualized Molecular Pancreatic Cancer Therapy (IMPaCT) trial. Clin Cancer Res 2015; 354: 1698–1705. [DOI] [PubMed] [Google Scholar]
- 2.Taly V, Pekin D, Benhaim L et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin Chem 2013; 59: 1722–1731. [DOI] [PubMed] [Google Scholar]
- 3.Lanman RB, Mortimer SA, Zill OA et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS One 2015; 10: e0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zill OA, Greene C, Sebisanovic D et al. Cell-free DNA next-generation sequencing in pancreatobiliary carcinomas. Cancer Discov 2015; 5: 1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan KC, Jiang P, Zheng YW et al. Cancer genome scanning in plasma: detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin Chem 2013; 59: 211–224. [DOI] [PubMed] [Google Scholar]
- 6.Murtaza M, Dawson SJ, Tsui DW et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013; 497: 108–112. [DOI] [PubMed] [Google Scholar]
- 7.Butler TM, Johnson-Camacho K, Peto M et al. Exome sequencing of cell-free DNA from metastatic cancer patients identifies clinically actionable mutations distinct from primary disease. PLoS One 2015; 10: e0136407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mouliere F, Robert B, Arnau Peyrotte E et al. High fragmentation characterizes tumour-derived circulating DNA. PLoS One 2011; 6: e23418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleischhacker M. Biology of circulating mRNA: still more questions than answers? Ann N Y Acad Sci 2006; 1075: 40–49. [DOI] [PubMed] [Google Scholar]
- 10.Mitchell PS, Parkin RK, Kroh EM et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA 2008; 105: 10513–10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Hefnawy T, Raja S, Kelly L et al. Characterization of amplifiable, circulating RNA in plasma and its potential as a tool for cancer diagnostics. Clin Chem 2004; 50: 564–573. [DOI] [PubMed] [Google Scholar]
- 12.Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol 2002; 2: 569–579. [DOI] [PubMed] [Google Scholar]
- 13.Skog J, Wurdinger T, van Rijn S et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 2008; 10: 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kahlert C, Kalluri R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl) 2013; 91: 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kahlert C, Melo SA, Protopopov A et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem 2014; 289: 3869–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thakur BK, Zhang H, Becker A et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res 2014; 24: 766–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forbes SA, Beare D, Gunasekaran P et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015; 43: D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alexandrov LB, Nik-Zainal S, Wedge DC et al. Signatures of mutational processes in human cancer. Nature 2013; 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Consortium G. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015; 348: 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Overwijk WW, Wang E, Marincola FM et al. Mining the mutanome: developing highly personalized Immunotherapies based on mutational analysis of tumors. J Immunother Cancer 2013; 1: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348: 69–74. [DOI] [PubMed] [Google Scholar]
- 22.Waddell N, Pajic M, Patch AM et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015; 518: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balia C, Galli A, Caligo MA. Effect of the overexpression of BRCA2 unclassified missense variants on spontaneous homologous recombination in human cells. Breast Cancer Res Treat 2011; 129: 1001–1009. [DOI] [PubMed] [Google Scholar]
- 24.Lowery MA, Kelsen DP, Stadler ZK et al. An emerging entity: pancreatic adenocarcinoma associated with a known BRCA mutation: clinical descriptors, treatment implications, and future directions. Oncologist 2011; 16: 1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henjes F, Bender C, von der Heyde S et al. Strong EGFR signaling in cell line models of ERBB2-amplified breast cancer attenuates response towards ERBB2-targeting drugs. Oncogenesis 2012; 1: e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran E, Turcotte S, Gros A et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014; 344: 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerlinger M, Horswell S, Larkin J et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014; 46: 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}