

Mutations in chromatin machinery define pediatric high-grade gliomas; efforts to define and target their functions are under way.
Keywords: Histone mutation, K27M, G34V/R, pediatric high-grade glioma, DIPG, methyltransferase, demethylase, PRC2, JMJD3, GSKJ4
Abstract
Pediatric central nervous system tumors are the most common solid tumor of childhood. Of these, approximately one-third are gliomas that exhibit diverse biological behaviors in the unique context of the developing nervous system. Although low-grade gliomas predominate and have favorable outcomes, up to 20% of pediatric gliomas are high-grade. These tumors are a major contributor to cancer-related morbidity and mortality in infants, children, and adolescents, with long-term survival rates of only 10 to 15%. The recent discovery of somatic oncogenic mutations affecting chromatin regulation in pediatric high-grade glioma has markedly improved our understanding of disease pathogenesis, and these findings have stimulated the development of novel therapeutic approaches targeting epigenetic regulators for disease treatment. We review the current perspective on pediatric high-grade glioma genetics and epigenetics, and discuss the emerging and experimental therapeutics targeting the unique molecular abnormalities present in these deadly childhood brain tumors.
INTRODUCTION
Pediatric high-grade gliomas are clinically and biologically distinct from adult gliomas. Historically, pediatric high-grade glioma was considered similar to secondary glioblastoma multiforme (GBM), an adult high-grade brain tumor that evolves from a less malignant precursor due to an accumulation of gene alterations (1). However, pediatric gliomas do not necessarily conform to this model, and recent advanced genomic analyses have defined unique epigenetic and genetic alterations that distinguish gliomagenesis in children and adults (2).
Pediatric high-grade gliomas located in the brainstem represent approximately 10% of all pediatric central nervous system (CNS) tumors (3), and 80% are restricted to the ventral pons. These tumors, known as diffuse intrinsic pontine gliomas (DIPGs), primarily affect very young children with peak incidence at 6 years of age and have the highest mortality of all childhood solid tumors. The median survival for a child with DIPG is only 9 months, and virtually all patients die within 2 years of diagnosis (3).
The clinical management of children with DIPG remains one of the most formidable challenges in pediatric neuro-oncology. Given its location, surgical resection of tumor is not an option, whereas systemic and local administration of chemotherapeutics, which are effective for adult and other pediatric gliomas, have proven ineffective for DIPG. To date, more than 250 clinical trials of standard and novel chemotherapeutics, administered using a variety of regimens, have been conducted, but none has demonstrated any improvement in patient survival (4). Currently, conventional therapy for DIPG is fractionated focal radiation therapy to a total dose of 54 to 60 Gy over a 6-week period. This provides a “honeymoon period” of temporary symptom relief and a delay in tumor progression in about 70 to 80% of patients. No further clinical benefit has been achieved using alternative radiation strategies or from combination therapies with radiation sensitizers (5, 6). Unfortunately, no effective salvage therapy is available at the time of tumor progression, though reirradiation may provide transient symptom improvement in some patients (7). The poor clinical response to therapeutic approaches that are more effective against other pediatric and adult gliomas suggests that DIPG is a biologically and molecularly distinct malignancy.
Historically, diagnosis of DIPG has been made based on magnetic resonance imaging; surgical biopsy to obtain tissue for histological verification was rarely performed (8). The resulting lack of readily available DIPG tissue samples for molecular analysis hindered the investigation of its tumor and molecular biology, limiting progress in treatment outcomes for DIPG patients, relative to that achieved for other pediatric and adult brain tumors (9). However, advances in neurosurgical and molecular analytical techniques have enabled recent genomic analyses of DIPG tissue samples. Associated studies have revealed distinct genetic alterations in DIPG, including Lys27Met (K27M) missense mutation in genes encoding histone H3 isoforms that occur in up to 80% of DIPGs. Histone H3 mutations have also been reported in pediatric high-grade glioma outside the brainstem, but are rarely detected in adult malignant gliomas, and H3 mutations therefore represent an important distinguishing characteristic for pediatric versus adult high-grade glioma (10–12). In DIPG, histone H3 mutation is associated with a clinically and biologically distinct subgroup of patients with a more aggressive clinical course, and poorer overall response to therapy (10).
Mutations in histone H3 in pediatric high-grade glioma can be placed in a growing category of epigenetic gene alterations whose primary effect is to alter gene transcription. Epigenetic regulation controls gene expression without affecting the sequence of the genetic code and is mediated by selective and reversible modifications of DNA and histone proteins that control the conformational transition between transcriptionally active and inactive chromatin states (13). These modifications are affected by enzymes, many of which have been revealed as cancer-associated mutation targets, with mutations invariably resulting in altered regulation of gene transcription. Epigenetic events such as methylation, acetylation, and phosphorylation can be therapeutically targeted, which indicates that pharmacologic inhibition of histone modifiers has intriguing potential as an approach for more effective treatment of cancer.
Molecular characterization of pediatric high-grade glioma
Advances in stereotactic neurosurgery are a major factor contributing to increased access to previously scarce DIPG tissue. In the last decade, several neurosurgical groups have demonstrated the relative safety and diagnostic utility of image-guided, stereotactic biopsy of brainstem and midline gliomas that, traditionally, had been avoided due to concern over procedure-associated morbidity. In the largest published series, Roujeau and colleagues reported no surgery-related deaths and low, transient morbidity in less than 4% of patients after performing more than 200 brainstem biopsies (14). Tissue specimens obtained via stereotactic biopsy are reliable for accurate histological and molecular analysis (15, 16), including comprehensive genome sequencing (17, 18), and yield viable, sustainable tumor cells in culture and in xenograft models.
Given the relative safety of stereotactic biopsy and high-quality genomic data from small tumor samples, multiple groups have begun to practice, and advocate for, routine stereotactic biopsy of DIPG. In 2013, experts in pediatric neuro-oncology and neurosurgery issued a consensus statement recommending upfront biopsy of DIPG for genomic analysis and patient stratification for molecularly targeted treatments, before using radiotherapy, in an effort to minimize confounding findings resulting from radiation-induced mutations (19). Presently, there are two ongoing clinical trials for patients with newly diagnosed DIPG who undergo biopsy for molecular characterization, with 24 participating institutions across the United States and Europe. One trial (www.clinicaltrials.gov, NCT02233049) stratifies patients by EGFR (epidermal growth factor receptor) and PTEN (phosphatase and tensin homolog) expression, and the other (NCT01182350) uses MGMT (O6-methylguanine-DNA methyltransferase) promoter methylation status and EGFR expression analysis as biomarkers to determine treatment. Although conceptually advanced in using molecular characterization of biopsy specimens to influence treatment decisions, it is nonetheless evident that the molecular characterizations for these studies are still based primarily on genetic lesions that commonly occur in adult gliomas.
Histone gene mutations and chromatin machinery
Histones are the major protein component of eukaryotic chromatin and are deposited during DNA synthesis. Histones H2A, H2B, H3, and H4 combine as an octamer to compose the nucleosome core, and associate with DNA via the linker histone H1. In mammals, histone H3 has three isoforms that share highly conserved amino acid sequences. Histone H3 isoforms are important for transcriptional regulation and telomere stabilization. H3.1 and H3.2 are replication-dependent and are synthesized and deposited on DNA during the S phase, whereas H3.3 synthesis and DNA deposition occur throughout the cell cycle (20–22). Regulatory posttranslational modifications of the N-terminal tail of histone H3 (including methylation, acetylation, and ubiquitylation of lysine residues; phosphorylation of serine or threonine residues; and methylation of arginine residues) have important effects on the processes of DNA replication, repair, and transcription (23, 24). The extent of methylation on histone H3 lysine residues (mono-, di-, or trimethylation) is an especially important determinant of DNA functional properties, with methylation of histone H3 lysine 27 (H3K27) being particularly influential in regulating the transcription of cancer-associated genes (25–27).
H3K27 methylation is determined by the activity of EZH2 methyltransferase, as the enzymatic subunit of polycomb repressive complex 2 (PRC2), combined with the demethylating activities of JMJD3/KDM6B and UTX/KDM6A (Fig. 1). EZH2 has been the focus of substantial preclinical investigation as a therapeutic target in the treatment of various types of human cancer, since the discovery of elevated EZH2 expression, as contributing to aggressive tumor biology, metastasis, and poor clinical outcomes in breast, prostate, lung, bladder, ovarian, and skin cancer, as well as in hematological malignancies (28–32). Thus far, EZH2 has not been explored as a target for treatment of pediatric high-grade glioma. Despite the importance of EZH2, H3K27 methylation requires other PRC2 components including EZH1 (functional homolog of EZH2), histone core accessory proteins (EED, SUZ12, and RbAP48), and PRC2-associated factors JARID2 and ASXL1 (Fig. 1) (29, 32). Of the enzymes that remove methyl groups from H3K27, somatic inactivating mutations and deletions of UTX have been reported in hematological malignancies and solid tumors (33–35), suggestive of a tumor suppressor role for UTX. Both JMJD3 and UTX function as part of a transcriptional activator complex that includes the MLL3/MLL4 H3K4 methyltransferases, indicating that these enzymes have a dual role involving removal of H3K27 methyl marks and addition of methyl groups to H3K4 (Fig. 1) (29, 36).
Fig. 1. Histone modifications by PRC2 methyltransferase and KDM demethylase activities, and associations with transcriptionally active versus inactive states.
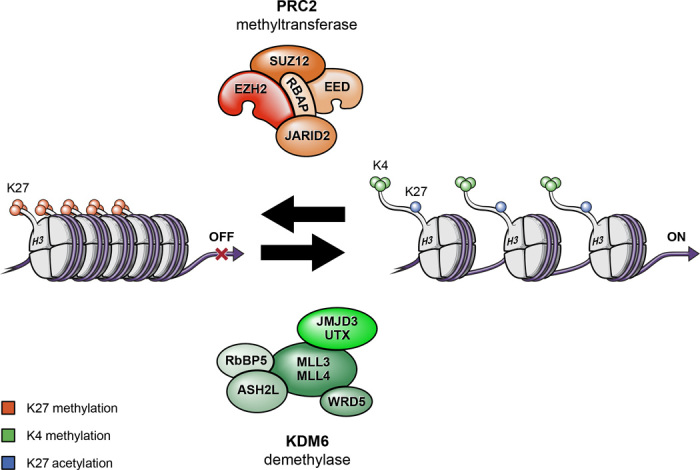
Histone proteins are modified by the PRC2 methyltransferase and by KDM demethylase. PRC2 increases methylation of Lys27, which promotes a more compact and transcriptionally repressed chromatin state. In contrast, KDM demethylase complex removes methyl groups from Lys27 and increases methylation of Lys4 that, in combination, promote an open and transcriptionally active chromatin state.
Recent studies of pediatric high-grade gliomas have identified gain-of-function mutations in H3 histone genes, specifically histone 3A (H3F3A) and histone H3b (HIST1H3B), encoding histone H3 variants H3.3 and H3.1, respectively (10–12, 37–39). Histone H3.3 mutations occur at two distinct residues, either lysine at position 27 or glycine at position 34 (40), and result in replacement of lysine 27 by methionine (K27M) or replacement of glycine 34 by valine or arginine (G34V/R). The former occurs in approximately 80% of DIPGs (11, 12), and most of the K27M mutations involve H3F3A, with the remainder involving HIST1H3B (1, 10, 12, 18, 40, 41). In contrast, G34V/R mutations are exclusive to H3F3A.
Histone gene mutations are associated with both tumor location and patient age. K27M mutant tumors are predominantly seen in midline locations including the thalamus, pons, and spinal cord (Fig. 2). In contrast, G34V/R mutations are restricted to tumors of the cerebral hemispheres (1, 12, 17, 38, 42) (Fig. 2) and are not detected in midline high-grade gliomas. This location-specific pattern of histone mutation suggests a different cell of origin and/or developmental stage for supratentorial versus midline high-grade glioma. For example, gene expression signatures of K27 mutant tumors were found to be associated with mid- to late-fetal stages of striatal and thalamic development (1, 38), whereas G34V/R signatures are more typical of expression signatures in early- to mid-fetal stages of neocortex and striatal development (38).
Fig. 2. Neuroanatomic and gene associations with histone mutations.
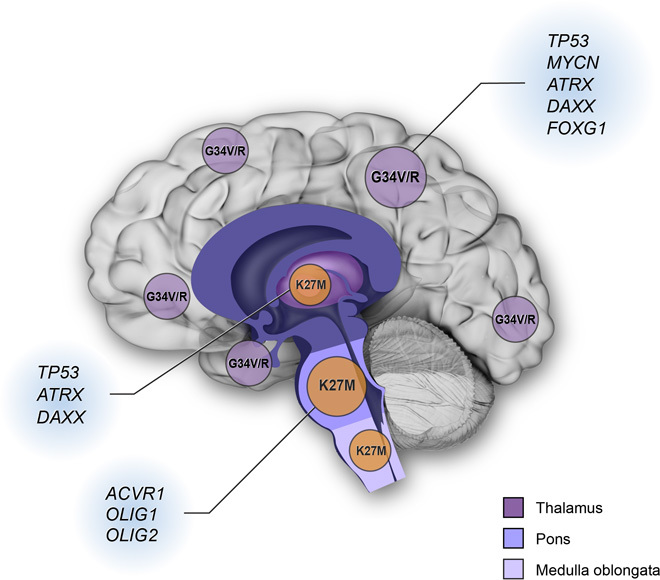
K27M mutations are predominantly found in the tumors occurring in midline locations (thalamus, pons, and medulla oblongata). G34V or G34R (G34V/R) mutations are found in cerebral cortical tumors. Other gene alterations associated with histone gene mutations also occur in location-specific patterns. For example, TP53 mutations overlap with H3F3A mutations in cortical and thalamic tumors. ATRX and DAXX mutations are strongly associated with cortical G34V/R tumors and K27M mutant thalamic tumors, respectively. ACVR1 mutations are frequently present in histone H3.1 K27M mutant DIPG. OLIG1 and OLIG2 are highly expressed in K27M mutant tumors, whereas FOXG1 expression is predominantly found in G34V/R mutant tumors.
As suggested above, the pattern of histone mutation in pediatric glioma also appears to be affected by age. For instance, H3.1 K27M mutant tumors tend to occur in very young children; H3.1 K27M additionally has frequent companion mutations, such as that of ACVR1 (14, 17, 18, 43). For tumors with mutant H3.3, K27M is more common in school-age children (median age, 11 years; range, 5 to 29), whereas G34V/R mutations are more prevalent in adolescents and young adults (median age, 20 years; range, 9 to 42) (10, 11, 44). Although primarily associated with pediatric DIPG and midline high-grade gliomas, K27M mutant thalamic tumors have also been reported in patients from 21 to 49 years old (45). Given these age- and location-specific distributions, patients with histone mutant glioma are encountered in both pediatric and adult neuro-oncology practices.
The detection of histone gene mutation has proven to be informative with respect to prognosis. As compared to patients with H3.1 mutant tumors, patients with H3.3 mutant tumors have a shorter survival, and among H3.3 mutant tumors, patients with the K27M mutation show significantly shorter overall survival than those with the G34V/R mutation (median of 12 months for K27M; median of 24 months for G34V/R) (1, 10, 17, 18, 38, 42). Although this association may be in part due to G34V/R tumor surgical accessibility, at least one study suggests that the prognosis for patients with K27M tumors is worse irrespective of location (1), thereby supporting the K27M tumors as an especially aggressive subtype of high-grade glioma.
The role of histone gene mutations in gliomagenesis
Because of its high frequency and poor prognosis, the histone H3K27M mutation has gained considerable attention regarding its role in tumor initiation, maintenance, and progression. At a molecular level, Lewis and others have identified a gain-of-function activity for this mutation by showing that H3K27M sequesters PRC2 histone methyltransferase and, in so doing, suppresses PRC2 function (Fig. 3) (37, 39, 46, 47). K27M-associated suppression of PRC2, in turn, results in a marked reduction of wild-type H3K27 methylation, to an extent that far exceeds the proportion of K27M to wild-type H3 in DIPG. This effect is made remarkable by the existence of 16 distinct histone H3–encoding genes in the human genome, and of the 32 histone H3–encoding alleles in a diploid cell; only one histone H3–encoding allele, of the two H3F3A genes, is mutated. Yet, amazingly, H3K27M encoded by the single mutant allele can drastically suppress di- and trimethylation of wild-type H3K27 produced from the 31 wild-type–encoding H3 genes in a K27M tumor cell (37, 39, 46–48). This marked reduction in wild-type H3K27 methylation is thought to result in an extensive transcriptional reprogramming of the tumor cell (39).
Fig. 3. Model for global reduction of H3K27 methylation in K27M DIPG, and effect of GSKJ4 on K27 methylation status.
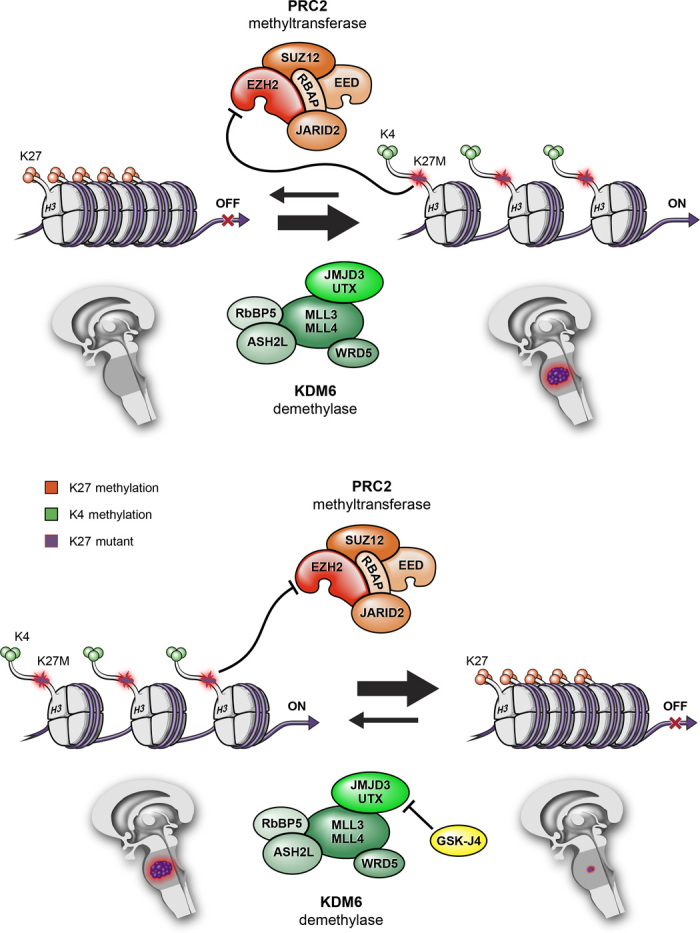
Histone H3K27M mutant protein sequesters PRC2 histone methyltransferase and functionally inactivates it, leading to a global reduction of K27 methylation, thereby promoting an open chromatin structure that favors increased gene transcription. Pharmacological inhibition of JMJD3 H3K27 demethylase by GSKJ4 increases K27 methylation. In so doing, GSKJ4 suppresses gene expression and reduces tumor growth.
The effect of K27M on PRC2 function and wild-type K27 methylation is not random, however, as the mutation results in increased and decreased gene expression. Despite a massive decrease in total chromatin H3 dimethylation (K27me2) and trimethylation (K27me3), and the associated suppression of transcription that comes with decreased K27 methylation, select genes in DIPG are highly expressed. Thus, it would seem that K27M both represses and activates gene expression, which prompts the related question, “Which genes are activated and which genes are repressed?” Although the answer to this question remains an area of intensive investigation, some clues are beginning to emerge. For instance, Chan et al. showed that genes with increased H3K27me3 in H3K27M mutant tumors are associated with cancer development pathways (39). In a study of human embryonic stem cell–derived neural progenitor cells, the combination of K27M with PDGFRA overexpression and short hairpin RNA (shRNA)–mediated p53 knockdown has been shown to promote transformation and confer tumorigenicity to cells implanted in the brainstem of immunocompromised mice (49). Together, these three genetic modifications produced very slow growing tumors that lack the typical histological features and proliferative capacity of high-grade glioma, which suggests that additional gene alterations may be required to accurately model for human DIPG (49). Nonetheless, these results suggest a specific oncogene, PDGFRA, and a specific tumor suppressor gene, p53, as key expression modification targets in K27M mutant tumors. Moreover, the results of this study showed that K27M expression was only tumor-promoting in neural progenitors derived from embryonic stem cells and not in undifferentiated embryonic stem cells or astrocytes, thereby supporting the cell context specificity of K27M-associated transformation.
In contrast to H3K27, H3G34 is not a known target of posttranslational modification. However, the G34R substitution has been shown to diminish K36 trimethylation in vitro (37) and K36 trimethylation that is known to be important to the regulation of gene expression and alternative splicing.
Gene alterations associated with histone gene mutations
Additional gene alterations are common in pediatric high-grade gliomas with histone gene mutations, including those affecting TP53, MYCN, α-thalassemia/mental retardation syndrome X-linked (ATRX), death domain–associated protein (DAXX), and activin receptor type 1 (ACVR1) (Fig. 2) (18, 50, 51). TP53 mutations overlap significantly with H3F3A mutations (~70% of K27M and nearly all with G34V/R mutation) (11, 17, 18, 41, 42). MYCN is up-regulated but not amplified in pediatric high-grade glioma with G34V/R, as compared to H3F3A wild-type tumors (38). Although MYCN has long been considered as an “undruggable” target, strategies have emerged to destabilize the MYCN protein and thereby inhibit its activity (52). The bromodomain inhibitor JQ1 has recently been shown to target MYCN-amplified tumors by blocking MYCN-dependent transcription (53), suggesting BET-bromodomain inhibitors as potential therapeutics for treating pediatric high-grade glioma with G34V/R mutation.
Mutations in the histone chaperones ATRX and DAXX affect the loading of histone H3.3 in heterochromatic regions of telomeres (54), and combined H3F3A mutations plus ATRX and/or DAXX mutation are strongly associated with telomerase-independent lengthening of telomeres (11). ATRX or DAXX mutations are reported in up to 50% of pediatric high-grade gliomas (Fig. 2) (55). For K27M mutant brainstem tumors, ATRX and DAXX mutations are present but are more common in older children and adolescents (10, 44). G34V/R mutation, in supratentorial high-grade gliomas, nearly always occurs with ATRX or DAXX mutation and additionally overlaps with TP53 mutation (Fig. 2) (11).
Recurrent mutations in ACVR1 (also known as ALK2) occur frequently in histone H3.1 K27M mutant DIPG (80%) but are not observed in other midline or cortical malignant gliomas (Fig. 2) (15, 17, 18, 43). These ACVR1 mutations have been shown to constitutively activate bone morphogenic protein (BMP)–dependent transforming growth factor–β (TGF-β) signaling, which results in SMAD transcription factor phosphorylation and elevated transcription of SMAD target genes.
The expression of oligodendrocyte lineage transcription factors 1 and 2 (OLIG1 and OLIG2), and the forebrain marker forkhead box G1 transcription factor (FOXG1), is also associated with histone mutations. H3.3 K27M tumors express high OLIG1 and OLIG2 and low FOXG1. In contrast, H3.3 G34V/R tumors express low OLIG1 and OLIG2 and high FOXG1 (1). Other gene alterations known to occur in pediatric high-grade gliomas include amplification of PDGFRA, MET, IGF1, CDK4, CDK6, and CCND1; mutation of PIK3CA and PIK3R1; and deletion of CDKN2A (17, 41, 42, 56). However, most of these alterations are not associated with histone gene mutations, and all are implicated in adult gliomas as well (2).
Epigenetic therapy for pediatric high-grade glioma
Epigenetic changes, either alone or in combination with gene mutations, drive tumor initiation and progression. Whereas targeting mutant and/or activated oncoproteins has been a popular cancer treatment concept for several years, therapeutic targeting of enzyme activities responsible for chromatin modifications and epigenetic regulation of gene expression is a relatively new strategy but has become of high interest in pediatric neuro-oncology (10, 57). For instance, there are two paths to increasing H3K27 methylation in K27M DIPG: either enhancing PRC2 methyltransferase activity or inhibiting K27 demethylase activity. Histone H3K27 is methylated by EZH2, a component of PRC2, and is demethylated by the KDM6 subfamily K27 demethylase JMJD3 and UTX (26, 29–32, 58–60). Support for one of these conceptual approaches has emerged, in association with a study whose results show antitumor activity from treating K27M DIPG with the JMJD3 K27 histone demethylase inhibitor GSKJ4, an investigational compound (Fig. 3) (61). By using GSKJ4 to treat patient-derived K27M mutant DIPG tumor cells and xenografts, JMJD3 inhibition was shown to increase K27me2 and K27me3 (57). GSKJ4 treatment–associated increases in K27me2 and K27me3 were, in turn, associated with a marked dose- and time-dependent inhibition of K27M mutant DIPG cell growth. In contrast, GSKJ4 had little effect against pediatric glioma cells with wild-type or G34V mutant H3F3A that lack K27M suppression of K27me2 and K27me3. In addition to the observed antiproliferative activity, GSKJ4 increased apoptosis of K27M mutant cells while having no apparent effect on apoptosis for tumor cells expressing wild type or G34V. Pharmacologic inhibition of JMJD3 in DIPG orthotopic xenografts reduced tumor growth and significantly extended animal survival, and analysis of treated tumors revealed decreased proliferation and increased apoptosis, relative to untreated control tumors. These results suggest that GSKJ4 antitumor activity is specific to K27M mutant tumors, both in vitro and in vivo, and this antitumor activity occurs in association with increasing K27me2 and K27me3 in tumor cells (Fig. 3) (57). Results from high-performance liquid chromatography revealed good penetration of GSKJ4 into the brain, including to the site of brainstem tumor development, following systemic administration of inhibitor, and further support the development of GSKJ4 or other histone demethylase inhibitors as potentially effective targeted therapy for DIPG patients.
It is significant that results from an additional study, involving T cell acute lymphoblastic leukemia (T-ALL), a hematological malignancy in which H3K27 methylation is reduced by loss-of-function EZH2 mutation, have also shown antitumor activity through use of the JMJD3 inhibitor GSKJ4 and, as observed for K27M DIPG, with an accompanying increase in K27M methylation (62). For both T-ALL and DIPG, inhibition of UTX, the other known H3K27 demethylase, did not impair proliferation. Additionally in T-ALL, the genes suppressed by GSKJ4 treatment overlapped significantly with genes upregulated by UTX knockdown. Collectively, the results from these studies suggest distinct chromatin-modifying roles for UTX and JMJD3 and additionally support JMJD3 as an emerging therapeutic epigenetic target for cancer treatment.
Whereas decreased EZH2 methyltransferase activity may be important to T-ALL and DIPG development, other cancers, including medulloblastoma, ependymoma, atypical teratoid/rhabdoid tumor, and lymphoma, are known to have increased EZH2 activity (63–66). This paradox of EZH2 function having either pro- or antitumor activity, may indicate that the effects of K27 methylation changes are cell context–dependent. It may also reflect our insufficient understanding of the relationships between histone modifications and cancer for establishing an integrated framework that provides logical and consistent explanation for all histone modification–cancer associations. The development of such a framework will likely require a detailed understanding of all histone modifications, rather than attempting explanations for tumor development based on the analysis of single modifications. For instance, H3K27 is also modified by acetylation, which is mutually exclusive of K27 methylation. Consistent with this, increased K27 acetylation has been shown to occur in concert with decreased K27me3 in K27M mutant glioma cells (37). Moreover, a recent study, using a Drosophila melanogaster model, showed that constitutive expression of H3K27M elevated H3K27 acetylation in conjunction with increased bromodomain-containing protein 1 (BRD1) and BRD4 localization to K27M mononucleosomes (67). These Drosophila K27M mutant models were shown to resemble Drosophila PRC2 loss-of-function phenotypes in which there is reduced H3K27 methylation and derepression of PRC2 target genes. The increased level of H3K27 acetylation in K27M cells suggests that K27 acetylation may also be responsible for glioma formation. Thus, neither K27 methylation nor acetylation can be studied in isolation, and the enzymatic activities responsible for all types of K27 modification need to be taken into account when investigating relationships between K27 modifications and tumor development.
The elevated level of H3K27 acetylation in DIPG suggests a potential patient benefit from being treated with histone deacetylase (HDAC) inhibitors. A substantial amount of preclinical data have been generated by multiple groups that indicate benefit from the use of a pan-HDAC inhibitor, vorinostat, to treat high-grade gliomas and other pediatric CNS tumors (68–70). Recently, a chemical screening applied to DIPG cell cultures identified panobinostat, a U.S. Food and Drug Administration–approved and potent HDAC inhibitor, as demonstrating the most antitumor activity against DIPG in vitro, an order of magnitude greater than vorinostat (71). Increased H3 acetylation and H3K27 trimethylation by the treatment of panobinostat suggest that polyacetylation of the H3 N-terminal tail can “detoxify” K27M-induced inhibition of PRC2 (72), and subsequently normalize the K27M gene expression signature and decrease the oncogenetic MYC target gene expression signature (71). A phase 1 clinical trial of panobinostat for progressive DIPG and high-grade glioma is currently in development (Pediatric Brain Tumor Consortium PBTC-047). A combination of panobinostat with GSKJ4 has shown a synergistic effect against DIPG cells (71), which supports a need for additional study of antitumor activities resulting from the simultaneous inhibition of multiple histone modifiers.
Histone gene mutation and future clinical trials
Clinical trials for children with brain cancer are beginning to incorporate new information involving pediatric glioma epigenetics into their study design. In the near future, it appears imminent that pediatric glioma patients will be stratified for treatment based on the presence or absence of a specific histone mutation and/or specific histone modifications. This stratification, in turn, will be facilitated by routine biopsy of tumor. Two ongoing clinical trials that include biopsy for patients with DIPG are expected to confirm the safety and feasibility of biopsy.
As is the case for most brain tumor clinical trials, single-agent epigenetic therapies are not anticipated to result in durable tumor response, but information from single-agent trials is nonetheless important for designing subsequent, second-generation trials involving combination approaches with additional epigenetic agents, and/or conventional cytotoxic chemotherapy. Clinical trial designs are also expected to include the use of approaches for monitoring tumor response to treatment, which requires the identification and validation of noninvasive biomarkers indicative of residual tumor status (for example, detection of cell-free tumor DNA), and for evaluating molecular effects of therapy through analysis of biopsy tissue obtained during and/or after treatment.
SUMMARY
In the past 3 years, our knowledge of pediatric high-grade glioma genetics and epigenetics has significantly improved. Highlighted in association with this improved understanding is the identification of histone mutations in several glioma subtypes, especially DIPG. Efforts to understand the function of these mutations and their complex interactions with other known oncogenes are under way. Further, elegant preclinical models have been developed and are being used to study novel epigenetic therapies. Clinical trials have begun to explore histone modifier inhibitors for antitumor activity, and for interactions with conventional genotoxic therapies, in an effort to improve outcomes for children affected by these devastating malignancies.
Acknowledgments
We thank C. David James for the critical review of the manuscript and M. Gallagher for the illustration of the figures. Funding: Supported by the Matthew Larson Foundation (R.H.), Bear Necessities Pediatric Cancer Foundation and Rally Foundation (R.H.), U.S. NIH grant RO1NS093079 (R.H.), John McNicholas Pediatric Brain Tumor Foundation (R.R.L., A.M.S., and R.H.), The Bagan Family Fellowship at the Neurosurgery Research and Education Foundation (A.M.S.), The Cure Starts Now Foundation (A.M.S.), Northwestern Memorial Faculty Foundation (A.M.S.), and Northwestern University Clinical and Translational Sciences Institute grants UL1TR001422-01 and 1KL2TR001424-0 (A.M.S.) Author contributions: A.M.S., R.R.L., and R.H. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data used to obtain the conclusions in this paper are available in the U.S. National Library of Medicine and NIH, or presented in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
REFERENCES AND NOTES
- 1.Sturm D., Witt H., Hovestadt V., Khuong-Quang D.-A., Jones D. T. W., Konermann C., Pfaff E., Tönjes M., Sill M., Bender S., Kool M., Zapatka M., Becker N., Zucknick M., Hielscher T., Liu X.-Y., Fontebasso A. M., Ryzhova M., Albrecht S., Jacob K., Wolter M., Ebinger M., Schuhmann M. U., van Meter T., Frühwald M. C., Hauch H., Pekrun A., Radlwimmer B., Niehues T., von Komorowski G., Dürken M., Kulozik A. E., Madden J., Donson A., Foreman N. K., Drissi R., Fouladi M., Scheurlen W., von Deimling A., Monoranu C., Roggendorf W., Herold-Mende C., Unterberg A., Kramm C. M., Felsberg J., Hartmann C., Wiestler B., Wick W., Milde T., Witt O., Lindroth A. M., Schwartzentruber J., Faury D., Fleming A., Zakrzewska M., Liberski P. P., Zakrzewski K., Hauser P., Garami M., Klekner A., Bognar L., Morrissy S., Cavalli F., Taylor M. D., van Sluis P., Koster J., Versteeg R., Volckmann R., Mikkelsen T., Aldape K., Reifenberger G., Collins V. P., Majewski J., Korshunov A., Lichter P., Plass C., Jabado N., Pfister S. M., Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22, 425–437 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Sturm D., Bender S., Jones D. T. W., Lichter P., Grill J., Becher O., Hawkins C., Majewski J., Jones C., Costello J. F., Iavarone A., Aldape K., Brennan C. W., Jabado N., Pfister S. M., Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 14, 92–107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donaldson S. S., Laningham F., Fisher P. G., Advances toward an understanding of brainstem gliomas. J. Clin. Oncol. 24, 1266–1272 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Warren K. E., Diffuse intrinsic pontine glioma: Poised for progress. Front. Oncol. 2, 205 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robison N. J., Kieran M. W., Diffuse intrinsic pontine glioma: A reassessment. J. Neurooncol. 119, 7–15 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Roos D. E., Smith J. G., Randomized trial on radiotherapy for paediatric diffuse intrinsic pontine glioma (DIPG). Radiother. Oncol. 113, 425 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Fontanilla H. P., Pinnix C. C., Ketonen L. M., Woo S. Y., Vats T. S., Rytting M. E., Wolff J. E., Mahajan A., Palliative reirradiation for progressive diffuse intrinsic pontine glioma. Am. J. Clin. Oncol. 35, 51–57 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Barkovich A. J., Krischer J., Kun L. A., Packer R. J., Zimmerman R. A., Freeman C. R., Wara W. M., Albright L., Allen J. C., Hoffman H. J., Brain stem gliomas: A classification system based on magnetic resonance imaging. Pediatr. Neurosurg. 16, 73–83 (1990). [DOI] [PubMed] [Google Scholar]
- 9.Phillips H. S., Kharbanda S., Chen R., Forrest W. F., Soriano R. H., Wu T. D., Misra A., Nigro J. M., Colman H., Soroceanu L., Williams P. M., Modrusan Z., Feuerstein B. G., Aldape K., Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Khuong-Quang D.-A., Buczkowicz P., Rakopoulos P., Liu X.-Y., Fontebasso A. M., Bouffet E., Bartels U., Albrecht S., Schwartzentruber J., Letourneau L., Bourgey M., Bourque G., Montpetit A., Bourret G., Lepage P., Fleming A., Lichter P., Kool M., von Deimling A., Sturm D., Korshunov A., Faury D., Jones D. T., Majewski J., Pfister S. M., Jabado N., Hawkins C., K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 124, 439–447 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartzentruber J., Korshunov A., Liu X.-Y., Jones D. T. W., Pfaff E., Jacob K., Sturm D., Fontebasso A. M., Quang D.-A. K., Tönjes M., Hovestadt V., Albrecht S., Kool M., Nantel A., Konermann C., Lindroth A., Jäger N., Rausch T., Ryzhova M., Korbel J. O., Hielscher T., Hauser P., Garami M., Klekner A., Bognar L., Ebinger M., Schuhmann M. U., Scheurlen W., Pekrun A., Frühwald M. C., Roggendorf W., Kramm C., Dürken M., Atkinson J., Lepage P., Montpetit A., Zakrzewska M., Zakrzewski K., Liberski P. P., Dong Z., Siegel P., Kulozik A. E., Zapatka M., Guha A., Malkin D., Felsberg J., Reifenberger G., von Deimling A., Ichimura K., Collins V. P., Witt H., Milde T., Witt O., Zhang C., Castelo-Branco P., Lichter P., Faury D., Tabori U., Plass C., Majewski J., Pfister S. M., Jabado N., Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Wu G., Broniscer A., McEachron T. A., Lu C., Paugh B. S., Becksfort J., Qu C., Ding L., Huether R., Parker M., Zhang J., Gajjar A., Dyer M. A., Mullighan C. G., Gilbertson R. J., Mardis E. R., Wilson R. K., Downing J. R., Ellison D. W., Zhang J., Baker S. J.; St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome Project , Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henikoff S., Nucleosome destabilization in the epigenetic regulation of gene expression. Nat. Rev. Genet. 9, 15–26 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Roujeau T., Machado G., Garnett M. R., Miquel C., Puget S., Geoerger B., Grill J., Boddaert N., Di Rocco F., Zerah M., Sainte-Rose C., Stereotactic biopsy of diffuse pontine lesions in children. J. Neurosurg. 107 (Suppl. 1), 1–4 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Puget S., Philippe C., Bax D. A., Job B., Varlet P., Junier M.-P., Andreiuolo F., Carvalho D., Reis R., Guerrini-Rousseau L., Roujeau T., Dessen P., Richon C., Lazar V., Le Teuff G., Sainte-Rose C., Geoerger B., Vassal G., Jones C., Grill J., Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLOS One 7, e30313 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geoerger B., Hargrave D., Thomas F., Ndiaye A., Frappaz D., Andreiuolo F., Varlet P., Aerts I., Riccardi R., Jaspan T., Chatelut E., Le Deley M.-C., Paoletti X., Saint-Rose C., Leblond P., Morland B., Gentet J.-C., Méresse V., Vassal G.; ITCC (Innovative Therapies for Children with Cancer) European Consortium , Innovative Therapies for Children with Cancer pediatric phase I study of erlotinib in brainstem glioma and relapsing/refractory brain tumors. Neuro Oncol. 13, 109–118 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fontebasso A. M., Papillon-Cavanagh S., Schwartzentruber J., Nikbakht H., Gerges N., Fiset P.-O., Bechet D., Faury D., De Jay N., Ramkissoon L. A., Corcoran A., Jones D. T. W., Sturm D., Johann P., Tomita T., Goldman S., Nagib M., Bendel A., Goumnerova L., Bowers D. C., Leonard J. R., Rubin J. B., Alden T., Browd S., Geyer J. R., Leary S., Jallo G., Cohen K., Gupta N., Prados M. D., Carret A.-S., Ellezam B., Crevier L., Klekner A., Bognar L., Hauser P., Garami M., Myseros J., Dong Z., Siegel P. M., Malkin H., Ligon A. H., Albrecht S., Pfister S. M., Ligon K. L., Majewski J., Jabado N., Kieran M. W., Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 46, 462–466 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor K. R., Mackay A., Truffaux N., Butterfield Y. S., Morozova O., Philippe C., Castel D., Grasso C. S., Vinci M., Carvalho D., Carcaboso A. M., de Torres C., Cruz O., Mora J., Entz-Werle N., Ingram W. J., Monje M., Hargrave D., Bullock A. N., Puget S., Yip S., Jones C., Grill J., Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 46, 457–461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker D. A., Liu J., Kieran M., Jabado N., Picton S., Packer R., St. Rose C.; CPN Paris 2011 Conference Consensus Group , A multi-disciplinary consensus statement concerning surgical approaches to low-grade, high-grade astrocytomas and diffuse intrinsic pontine gliomas in childhood (CPN Paris 2011) using the Delphi method. Neuro Oncol. 15, 462–468 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maze I., Noh K.-M., Allis C. D., Histone regulation in the CNS: Basic principles of epigenetic plasticity. Neuropsychopharmacology 38, 3–22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elsaesser S. J., Goldberg A. D., Allis C. D., New functions for an old variant: No substitute for histone H3.3. Curr. Opin. Genet. Dev. 20, 110–117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skene P. J., Henikoff S., Histone variants in pluripotency and disease. Development 140, 2513–2524 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Mosammaparast N., Shi Y., Reversal of histone methylation: Biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 79, 155–179 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Barski A., Cuddapah S., Cui K., Roh T.-Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K., High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Shen X., Liu Y., Hsu Y.-J., Fujiwara Y., Kim J., Mao X., Yuan G.-C., Orkin S. H., EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol. Cell 32, 491–502 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agger K., Cloos P. A. C., Christensen J., Pasini D., Rose S., Rappsilber J., Issaeva I., Canaani E., Salcini A. E., Helin K., UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449, 731–734 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Broniscer A., Baker J. N., Baker S. J., Chi S. N., Geyer J. R., Morris E. B., Gajjar A., Prospective collection of tissue samples at autopsy in children with diffuse intrinsic pontine glioma. Cancer 116, 4632–4637 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deb G., Singh A. K., Gupta S., EZH2: Not EZHY (easy) to deal. Mol. Cancer Res. 12, 639–653 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ezponda T., Licht J. D., Molecular pathways: Deregulation of histone H3 lysine 27 methylation in cancer—Different paths, same destination. Clin. Cancer Res. 20, 5001–5008 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simon J. A., Lange C. A., Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 647, 21–29 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Kleer C. G., Cao Q., Varambally S., Shen R., Ota I., Tomlins S. A., Ghosh D., Sewalt R. G. A. B., Otte A. P., Hayes D. F., Sabel M. S., Livant D., Weiss S. J., Rubin M. A., Chinnaiyan A. M., EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 100, 11606–11611 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varambally S., Dhanasekaran S. M., Zhou M., Barrette T. R., Kumar-Sinha C., Sanda M. G., Ghosh D., Pienta K. J., Sewalt R. G. A. B., Otte A. P., Rubin M. A., Chinnaiyan A. M., The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 624–629 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Van der Meulen J., Speleman F., Van Vlierberghe P., The H3K27me3 demethylase UTX in normal development and disease. Epigenetics 9, 658–668 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gui Y., Guo G., Huang Y., Hu X., Tang A., Gao S., Wu R., Chen C., Li X., Zhou L., He M., Li Z., Sun X., Jia W., Chen J., Yang S., Zhou F., Zhao X., Wan S., Ye R., Liang C., Liu Z., Huang P., Liu C., Jiang H., Wang Y., Zheng H., Sun L., Liu X., Jiang Z., Feng D., Chen J., Wu S., Zou J., Zhang Z., Yang R., Zhao J., Xu C., Yin W., Guan Z., Ye J., Zhang H., Li J., Kristiansen K., Nickerson M. L., Theodorescu D., Li Y., Zhang X., Li S., Wang J., Yang H., Wang J., Cai Z., Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 43, 875–878 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ross J. S., Wang K., Al-Rohil R. N., Nazeer T., Sheehan C. E., Otto G. A., He J., Palmer G., Yelensky R., Lipson D., Ali S., Balasubramanian S., Curran J. A., Garcia L., Mahoney K., Downing S. R., Hawryluk M., Miller V. A., Stephens P. J., Advanced urothelial carcinoma: Next-generation sequencing reveals diverse genomic alterations and targets of therapy. Mod. Pathol. 27, 271–280 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Agger K., Cloos P. A. C., Rudkjær L., Williams K., Andersen G., Christensen J., Helin K., The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A–ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 23, 1171–1176 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis P. W., Müller M. M., Koletsky M. S., Cordero F., Lin S., Banaszynski L. A., Garcia B. A., Muir T. W., Becher O. J., Allis C. D., Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bjerke L., Mackay A., Nandhabalan M., Burford A., Jury A., Popov S., Bax D. A., Carvalho D., Taylor K. R., Vinci M., Bajrami I., McGonnell I. M., Lord C. J., Reis R. M., Hargrave D., Ashworth A., Workman P., Jones C., Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 3, 512–519 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan K.-M., Fang D., Gan H., Hashizume R., Yu C., Schroeder M., Gupta N., Mueller S., James C. D., Jenkins R., Sarkaria J., Zhang Z., The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 27, 985–990 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waldmann T., Schneider R., Targeting histone modifications—Epigenetics in cancer. Curr. Opin. Cell Biol. 25, 184–189 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Buczkowicz P., Hoeman C., Rakopoulos P., Pajovic S., Letourneau L., Dzamba M., Morrison A., Lewis P., Bouffet E., Bartels U., Zuccaro J., Agnihotri S., Ryall S., Barszczyk M., Chornenkyy Y., Bourgey M., Bourque G., Montpetit A., Cordero F., Castelo-Branco P., Mangerel J., Tabori U., Ho K. C., Huang A., Taylor K. R., Mackay A., Bendel A. E., Nazarian J., Fangusaro J. R., Karajannis M. A., Zagzag D., Foreman N. K., Donson A., Hegert J. V., Smith A., Chan J., Lafay-Cousin L., Dunn S., Hukin J., Dunham C., Scheinemann K., Michaud J., Zelcer S., Ramsay D., Cain J., Brennan C., Souweidane M. M., Jones C., Allis C. D., Brudno M., Becher O., Hawkins C., Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 46, 451–456 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu G., Diaz A. K., Paugh B. S., Rankin S. L., Ju B., Li Y., Zhu X., Qu C., Chen X., Zhang J., Easton J., Edmonson M., Ma X., Lu C., Nagahawatte P., Hedlund E., Rusch M., Pounds S., Lin T., Onar-Thomas A., Huether R., Kriwacki R., Parker M., Gupta P., Becksfort J., Wei L., Mulder H. L., Boggs K., Vadodaria B., Yergeau D., Russell J. C., Ochoa K., Fulton R. S., Fulton L. L., Jones C., Boop F. A., Broniscer A., Wetmore C., Gajjar A., Ding L., Mardis E. R., Wilson R. K., Taylor M. R., Downing J. R., Ellison D. W., Zhang J., Baker S. J.; St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome Project , The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 46, 444–450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Becher O. J., Wechsler-Reya R. J., For pediatric glioma, leave no histone unturned. Science 346, 1458–1459 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Gerges N., Fontebasso A. M., Albrecht S., Faury D., Jabado N., Pediatric high-grade astrocytomas: A distinct neuro-oncological paradigm. Genome Med. 5, 66 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aihara K., Mukasa A., Gotoh K., Saito K., Nagae G., Tsuji S., Tatsuno K., Yamamoto S., Takayanagi S., Narita Y., Shibui S., Aburatani H., Saito N., H3F3A K27M mutations in thalamic gliomas from young adult patients. Neuro Oncol. 16, 140–146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bender S., Tang Y., Lindroth A. M., Hovestadt V., Jones D. T. W., Kool M., Zapatka M., Northcott P. A., Sturm D., Wang W., Radlwimmer B., Højfeldt J. W., Truffaux N., Castel D., Schubert S., Ryzhova M., Şeker-Cin H., Gronych J., Johann P. D., Stark S., Meyer J., Milde T., Schuhmann M., Ebinger M., Monoranu C.-M., Ponnuswami A., Chen S., Jones C., Witt O., Collins V. P., von Deimling A., Jabado N., Puget S., Grill J., Helin K., Korshunov A., Lichter P., Monje M., Plass C., Cho Y.-J., Pfister S. M., Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Venneti S., Garimella M. T., Sullivan L. M., Martinez D., Huse J. T., Heguy A., Santi M., Thompson C. B., Judkins A. R., Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol. 23, 558–564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ederveen T. H. A., Mandemaker I. K., Logie C., The human histone H3 complement anno 2011. Biochim. Biophys. Acta 1809, 577–586 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Funato K., Major T., Lewis P. W., Allis C. D., Tabar V., Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346, 1529–1533 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bax D. A., Mackay A., Little S. E., Carvalho D., Viana-Pereira M., Tamber N., Grigoriadis A. E., Ashworth A., Reis R. M., Ellison D. W., Al-Sarraj S., Hargrave D., Jones C., A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin. Cancer Res. 16, 3368–3377 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paugh B. S., Qu C., Jones C., Liu Z., Adamowicz-Brice M., Zhang J., Bax D. A., Coyle B., Barrow J., Hargrave D., Lowe J., Gajjar A., Zhao W., Broniscer A., Ellison D. W., Grundy R. G., Baker S. J., Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J. Clin. Oncol. 28, 3061–3068 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barone G., Anderson J., Pearson A. D. J., Petrie K., Chesler L., New strategies in neuroblastoma: Therapeutic targeting of MYCN and ALK. Clin. Cancer Res. 19, 5814–5821 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Puissant A., Frumm S. M., Alexe G., Bassil C. F., Qi J., Chanthery Y. H., Nekritz E. A., Zeid R., Gustafson W. C., Greninger P., Garnett M. J., McDermott U., Benes C. H., Kung A. L., Weiss W. A., Bradner J. E., Stegmaier K., Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 3, 308–323 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szenker E., Ray-Gallet D., Almouzni G., The double face of the histone variant H3.3. Cell Res. 21, 421–434 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pathak P., Jha P., Purkait S., Sharma V., Suri V., Sharma M. C., Faruq M., Suri A., Sarkar C., Altered global histone-trimethylation code and H3F3A-ATRX mutation in pediatric GBM. J. Neurooncol 121, 489–497 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Dawson M. A., Kouzarides T., Cancer epigenetics: From mechanism to therapy. Cell 150, 12–27 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Hashizume R., Andor N., Ihara Y., Lerner R., Gan H., Chen X., Fang D., Huang X., Tom M. W., Ngo V., Solomon D., Mueller S., Paris P. L., Zhang Z., Petritsch C., Gupta N., Waldman T. A., James C. D., Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 20, 1394–1396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hübner M. R., Spector D. L., Role of H3K27 demethylases Jmjd3 and UTX in transcriptional regulation. Cold Spring Harb. Symp. Quant. Biol. 75, 43–49 (2010). [DOI] [PubMed] [Google Scholar]
- 59.Kooistra S. M., Helin K., Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 13, 297–311 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Cloos P. A. C., Christensen J., Agger K., Helin K., Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev. 22, 1115–1140 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kruidenier L., Chung C.-w., Cheng Z., Liddle J., Che K., Joberty G., Bantscheff M., Bountra C., Bridges A., Diallo H., Eberhard D., Hutchinson S., Jones E., Katso R., Leveridge M., Mander P. K., Mosley J., Ramirez-Molina C., Rowland P., Schofield C. J., Sheppard R. J., Smith J. E., Swales C., Tanner R., Thomas P., Tumber A., Drewes G., Oppermann U., Patel D. J., Lee K., Wilson D. M., A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 488, 404–408 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ntziachristos P., Tsirigos A., Welstead G. G., Trimarchi T., Bakogianni S., Xu L., Loizou E., Holmfeldt L., Strikoudis A., King B., Mullenders J., Becksfort J., Nedjic J., Paietta E., Tallman M. S., Rowe J. M., Tonon G., Satoh T., Kruidenier L., Prinjha R., Akira S., Van Vlierberghe P., Ferrando A. A., Jaenisch R., Mullighan C. G., Aifantis I., Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 514, 513–517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang X., Dubuc A. M., Ramaswamy V., Mack S., Gendoo D. M. A., Remke M., Wu X., Garzia L., Luu B., Cavalli F., Peacock J., López B., Skowron P., Zagzag D., Lyden D., Hoffman C., Cho Y.-J., Eberhart C., MacDonald T., Li X.-N., Van Meter T., Northcott P. A., Haibe-Kains B., Hawkins C., Rutka J. T., Bouffet E., Pfister S. M., Korshunov A., Taylor M. D., Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol. 129, 449–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dubuc A. M., Remke M., Korshunov A., Northcott P. A., Zhan S. H., Mendez-Lago M., Kool M., Jones D. T. W., Unterberger A., Morrissy A. S., Shih D., Peacock J., Ramaswamy V., Rolider A., Wang X., Witt H., Hielscher T., Hawkins C., Vibhakar R., Croul S., Rutka J. T., Weiss W. A., Jones S. J. M., Eberhart C. G., Marra M. A., Pfister S. M., Taylor M. D., Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 125, 373–384 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mack S. C., Witt H., Piro R. M., Gu L., Zuyderduyn S., Stütz A. M., Wang X., Gallo M., Garzia L., Zayne K., Zhang X., Ramaswamy V., Jäger N., Jones D. T. W., Sill M., Pugh T. J., Ryzhova M., Wani K. M., Shih D. J. H., Head R., Remke M., Bailey S. D., Zichner T., Faria C. C., Barszczyk M., Stark S., Seker-Cin H., Hutter S., Johann P., Bender S., Hovestadt V., Tzaridis T., Dubuc A. M., Northcott P. A., Peacock J., Bertrand K. C., Agnihotri S., Cavalli F. M. G., Clarke I., Nethery-Brokx K., Creasy C. L., Verma S. K., Koster J., Wu X., Yao Y., Milde T., Sin-Chan P., Zuccaro J., Lau L., Pereira S., Castelo-Branco P., Hirst M., Marra M. A., Roberts S. S., Fults D., Massimi L., Cho Y. J., Van Meter T., Grajkowska W., Lach B., Kulozik A. E., von Deimling A., Witt O., Scherer S. W., Fan X., Muraszko K. M., Kool M., Pomeroy S. L., Gupta N., Phillips J., Huang A., Tabori U., Hawkins C., Malkin D., Kongkham P. N., Weiss W. A., Jabado N., Rutka J. T., Bouffet E., Korbel J. O., Lupien M., Aldape K. D., Bader G. D., Eils R., Lichter P., Dirks P. B., Pfister S. M., Korshunov A., Taylor M. D., Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506, 445–450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Okosun J., Bödör C., Wang J., Araf S., Yang C.-Y., Pan C., Boller S., Cittaro D., Bozek M., Iqbal S., Matthews J., Wrench D., Marzec J., Tawana K., Popov N., O’Riain C., O’Shea D., Carlotti E., Davies A., Lawrie C. H., Matolcsy A., Calaminici M., Norton A., Byers R. J., Mein C., Stupka E., Lister T. A., Lenz G., Montoto S., Gribben J. G., Fan Y., Grosschedl R., Chelala C., Fitzgibbon J., Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet. 46, 176–181 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Herz H.-M., Morgan M., Gao X., Jackson J., Rickels R., Swanson S. K., Florens L., Washburn M. P., Eissenberg J. C., Shilatifard A., Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 345, 1065–1070 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kitange G. J., Mladek A. C., Carlson B. L., Schroeder M. A., Pokorny J. L., Cen L., Decker P. A., Wu W., Lomberk G. A., Gupta S. K., Urrutia R. A., Sarkaria J. N., Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin. Cancer Res. 18, 4070–4079 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Singh M. M., Johnson B., Venkatarayan A., Flores E. R., Zhang J., Su X., Barton M., Lang F., Chandra J., Preclinical activity of combined HDAC and KDM1A inhibition in glioblastoma. Neuro. Oncol. 17, 1463–1473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Milde T., Kleber S., Korshunov A., Witt H., Hielscher T., Koch P., Kopp H.-G., Jugold M., Deubzer H. E., Oehme I., Lodrini M., Gröne H.-J., Benner A., Brüstle O., Gilbertson R. J., von Deimling A., Kulozik A. E., Pfister S. M., Martin-Villalba A., Witt O., A novel human high-risk ependymoma stem cell model reveals the differentiation-inducing potential of the histone deacetylase inhibitor Vorinostat. Acta Neuropathol. 122, 637–650 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grasso C. S., Tang Y., Truffaux N., Berlow N. E., Liu L., Debily M.-A., Quist M. J., Davis L. E., Huang E. C., Woo P. J., Ponnuswami A., Chen S., Johung T. B., Sun W., Kogiso M., Du Y., Qi L., Huang Y., Hütt-Cabezas M., Warren K. E, Le Dret L., Meltzer P. S., Mao H., Quezado M., van Vuurden D. G., Abraham J., Fouladi M., Svalina M. N., Wang N., Hawkins C., Nazarian J., Alonso M. M., Raabe E. H., Hulleman E., Spellman P. T, Li X.-N., Keller C., Pal R., Grill J., Monje M., Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 21, 555–559 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brown Z. Z., Müller M. M., Jain S. U., Allis C. D., Lewis P. W., Muir T. W., Strategy for "detoxification" of a cancer-derived histone mutant based on mapping its interaction with the methyltransferase PRC2. J. Am. Chem. Soc. 136, 13498–13501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]