

Abstract
Undesirable truncated recombinant protein products pose a special expression and purification challenge because such products often share similar chromatographic properties as the desired full length protein. We describe here our observation of both full length and a truncated form of a yeast protein (Gcn5) expressed in E. coli, and the reduction or elimination of the truncated form by mutating a cryptic Shine-Dalgarno or START codon within the Gcn5 coding region. Unsuccessful attempts to engineer in a cryptic translation initiation site into other recombinant proteins suggest that cryptic Shine-Dalgarno or START codon sequences are necessary but not sufficient for cryptic translation in E. coli.
Keywords: recombinant protein expression, cryptic initiation, E. coli expression
Introduction
Heterologous overexpression in E. coli is a common technique for producing recombinant proteins [1-3]. Although this mature technology has many advantages such as speed, low cost and simplicity, complications can include insoluble or inactive products, low levels of expression or the occurrence of truncated products. Truncated products may pose a particular problem for the subsequent purification of the recombinant protein because the truncated protein is shorter but otherwise identical in amino acid sequence to the desired full length protein. Truncated polypeptides produced in E. coli likely result from limited proteolysis of the heterologous protein or from improper initiation of translation.
Our understanding of the mechanism of translational initiation has been significantly enhanced by recent important structural investigations of the ribosome [4-8]. The key steps in prokaryotic translational initiation are the binding of mRNA containing a Shine-Dalgarno or ribosomal binding sequence and a START codon to a complex of the 30S ribosomal subunit, initiation factors and formyl-Met tRNA, adaptation of the mRNA to the ribosome 30S subunit mRNA channel which exposes the START codon to bind to the fMet-tRNA, and subsequent binding of the 50S ribosomal subunit.
The prokaryotic Shine-Dalgarno sequence forms the ribosome binding site on the mRNA through base pairing with the complementary sequence at the 3′ end of the ribosomal 16S rRNA [9]. In E. coli, the consensus Shine-Dalgarno sequence is AGGAGGT but a Shine-Dalgarno site does not need to match this consensus to be functional [10,11]. The 3′ edge of this sequence is usually located 3 to 7 nucleotides from the first base of the START codon. Although ATG is the canonical START codon, it accounts for only 83% of the START codons in E. coli genes [12]. The GTG codon, which otherwise codes for Val, is found at the start of 14% of E. coli genes, while the TTG Leu codon is found at the start of 3% of E. coli genes. The ATG, GTG or TTG START codons are all recognized by the formyl-Met tRNA, resulting in a formyl-Met at the N-terminus of the newly synthesized polypeptide.
Given the relatively degenerate sequence requirements for the Shine-Dalgarno site and the START codon and the variable distance possible between these two sequence elements [10,13], one might expect these sequences to be found internal to coding regions in addition to the canonical location at the 5′ end of the coding region. For example, such potential cryptic initiation sites might be found in the coding regions of heterologous genes since there would be no evolutionary pressure to avoid such occurrences. If such cryptic initiation sites occurred in frame with the reading frame of the full length gene, a truncated protein product could result.
We describe here our finding that a cryptic initiation site comprised of a near consensus Shine-Dalgarno site coupled with a GTG START codon accounted for a truncated product when a particular yeast gene (Gcn5, a histone acetyltransferase) was expressed in E. coli. The truncated product could be nearly eliminated by silent mutations that removed the cryptic initiation sequence. However, installing a consensus Shine-Dalgarno sequence spaced appropriately from a GTG potential START codon in a different context was not sufficient to cause translation of a truncated product.
Results
Truncated yeast Gcn5 polypeptide coexpressed in E. coli
We have previously described methods to express and purify recombinant protein complexes by coexpression from polycistronic vectors in E. coli [14-16]. While we were purifying recombinant yeast Ada2/Ada3/Gcn5 SAGA histone acetyltransferase subcomplexes produced by coexpression, we observed a 41 kD polypeptide which copurified with the desired Ada2/Ada3/Gcn5 complexes (hexahistidine tagged on the Ada3 subunit) over multiple chromatography steps including metal affinity, cation-exchange, anion-exchange and size exclusion chromatography (data not shown). For example, the 41 kD polypeptide was present when we coexpressed a particular deletion Ada2/Ada3/Gcn5 variant and partially purified the complex by metal affinity chromatography (Fig. 1). The same truncated Gcn5 product was observed with the BL21(DE3)pLysS or BL21-CodonPlus(DE3)-RIL E. coli host strains, but for unknown reasons, significantly better purification over Talon metal affinity resin was obtained using BL21-CodonPlus(DE3)-RIL cells [17].
Figure 1.
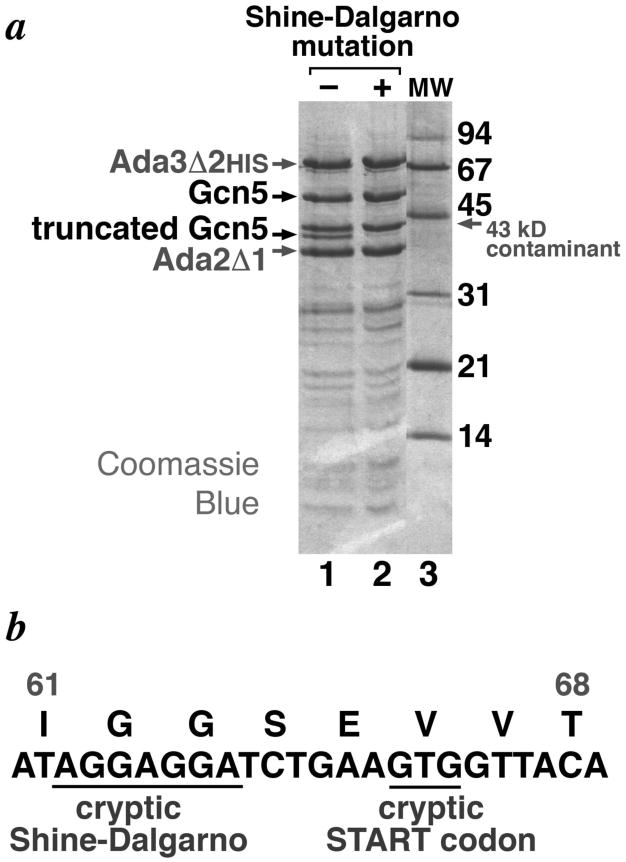
Truncated Gcn5 product in recombinant yeast Ada2/Ada3/Gcn5 complex expressed in E. coli. (a) SDS-PAGE gel of metal-affinity purified yeast Ada2Δ1/Ada3Δ2HIS/Gcn5 complex expressed in E. coli BL21-CodonPlus(DE3)-RIL cells of wild-type Gcn5 sequence (lane 1) and Gcn5 sequence mutated to remove cryptic Shine-Dalgarno sequence in codons 62 and 63 (lane 2). The prominent 43 kD contaminant was determined to be E. coli translation elongation factor EFTu [17]. Molecular weight markers are show in lane 3 (b) Amino acid and DNA sequence around cryptic Shine Dalgarno and cryptic START codon in native yeast Gcn5.
Since it was unlikely that an E. coli contaminant would copurify through all these chromatographic procedures, we suspected that the 41 kD polypeptide was a proteolytic degradation product of Ada3 or Gcn5 (the 41 kD polypeptide was unlikely to be a proteolytic product of Ada2 since it migrated slower on SDS-PAGE than the Ada2Δ1 deletion). To determine the identity of the 41 kD polypeptide, we therefore analyzed this polypeptide by N-terminal protein sequencing. The sequencing results indicated that the 41 kD polypeptide contained an N-terminus starting from Gcn5 position 67, although the limited sample amount and/or potential variability due to partial processing by methionine aminopeptidase precluded unambiguous identification of the first three amino acids (the following 5 residues were identified unambiguously and matched the corresponding residues in the Gcn5 sequence). We concluded that the 41 kD polypeptide corresponds to Gcn5 from Val67 through to its C-terminus, i.e. Gcn5(67-439) because of the good agreement between the expected and observed molecular weights (41.4 kD vs 41 kD). We surmised that truncation of the Gcn5 polypeptide occurred in the cell and not during purification because the 41 kD polypeptide was observed in Western blots of crude cell extracts prepared by boiling the recombinant E. coli cells in SDS-PAGE loading buffer. The explanation we favored at this time was that the truncated product resulted from proteolysis in vivo.
Cryptic initiation of translation produces truncated yeast Gcn5 coexpressed in E. coli
An alternate interpretation of the previous observations was prompted by discussions with David Garboczi (NIAID, NIH) who had noted translation from cryptic initiation sites in E. coli, including ones from Val GTG codons. These discussions spurred us to examine the coding sequence around the truncated Gcn5 product. We found the sequence AGGAGGA, a near perfect match to the consensus E. coli Shine-Dalgarno AGGAGGT, positioned 6 bp upstream of GTG, the codon for Val67. This suggested the possibility that the truncated product, Gcn5(67-439) resulted not from proteolysis but instead from initiation of translation from a cryptic initiation site at Val67.
To test this hypothesis, we engineered translationally silent mutations in yeast Gcn5 that removed the cryptic Shine-Dalgarno site, the cryptic START GTG codon or both. We coexpressed these translationally silent Gcn5 mutants together with Ada2 and hexahistidine tagged Ada3, and prepared crude extracts as well as partially purified the complex by metal affinity chromatography. The crude extract samples showed us truncations that presumably occurred in the cell and not during the purification process, while the metal affinity purified samples allowed us to distinguish between Gcn5 polypeptides in the tagged complex from polypeptides in the crude extract that cross-react with the anti-Gcn5 antibodies used for the Western blot. As Fig. 2 shows, the truncated Gcn5 product is found in both the crude extract and in the metal affinity purified complex (Fig. 2, lanes 1 and 2), consistent with it copurifying with the Ada2/Ada3/Gcn5 complex tagged on the Ada3 subunit. When the cryptic Shine-Dalgarno site was removed by silently mutating the natural Gcn5 AGGAGGA sequence to AGGCGGC, the band corresponding to the truncated Gcn5 polypeptide is almost completely removed and only a faint band at this position on the gel remains in both the crude extract and in the partially purified complex (Fig. 2 lanes 3 and 4). Mutating the cryptic START GTG site to a GTT codon appears to be even more effective at eliminating the truncated product since even the faint band observed with the Shine-Dalgarno mutation is not evident (Fig. 2 lanes 5 and 6). Mutating both the cryptic Shine-Dalgarno and the START GTG sites produced results similar to only mutating the START GTG site (Fig. 2 lanes 7 and 8). These results support the hypothesis that initiation from a cryptic site is responsible for the truncated Gcn5 product in our recombinant Ada2/Ada3/Gcn5 complexes, and further that the truncated Gcn5 product can be significantly reduced by silent mutations that remove the cryptic initiation site.
Figure 2.
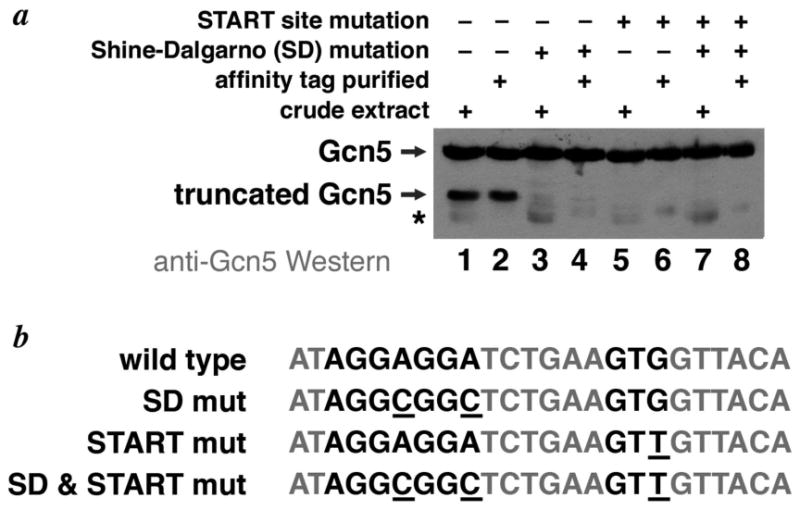
Anti-Gcn5 Western analysis of variant yeast Ada2Δ1/Ada3Δ3HIS/Gcn5 complexes coexpressed in E. coli. (a) Gcn5 in crude extracts (lanes 1, 3, 5, 7) or metal affinity purified (lanes, 2, 4, 6, 8) complexes coexpressed in BL21-CodonPlus(DE3)-RIL was detected using aniti-Gcn5 antibodies. The wild-type Gcn5 sequence was used in lanes 1 & 2, while the Gcn5 sequence mutated to remove the cryptic Shine-Dalgarno mutation or START codon or both elements are shown in lanes 3 & 4, lanes 5 & 6 and lanes 7&8 respectively. The truncated Gcn5 polypeptide is greatly reduced by the Shine-Dalgarno mutation, and is not detected in the START mutation sample. The position of a contaminating band that cross reacts with the anti-Gcn5 antibodies is indicated by an asterisk (*). (b) Amino acid and DNA sequences for the wild type, Shine-Dalgarno (SD), START mutation and combination Shine-Dalgarno & START mutations
Cryptic initiation produces truncated yeast Gcn5 expressed on its own in E. coli
The truncated Gcn5 product resulting from cryptic initiation was observed during coexpression of the Ada2/Ada3/Gcn5 complex by polycistronic expression in E. coli, a relatively specialized method. We therefore also investigated whether the cryptic initiation of yeast Gcn5 would occur if Gcn5 were overexpressed on its own in a more traditional monocistronic expression vector in E. coli. We find that both the full length and the truncated Gcn5 products are induced when Gcn5 is expressed on its own (Fig. 3 lane 1). Furthermore, mutating the cryptic Shine-Dalgarno sequence also significantly reduces the amount of truncated Gcn5 product (Fig. 3 lane 2). While both the cryptic Shine-Dalgarno and GTG START codon are mutated, we still observe a faint band at the position of the truncated Gcn5 polypeptide whereas this band was not detectable even on an overexposed Western blot when the Gcn5 construct was coexpressed in the Ada2/Ada3/Gcn5 complex (compare Fig. 2 lane 7 and Fig. 3 lane 3). These results demonstrate that the cryptic initiation of Gcn5 was not an artifact of polycistronic expression.
Figure 3.
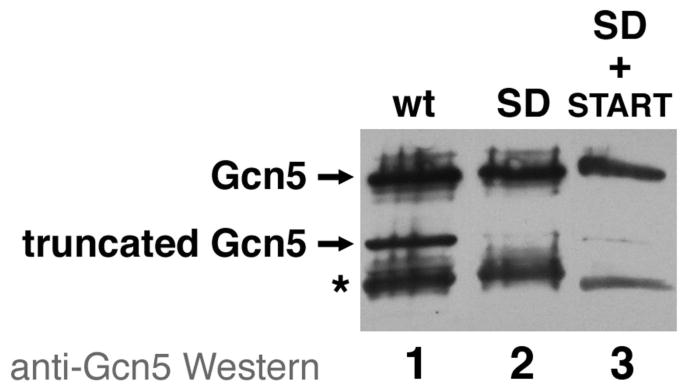
Anti-Gcn5 Western analysis of yeast Gcn5 protein expressed in E. coli. Gcn5 in crude extracts for wild-type, mutated to remove cryptic Shine-Dalgarno sequence or both cryptic Shine-Dalgarno and START sequences are shown in lanes 1, 2, and 3 respectively. The positions of full length and truncated Gcn5 are shown, as is the position of a prominent cross-reactive protein with an asterisk (*). The truncated Gcn5 polypeptide is greatly reduced by the Shine-Dalgarno mutation on its own in combination with the START codon mutation.
A potential Shine-Dalgarno and START sequence is not sufficient to cause cryptic initiation
Our finding that the truncated Gcn5 product is largely removed by the elimination of the cryptic initiation site indicates that the cryptic Shine-Dalgarno and START sites are necessary for the observed cryptic initiation of translation. We were interested to determine if the presence of potential Shine-Dalgarno and START sequences were sufficient to cause cryptic initiation. We tested this using two different proteins. The first, dihydrofolate reductase (DHFR), naturally contains a sequence which can be silently mutated to a consensus Shine-Dalgarno site spaced 6 bp upstream of a GTG potential START codon. While the second protein we investigated, glutathione S-transferase (GST), does not normally contain an equivalent sequence, a glycine-rich linker [18] engineered at its C-terminus provides a different context for a consensus Shine-Dalgarno site also spaced 6 bp upstream of a GTG codon.
Our results suggest that the presence of a consensus Shine-Dalgarno sequence 6 bp upstream of a GTG codon is not sufficient to cause cryptic intiation. We mutated a C-terminally HIS-tagged DHFR expression construct to create a potential cryptic site variant. If cryptic initiation occurs, the resulting polypeptide would have an expected molecular weight of 8.3 kD compared to the full length protein at 19.0 kD (it should be noted that the full length DHFRHIS polypeptide migrates anomalously slowly at a position expected for a 24 kD polypeptide vs its expected 19 kD molecular weight). Since both the full length and a truncated polypeptide resulting from cryptic initiation will contain the C-terminal HIS affinity tag, both are therefore expected to bind metal affinity resin and to bind anti-HIS antibodies. Fig. 4 shows that no such truncated polypeptide of the expected size is expressed when potential cryptic site mutations are introduced into DHFRHIS (Fig. 4, lanes 1 and 3). A 15 kD band is present for wildtype DHFRHIS, but this is presumably a contaminant that cross-reacts with the anti-HIS antibodies used to detect the HIS tagged polypeptide since this contaminant does not appear in the metal affinity purified fraction (Fig 4, lanes 1 and 2). Thus, the engineered internal Shine-Dalgarno and GTG START codon are not sufficient to cause cryptic initiation in this experiment.
Figure 4.
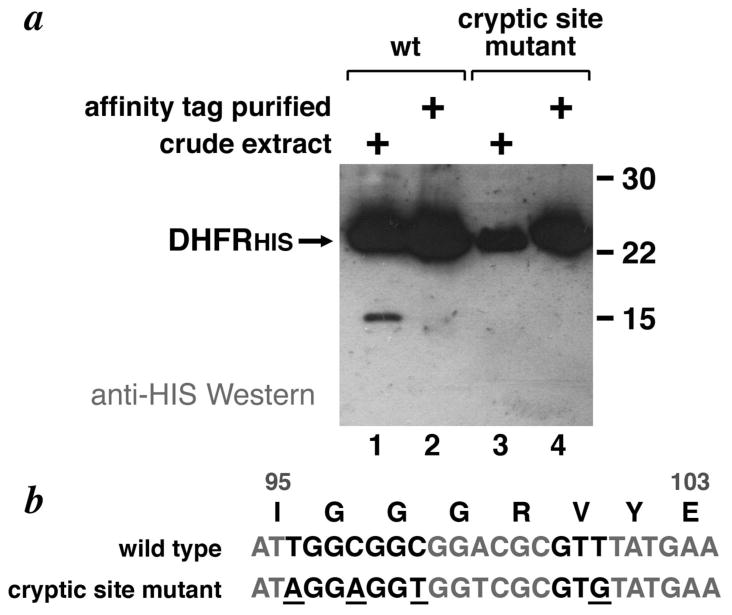
Anti-HIS Western analysis of wild-type and engineered cryptic site DHFR variant protein expressed in E. coli. (a) C-terminally HIS-tagged DHFR in crude extracts (lanes 1, 3) or metal affinity purified (lanes, 2, 4) complexes coexpressed in BL21(DE3)pLysS was detected using anti-DHFR antibodies. The wild-type DHFR sequence was used in lanes 1 & 2, while the DHFR sequence mutated to remove the cryptic Shine-Dalgarno mutation or START codon or both elements are shown in lanes 3 & 4 respectively. A polypeptide corresponding to translation from the engineered cryptic translation initiation site was not detected (b) Amino acid and DNA sequences for the wild type and cryptic initiation site DFHR mutant.
For the second experiment, we expressed a fusion protein containing GST as an N-terminal fusion to human RCC1 (regulator of chromosomal condensation) together with a C-terminal HIS tag. The full length protein GSThRCC1HIS would have an expected molecular weight of 72 kD whereas the product of cryptic initiation from the engineered cryptic Shine-Delgarno and GTG START codon would have an expected molecular weight of 46.3 kD. The full length protein is observed around its expected position on a Western blot (Fig. 5, lanes 1 and 2). A prominent band is observed near 46 kD, but this band is present for both the wild type and the mutant containing the engineered internal Shine-Dalgarno and GTG START codon sites (Fig. 5, lanes 1 and 3). Since this polypeptide is present both in the crude extract as well as the metal affinity purified fraction and is detected by anti-HIS antibodies, it likely corresponds to a C-terminal truncation of the full length GSThRCC1HIS (Fig. 5, lanes 1 to 4). However, given that the polypeptide is also present in the unmutated construct, its presence does not depend on the engineered internal initiation site. Therefore, we are unable to determine for this construct whether internal Shine-Dalgarno and GTG START codon sites are sufficient to cause cryptic initiation.
Figure 5.
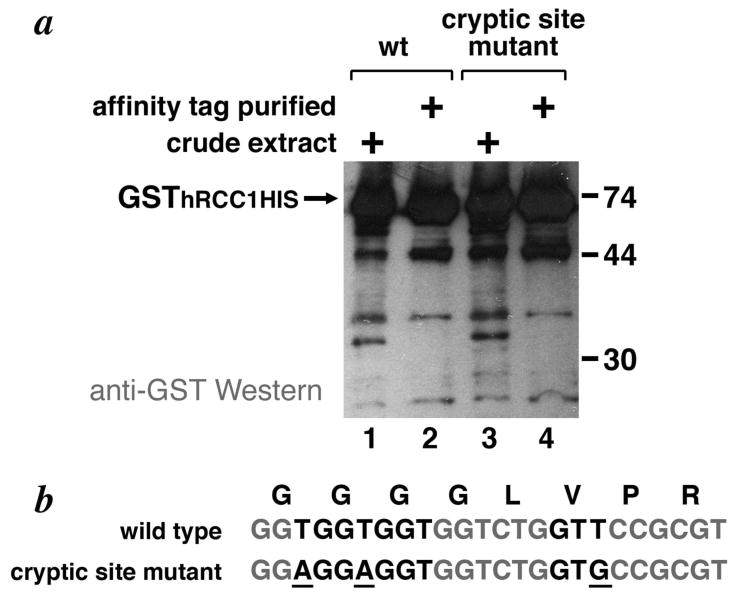
Anti-GST Western analysis of wild-type and engineered cryptic site GSThRCC1HIS variant protein expressed in E. coli. (a) C-terminally HIS-tagged GSThRCC1 in crude extracts (lanes 1, 3) or metal affinity purified (lanes, 2, 4) complexes coexpressed in BL21(DE3)pLysS was detected using anti-GST antibodies. The wild-type GST sequence was used in lanes 1 & 2, while the GST sequence mutated to remove the cryptic Shine-Dalgarno mutation or START codon or both elements are shown in lanes 3 & 4 respectively. from the engineered cryptic translation initiation site was not detected (b) Amino acid and DNA sequences for the wild type and cryptic initiation site GST mutant.
Discussion
Our results show that a truncated heterologous expressed polypeptide can result from cryptic translational initiation in E. coli, and that amount of the truncated product can be greatly reduced by translationally silent mutations to remove the cryptic initiation sites. We also find that although the cryptic initiation sites may be necessary for cryptic initiation, such sites are not sufficient to cause cryptic initiation since creating consensus Shine-Dalgarno spaced appropriately from a potential START codon did not produce a truncated polypeptide in at least one of the two constructs we created.
Our experiments have several implications for protein overexpression in E. coli. Firstly, we have documented that cryptic initiation of translation from a naturally occurring eukaryotic sequence can produce a truncated recombinant polypeptide. We were not able to remove the truncated product by protein purification presumably because we were coexpressing a three protein complex and the desired and truncated complex were not sufficiently distinct to fractionate. We had initially and mistakenly interpreted the truncated product as resulting from proteolysis in vivo. Secondly, the cryptic translation initiated at a GTG START codon. Although ATG is commonly recognized as a START codon, GTG is present as the START codon in 14% of E. coli genes. Thus, any search for potential cryptic START sites should consider GTG in addition to ATG codons.
Our finding that the presence of a consensus Shine-Dalgarno sequence spaced appropriately from a potential START codon is not sufficient to cause cryptic initiation reminds us that other sequence considerations can play significant roles in translational initiation [10,11,19]. The simplest and most straightforward possibility is that secondary structure of the mRNA around the translation initiation sequences contributes to efficiency of translational initiation. It is possible, for example, that the DHFR construct used in Fig. 4 produced a mRNA which formed secondary structure near the engineered internal Shine-Dalgarno and START codon. However, use of the MFOLD software [20] to analyze potential RNA secondary structures for each of the sequences investigated in this study failed to identify simple explanations for our results (data not shown). In any case, our results show that the mere presence of an internal Shine-Dalgarno site and START codon does not necessarily lead to cryptic initiation.
While our findings have highlighted the possibility of internal cryptic initiation of heterologous proteins overexpressed in E. coli, what remains to be determined is how frequently such cryptic initiation occurs and how commonly potential cryptic sites are present. These are not simple questions to answer in part (a) because even nonconsensus Shine-Dalgarno sequences can act as ribosomal binding sites, (b) because of the variable distance possible between the Shine-Dalgarno site and the START codon, and (c) because multiple START codons (ATG, GTG, TTG, UTG) can be used. The occurrence of translation initiation due to cryptic sites in E. coli has been rarely noted in the scientific literature, but there are at least two reports to indicate ours is not an isolated instance. Swaminathan et al observed expression of a truncated product for the eukaryotic Chlorella virus IL-3A restriction endonuclease, R.CviJI, due to cryptic initiation from a GTG Val codon spaced 6 bp downstream of a possible GAAAAAA Shine-Dalgarno sequence [21]. Initiation of translation from a GTG Val codon near the N-terminus was observed when the bacteria Pantoea ananatis lycopene cyclase gene was expressed in E. coli [22]. It would therefore be prudent to consider cryptic initiation as a possible issue when overexpressing proteins in E. coli.
Materials and Methods
Expression vectors: The polycistronic expression vector for the Ada2/Ada3/Gcn5 complex used in Fig. 1, pST44-yAda3Δ2HIS-yAda2Δ1-yGcn5, was created following methods described previously [14-16]. Each of the individual genes was subcloned into an appropriate T7 promoter based pST50Tr transfer vector before subcloning the translational cassette into the pST44 polycistronic vector. Since the transfer vectors are also monocistronic expression vectors, the transfer vector for Gcn5 (pST50Trc3-yGcn5) was used as the expression vector for the experiment in Fig. 3. The expression vector for C-terminally hexahistidine tagged DHFR has been described previously [16]. The plasmid pST50Trc2-GSTNhRCC1HIS used in Fig. 5 was created by subcloning the BamHI-BsrGI insert fragment from pST50Tr-hRCC1HIS (unpublished, J.R. England and S. Tan) into BamHI-BsrGI pST50Trc2-GST vector fragment (unpublished, S. Tan). Standard cloning procedures were used to create all expression vectors.
QuikChange mutagenesis was employed to introduce the Shine-Dalgarno and/or START site mutations into the appropriate expression vectors. The mutagenesis oligonucleotide sequences are provided in Supplementary Table 1. The coding region of all expression plasmids was validated by DNA sequencing.
Expression and metal affinity purification: Small scale (100 ml) expression in E. coli and small scale purification of the tagged proteins or protein complexes were performed as described [14]. Western blotting was performed using anti-Gcn5 antibodies (custom antibodies by Cocalico Biologicals, Reamstown, PA) or anti-HIS antibodies (GenScript, Piscataway, NJ).
Supplementary Material
Supplementary Table 1: Site-directed mutagenesis oligonucleotides
Highlights.
We observed a truncated version of a yeast protein expressed in E. coli.
The truncated protein resulted from cryptic initiation from a GTG codon.
Mutating the cryptic Shine Dalgarno site eliminated the truncated protein.
Introducing cryptic sites for other proteins did not produce truncated proteins.
Acknowledgments
We thank David Garboczi (NIH) for sharing unpublished results. We also thank members of the Tan Laboratory and the Penn State Center for Eukaryotic Gene Regulatio for discussions. This was supposed by the National Institute of Health grants GM060489 and GM088236 to S.T.
References
- 1.Jana S, Deb JK. Strategies for efficient production of heterologous proteins in Escherichia coli. Appl Microbiol Biotechnol. 2005;67:289–298. doi: 10.1007/s00253-004-1814-0. [DOI] [PubMed] [Google Scholar]
- 2.Peti W, Page R. Strategies to maximize heterologous protein expression in Escherichia coli with minimal cost. Protein Expr Purif. 2007;51:1–10. doi: 10.1016/j.pep.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 3.Sørensen HP, Mortensen KK. Advanced genetic strategies for recombinant protein expression in Escherichia coli. J Biotechnol. 2005;115:113–128. doi: 10.1016/j.jbiotec.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Laursen BS, Sørensen HP, Mortensen KK, Sperling-Petersen HU. Initiation of protein synthesis in bacteria. Microbiol Mol Biol Rev. 2005;69:101–123. doi: 10.1128/MMBR.69.1.101-123.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simonetti A, Marzi S, Jenner L, Myasnikov A, Romby P, Yusupova G, et al. A structural view of translation initiation in bacteria. Cell Mol Life Sci. 2009;66:423–436. doi: 10.1007/s00018-008-8416-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Julián P, Milon P, Agirrezabala X, Lasso G, Gil D, Rodnina MV, et al. The Cryo-EM structure of a complete 30S translation initiation complex from Escherichia coli. PLoS Biol. 2011;9:e1001095. doi: 10.1371/journal.pbio.1001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fischer N, Neumann P, Konevega AL, Bock LV, Ficner R, Rodnina MV, et al. Structure of the E. coli ribosome-EF-Tu complex at <3 Å resolution by Cs-corrected cryo-EM. Nature. 2015;520:567–570. doi: 10.1038/nature14275. [DOI] [PubMed] [Google Scholar]
- 8.Noeske J, Wasserman MR, Terry DS, Altman RB, Blanchard SC, Cate JHD. High-resolution structure of the Escherichia coli ribosome. Nat Struct Mol Biol. 2015;22:336–341. doi: 10.1038/nsmb.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shine J, Dalgarno L. The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc Natl Acad Sci USA. 1974;71:1342–1346. doi: 10.1073/pnas.71.4.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gold L. Posttranscriptional regulatory mechanisms in Escherichia coli. Annu Rev Biochem. 1988;57:199–233. doi: 10.1146/annurev.bi.57.070188.001215. [DOI] [PubMed] [Google Scholar]
- 11.Stormo GD, Schneider TD, Gold LM. Characterization of translational initiation sites in E. coli. Nucleic Acids Res. 1982;10:2971–2996. doi: 10.1093/nar/10.9.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 13.Chen H, Bjerknes M, Kumar R, Jay E. Determination of the optimal aligned spacing between the Shine-Dalgarno sequence and the translation initiation codon of Escherichia coli mRNAs. Nucleic Acids Res. 1994;22:4953–4957. doi: 10.1093/nar/22.23.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selleck W, Tan S. Recombinant protein complex expression in E. coli. Curr Protoc Protein Sci. 2008 doi: 10.1002/0471140864.ps0521s52. Chapter 5. Unit 5.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan S. A modular polycistronic expression system for overexpressing protein complexes in Escherichia coli. Protein Expr Purif. 2001;21:224–234. doi: 10.1006/prep.2000.1363. [DOI] [PubMed] [Google Scholar]
- 16.Tan S, Kern RC, Selleck W. The pST44 polycistronic expression system for producing protein complexes in Escherichia coli. Protein Expr Purif. 2005;40:385–395. doi: 10.1016/j.pep.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 17.Barrios A, Selleck W, Hnatkovich B, Kramer R, Sermwittayawong D, Tan S. Expression and purification of recombinant yeast Ada2/Ada3/Gcn5 and Piccolo NuA4 histone acetyltransferase complexes. Methods. 2007;41:271–277. doi: 10.1016/j.ymeth.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guan KL, Dixon JE. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- 19.Schneider TD, Stormo GD, Gold L, Ehrenfeucht A. Information content of binding sites on nucleotide sequences. J Mol Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- 20.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swaminathan N, Mead DA, McMaster K, George D, Van Etten JL, Skowron PM. Molecular cloning of the three base restriction endonuclease R.CviJI from eukaryotic Chlorella virus IL-3A. Nucleic Acids Res. 1996;24:2463–2469. doi: 10.1093/nar/24.13.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SW, Jung WH, Ryu JM, Kim JB, Jang HW, Jo YB, et al. Identification of an alternative translation initiation site for the Pantoea ananatis lycopene cyclase (crtY) gene in E. coli and its evolutionary conservation. Protein Expr Purif. 2008;58:23–31. doi: 10.1016/j.pep.2007.11.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Site-directed mutagenesis oligonucleotides