

Abstract
Background
Whether knowledge of genetic risk for coronary heart disease (CHD) affects health-related outcomes is unknown. We investigated whether incorporating a genetic risk score (GRS) in CHD risk estimates lowers low-density lipoprotein cholesterol (LDL-C) levels.
Methods and Results
Participants (n=203, 45–65 years old, at intermediate risk for CHD, and not on statins) were randomized to receive their 10-year probability of CHD based either on a conventional risk score (CRS) or CRS + GRS (+GRS). Participants in the +GRS group were stratified as having high (+H-GRS) or average/low (+L-GRS) GRS. Risk was disclosed by a genetic counselor followed by shared decision-making regarding statin therapy with a physician. We compared the primary endpoint of LDL-C levels at 6 months and assessed whether any differences were due to changes in dietary fat intake, physical activity levels or statin use. Participants (mean age 59.4±5 years, 48% men, mean 10-year CHD risk 8.5±4.1%) were allocated to receive either CRS (n=100) or +GRS (n=103). At the end of the study period, the +GRS group had a lower LDL-C than the CRS group (96.5±32.7 vs. 105.9±33.3 mg/dL; P=0.04). +H-GRS participants had lower LDL-C levels (92.3±32.9 mg/dL) than CRS participants (P=0.02) but not +L-GRS participants (100.9±32.2 mg/dL; P=0.18). Statins were initiated more often in the +GRS group than in the CRS group (39% vs. 22%, P<0.01). No significant differences in dietary fat intake and physical activity levels were noted.
Conclusions
Disclosure of CHD risk estimates that incorporated genetic risk information led to lower LDL-C levels than disclosure of CHD risk based on conventional risk factors alone.
Clinical Trial Registration Information
ClinicalTrials.gov. Identifier: NCT01936675.
Keywords: Randomized clinical trial, coronary heart disease, genetic risk disclosure, genetic risk score, genomic medicine, prevention, single-nucleotide polymorphism, statins
As genetic testing becomes widely available, its use for estimating risk of common diseases is becoming of increasing scientific and public health interest.1 Genome wide association studies (GWAS) have identified multiple loci associated with coronary heart disease (CHD).2, 3 The majority of these loci are associated with CHD independent of conventional risk factors and could potentially improve the accuracy of CHD risk estimates. Several studies have investigated the association of a genetic risk score (GRS) based on multiple CHD susceptibility single-nucleotide polymorphisms (SNPs) with incident CHD events.4–10 Most of the studies reported that a GRS is associated with adverse CHD events.4, 6–10 Incorporating CHD genetic risk information in clinical practice may refine risk estimates and aid in prevention of CHD, concordant with recent calls to promote the practice of precision medicine.11
Whether knowledge of genetic risk for CHD influences health-related outcomes remains unknown. We conducted a clinical trial to investigate whether disclosing a GRS for CHD leads to lowering of low-density lipoprotein cholesterol (LDL-C) levels. The GRS was incorporated into the CHD risk estimates in combination with a conventional risk score (CRS) yielding a genetically-informed risk score (+GRS).12 We assessed whether disclosure of genetic risk for CHD affects LDL-C levels, and whether any differences were due to changes in dietary fat intake, physical activity levels or statin initiation. We tested the following hypotheses: 1) in patients randomized to receive +GRS, LDL-C levels at the end of the study period would be lower than in participants randomized to receive CRS alone; 2) +GRS participants with a ‘high’ GRS would have lower LDL-C levels than +GRS participants with ‘average/low’ GRS and those randomized to receive CRS alone.
A major challenge in implementing genomic medicine is the integration of genomic information into the electronic health record (EHR).13 Genotyping was performed in a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory and results were placed into the EHR. Additionally, a decision aid was modified to include genetic risk information and allow integration of such information into the EHR to facilitate shared decision making regarding statin therapy.14, 15
Methods
Study Design
Study participants were drawn from the Mayo Clinic Biobank (n=29,352 at the time of study initiation) which recruits patients from the outpatient setting at Mayo.16 We identified 2026 participants who met the following eligibility criteria: age 45–65 years, non-Hispanic White ethnicity, no history of atherosclerotic cardiovascular disease, not on statins, at intermediate risk for CHD (10 year CHD risk 5–20%), and residents of Olmsted County Minnesota. To maximize the information yield from the study, we performed an initial screening genotyping of 28 CHD susceptibility SNPs (Supplemental Table 1) that are not associated with blood pressure or lipid levels,3 in a random sample of 1000 participants who met eligibility criteria16 (Figure 1).
Figure 1.
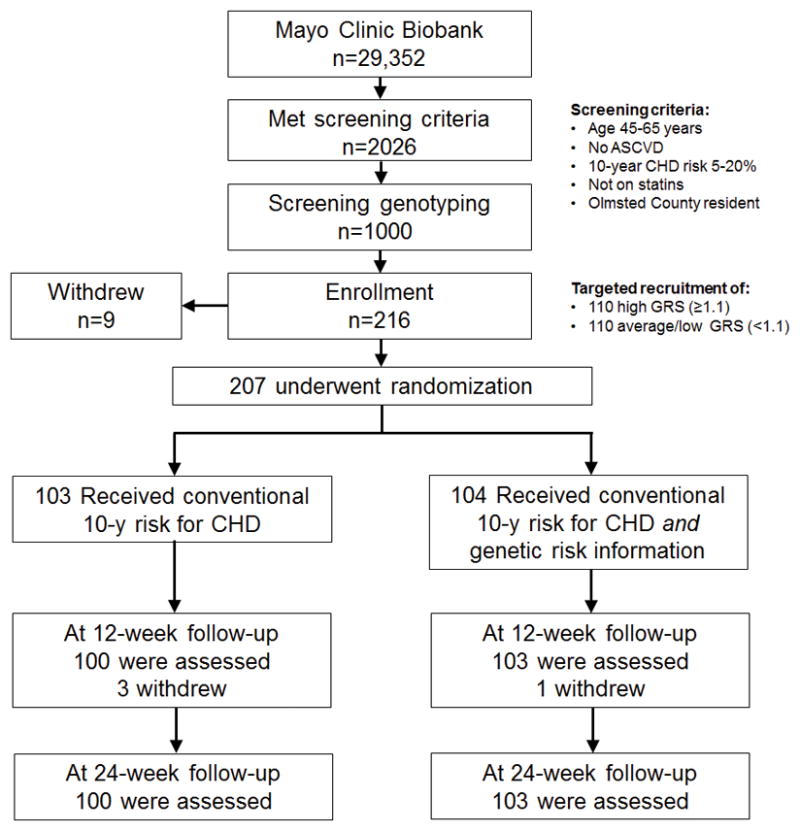
Flow diagram of the MI-GENES Clinical Trial. Of the 2026 individuals from the Mayo Biobank who met the eligibility criteria, a random subset of 1000 was genotyped. Genotyping results passed quality control measures in 968 individuals. Recruitment was based on screening genotyping results in order to achieve the targeted enrollment goals of ~110 individuals with high GRS (≥1.1) and ~110 with average/low GRS (<1.1) with the expectation that approximately 10–20 study participants may withdraw from the study or be lost to follow-up. Participants who withdrew from the study stated that they could not fit the study visits into their schedule. (ASCVD: atherosclerotic cardiovascular disease; CHD: coronary heart disease; GRS: genetic risk score).
A GRS for each individual was calculated as previously described, taking into account the average genetic risk in the population.17 In brief, we assumed an additive genetic model in which the genotypes are coded ‘0’ for non-risk allele homozygotes, ‘1’ for heterozygotes, and ‘2’ for risk-allele homozygotes. A weighted GRS was calculated by multiplying the logarithm of the odds ratio for a particular SNP by 0, 1, or 2 according to the number of risk alleles carried by each person. We used a GRS of ≥1.1, i.e., a 10% or greater increase in risk for CHD than would be predicted by a CRS, to classify individuals as having ‘high’ GRS. Those with a GRS of <1.1 were classified as having ‘average/low’ GRS.
Characteristics of the individuals who comprised the recruitment pool for the study are summarized in Supplemental Table 2. The initial screening genotyping allowed us to perform a targeted enrollment of equal numbers of high GRS and average/low GRS individuals. We were able to enroll 216 of a target of 220 participants for the study. A study coordinator invited these patients by phone to participate in the study and subsequently confirmed eligibility and obtained written informed consent. Individuals who agreed to participate underwent a blood draw for genotyping of 28 CHD susceptibility SNPs in a CLIA-certified laboratory using the TaqMan® procedure (Roche Molecular Diagnostics, Branchburg, NJ). A GRS was calculated17 and the CRS was then multiplied by the GRS to generate a genotype-informed probability of adverse CHD events over the next 10 years (+GRS). The 10-year probability of CHD was calculated at the first study visit as previously described.12 Additional information about the screening genotyping and GRS calculation can be found in the online-only Data Supplement.
Risk factors for CHD including family history were assessed at the first study visit. Participants returned 6–10 weeks later (Visit 2) once CLIA genotyping and calculation of a GRS was completed (n=207). At this visit, patients were randomized (1:1) to receive a conventional risk score (CRS, n=103) vs. a combined conventional and genetic risk score (+GRS, n=104). Study participants then underwent a 30-min CHD risk counseling session, followed by a visit with a physician for shared decision making regarding statin use. Three months following the disclosure of CHD risk, participants returned (Visit 3) for measurement of fasting lipid levels and assessment of dietary fat intake and physical activity levels. The final study visit (Visit 4) occurred three months after Visit 3. Apart from incorporating the GRS into the CRS in one arm of the study (+GRS), randomized patients received identical exposure to education about CHD risk reduction and preventive measures. The study was approved by the Mayo Clinic institutional review board and was registered at ClinicalTrials.gov (NCT01936675). Study methods and protocol have been previously described.18
Outcome Measures
The primary outcome was change in LDL-C level 6 months after disclosure of CHD risk (for ease of interpretation we present comparison of the actual LDL-C levels at the 6 month time point, the inferences being the same). Behaviors related to cardiovascular health including dietary fat intake and physical activity levels were assessed at baseline and subsequent study visits. Differences in the study arms as a result of disclosing CHD risk were assessed at 6 months after disclosure. To assess whether disclosure of genetic risk led to an increase in anxiety, anxiety levels were assessed at baseline and subsequent study visits.
Sample Size and Power
In general, studies have shown a 5–15% decrease in LDL-C with diet and lifestyle changes and a ~30% decrease in LDL-C with statin therapy.19, 20 Assuming the standard deviation of LDL-C change in the entire group to be 25 mg/dL, we had sufficient power to detect an LDL-C change of 15 mg/dL and to test the hypotheses that: 1) patients randomized to receive +GRS would have lower LDL-C levels than patients randomized to CRS; and 2) +GRS participants with a high GRS (≥1.1) would have lower LDL-C levels than +GRS participants with average/low GRS (<1.1) and those randomized to receive CRS alone.
Randomization
The second study visit was scheduled 6–10 weeks after the initial visit to allow for completion of genotyping and calculation of GRS. Patients were randomized (1:1) by means of a computer-generated random sequence stratifying for age, sex, and family history for CHD.21 The +GRS arm received genetically informed 10-year CHD risk and the CRS arm received conventional risk factor information alone.
Disclosure of CHD Risk
The CHD risk estimate was disclosed by the genetic counselor during a 30-min semi-scripted session. Patients randomized to +GRS were shown a pictograph that incorporated the revised 10-year CHD risk based on the genotypes of the 28 CHD susceptibility SNPs. The control group was shown a pictograph based on the CRS. The pictograph depicted 100 people “like the participant” and indicated how many in the next 10 years could be expected to experience an adverse CHD event and how many would not. The genetic counselor helped participants interpret and understand their results, highlighting the probabilistic nature of the genetic testing and that lifestyle factors such as diet, exercise, and smoking are major risk factors for developing CHD. The counselor encouraged participants to sign an action plan for behavioral change that included increased physical activity and reduced dietary fat intake and smoking cessation if the participant was a smoker. Participants were provided with a Frequently Asked Questions sheet that reiterated the key points conveyed by the genetic counselor at the visit. Fidelity of the scripts was assessed by analysis of video-recorded encounters. Having one genetic counselor (T.M.K) disclose CHD risk estimates to all study participants helped ensure that risk was disclosed similarly to all study participants (in their respective randomization groups).
Shared Decision Making Regarding Statin Therapy
Following the visit with the genetic counselor, each patient saw a physician in the Mayo Cardiovascular Health Clinic. During the patient-physician encounter the focus was on shared decision making regarding the need for statin therapy. The physicians (n=6) underwent a training session in the use of a Statin Choice decision aid14 modified to include the GRS (migenesstudy.mayoclinic.org) to disclose CHD risk and help patients understand the benefits and downsides of taking a statin medication to reduce CHD risk (Supplemental Figures 1–4). The participant and clinician navigated through pictograms that display the 10-year probability of CHD as well as the potential benefit of using statin medications. Consistency of the disclosure process was assured by following a checklist maintained by the study coordinator for both study arms and by review of videotaped encounters. A risk report describing conventional vs. genetics-informed CHD risk was deposited in the electronic health record according to the participant’s randomization group. New statin prescriptions were recorded by review of the EHRs. The online-only Data Supplement includes further details regarding the genomic decision aid, integration into the EHR, and the disclosure process of CHD risk estimates.
Dietary Fat Intake, Physical Activity and Anxiety Levels
We used validated surveys to assess whether disclosure of CHD risk led to changes in dietary fat intake, physical activity and anxiety levels. The percentage energy from fat (PFat) screener22 was adapted to estimate changes in fat consumption and the telephonic assessment of physical activity (TAPA) questionnaire23 was adapted to assess changes in physical activity. Anxiety level was measured at baseline and follow up using the validated State and Trait Anxiety Inventory for adults (STAI).24 Further details are provided in the online-only Data Supplement (Methods – survey instruments).
Follow-up
Three and six months after disclosure of CHD risk, participants returned to undergo assessment of fasting plasma lipid levels and fill out study questionnaires. Recruitment started in October 2013 and ended in May 2014. Acquisition of Visit 4 data was completed in December 2014.
Statistical Methods
All study data were analyzed using R software (version 3.1.2). Data analysts were blinded to allocation. Descriptive data were provided for all measures, using frequencies (%) for categorical variables and mean (±SD) for continuous variables. Simple group comparisons were made using either the Chi-Square or Fisher’s exact test as appropriate for binary variables and t-tests for continuous outcomes such as LDL-C levels.
We analyzed the primary outcome – LDL-C levels at 6 months after CHD risk disclosure – in the randomized treatment groups, CRS and +GRS. We conducted pre-specified secondary analyses comparing 3 groups: CRS, +H-GRS, and +L-GRS. Since overall hypothesis testing was based on the original 2 randomized groups, these secondary between-group analyses were each conducted at the nominal 0.05 level of significance without correction for multiple comparisons. We also compared change in LDL-C levels from baseline in the study groups. Finally, since LDL-C levels were measured at 3 and 6 months, we assessed the between group difference in the slope of LDL-C after randomization, in a mixed effects model with uncorrelated random effects for sample intercepts and the effect of time.
We included dietary fat intake, physical activity as well as statin use in models of the primary LDL-C endpoint to determine their influence on LDL-C levels. We also assessed whether incorporation of a GRS into conventional risk estimates led to an increase in anxiety levels. Family history of CHD was also analyzed as a predictor of LDL-C levels at follow up and the secondary endpoints, independent of randomized group status.
Results
The study flow from initial screening of Mayo BioBank participants through recruitment and the final study cohort is summarized in Figure 1. Characteristics of the participants randomized (n=203) are summarized in Table 1. No significant differences were present between the study groups for any of the characteristics listed. Baseline characteristics of +H-GRS and +L-GRS participants are provided in Supplemental Table 3.
Table 1.
Participant characteristics (n=203).
| CRS n=100 |
+GRS n=103 |
|
|---|---|---|
| Age, years | 59.4±5.3 | 59.4±4.9 |
| Male sex, n (%) | 49 (49.0%) | 48 (46.6%) |
| Ever smoker, n (%) | 41 (41.0%) | 32 (31.1%) |
| Family history of CHD, n (%) | 30 (30.0%) | 25 (24.3%) |
| Body mass index, kg/m2 | 30.5±7.0 | 30.2±6.1 |
| Systolic blood pressure, mmHg | 130.1±14.2 | 131.9±17.6 |
| Total cholesterol, mg/dL | 200.8±30.2 | 203.3±27.6 |
| LDL-C, mg/dL | 118.8±23.9 | 119.8±26.4 |
| HDL-C, mg/dL | 55.0±15.6 | 56.4±16.8 |
| Triglycerides, mg/dL | 134.1±70.2 | 132.7±78.8 |
| College education or higher, n (%) | 67 (67.0%) | 58 (56.3%) |
| GRS | 1.11±0.31 | 1.14±0.29 |
| CRS, 10 year predictability of CHD (%) | 8.48±3.76 | 8.56±4.47 |
| * Dietary fat intake score | 34±2.6 | 33.6±2.4 |
| † Physical activity score | 4.68±1.43 | 4.87±1.57 |
| ‡ Anxiety trait score | 31.1±7.8 | 30.9±7.6 |
| Anxiety state score | 27.9±7.5 | 28.8±9 |
Continuous traits are presented as mean ± standard deviation and categorical variables as percentage.
Dietary fat intake score is based on the percentage energy from fat (PFat) screener22;
physical activity score based on assessment of physical activity (TAPA) questionnaire23;
anxiety scores based on the State and Trait Anxiety Inventory for adults (STAI).24 CRS: conventional risk score; GRS: genetic risk score; +GRS: combined conventional and genetic risk score; LDL-C: low-density lipoprotein cholesterol; HDL-C: high-density lipoprotein cholesterol. To convert total, LDL and HDL-cholesterol to mmol/L, multiply by 0.0259; triglycerides to mmol/L, by 0.0113.
At the end of the study period, the LDL-C in the +GRS group was 9.4 mg/dL lower than the CRS group (96.5±32.7 vs. 105.9±33.3 mg/dL; P=0.04). +H-GRS participants had a 13.6 mg/dL lower LDL-C level (92.3±32.9 mg/dL) than CRS participants (P=0.02) and a 8.6 mg/dL lower LDL-C than +L-GRS participants (100.9±32.2 mg/dL; P=0.18) (Table 2 and Figure 2). When the values at 6 months after CHD disclosure were compared to baseline values, the mean LDL-C change was −13.6±31.3 mg/dL in the CRS group vs. −23.3±33.6 mg/dL in the +GRS group (P=0.03). The overall downward longitudinal trend in LDL-C was significantly greater in +GRS participants than in CRS participants (P=0.04). The downward trend in LDL-C in +H-GRS participants was significantly greater than in CRS participants (P=0.007) and tended to be greater than in the +L-GRS participants (P=0.07). The estimated slopes (±SE) representative of LDL-C change per 30 days were −1.8±0.4 mg/dL and −3.0±0.4 mg/dL in the CRS and +GRS groups, respectively (Figure 2). Supplemental Table 4 summarizes results of expanded LDL-C comparisons between the study groups.
Table 2.
Baseline and follow up LDL-C levels and statin use.
| Group | Baseline | 3 months after CHD risk disclosure | 6 months after CHD risk disclosure | |
|---|---|---|---|---|
| LDL-C (mg/dL) | CRS | 118.79 (23.94) | 100.75 (32.69) | 105.86 (33.31) |
| +GRS | 119.77 (26.39) | 93.52 (31.10) | 96.48 (32.71)A | |
| +L-GRS | 119.54 (25.75) | 98.68 (28.65) | 100.92 (32.24) | |
| +H-GRS | 119.98 (27.23) | 88.66 (32.78) | 92.28 (32.90)B | |
| Statin Use | CRS | 0 (0%) | 23 (23.7%) | 21 (21.9%) |
| +GRS | 0 (0%) | 41 (40.2%) | 40 (39.2%)A | |
| +L-GRS | 0 (0%) | 14 (28.6%) | 14 (28.6%) | |
| +H-GRS | 0 (0%) | 27 (50.9%) | 26 (49.1%)B,C |
CHD: coronary heart disease; CRS: conventional risk score; +GRS: combined conventional and genetic risk score; +H-GRS: participants randomized to +GRS with a GRS ≥1.1; +L-GRS: participants randomized to +GRS with a GRS <1.1; LDL-C: low-density lipoprotein cholesterol. LDL-C levels are presented as mean (SD). To convert LDL-C to mmol/L, multiply by 0.0259.
+GRS≠CRS at P<0.05;
+H-GRS≠CRS at P<0.05;
+H-GRS≠+L-GRS at P<0.05
Figure 2.

Change in LDL-C levels from baseline over the follow up period overall and in categories of GRS. LDL-C levels at 6 months post-disclosure were lower in +GRS participants than CRS participants. The overall downward longitudinal trend in LDL-C was significantly greater in +GRS participants than CRS participants. LDL-C levels at 6 months post-disclosure were lower in +H-GRS participants than CRS participants. The downward trend in LDL-C was significantly greater in +H-GRS participants than CRS participants. There was a trend towards a greater reduction in LDL-C levels in +H-GRS vs. +L-GRS. *denotes six month statistical significance at 0.05 level.
No significant differences in dietary fat intake, physical activity levels and anxiety levels six months after CHD risk disclosure were observed between CRS and +GRS participants (Figure 3 and Supplemental Tables 5 and 6). Statin use at the final visit was significantly higher in the +GRS group than in the CRS group (39.2% vs. 21.9%; P<0.01) (Table 2). A higher proportion of +H-GRS participants (49.1%) were on statins than CRS (21.9%, P<0.01) and +L-GRS (28.6%, P=0.03) participants. After adjustment for statin initiation, group randomization was not significantly associated with the end of study LDL-C levels (P=0.74).
Figure 3.

Dietary fat intake, physical activity and anxiety levels in the study groups. There was no difference between CRS and +GRS group in either dietary fat intake, physical activity, or anxiety levels 6 months post-disclosure. Dietary fat intake scores ranged between 0 (no fat intake) to 110 indicative of very high dietary fat intake. Physical activity scores ranged between 7 (active) and 1 (sedentary). Anxiety scores ranged between 20 (least anxious) up to 80 (highly anxious).
Family history of CHD was considered as a potential predictor variable of LDL-C levels. The mean GRS tended to be higher in patients with a family history of CHD than those without such history (1.19 vs.1.10, P=0.09). Family history was not associated with the 6-month LDL-C by itself (P=0.48) nor in combination with group randomization (P=0.40). However, family history was a borderline significant predictor of statin use at 6 months, independent of allocation to +GRS or CRS. (OR: 1.92, 95% CI=0.97,3.79; P= 0.06). An interaction term between family history and group allocation was not significant (P=0.40).
Discussion
Our goal in this study was to investigate whether disclosure of genetic risk of CHD influences LDL-C levels, lifestyle behavior as well as shared decision making regarding statin therapy. We included individuals at intermediate risk for CHD as decisions regarding statin initiation are often complex and motivating patients to change diet and lifestyle can be challenging. Disclosing CHD risk estimates that included genetic risk information in addition to conventional risk factors led to lower LDL-C levels six months after disclosure of risk. +H-GRS participants had significantly lower LDL-C levels than CRS participants and tended to have lower LDL-C levels than +L-GRS. The differences in LDL-C levels were due to higher proportion of participants in the +GRS arm being started on a statin medication. Disclosure of a GRS for CHD did not lead to significant differences in dietary fat intake, physical activity or anxiety levels, at the end of the study.
The lower LDL-C level in patients allocated to receive +GRS was due to a higher proportion starting statin therapy after shared decision making with a physician. Recent guidelines15 emphasize the need for shared decision making when considering statin medications for lowering CHD risk. We modified an existing decision aid14 to incorporate genetic risk information and facilitate shared decision making in the setting of disclosure of CHD genetic risk. Such visual depictions help patients as well as physicians to better comprehend statistical probabilities related to risk of disease.25 Armed with appropriate resources and a genomic decision aid embedded in the EHR, study participants and non-geneticist physicians were able to use genetic risk information in the shared-decision making process.
Participants in the +GRS group were more likely to receive statins than the CRS group. Increased statin prescription in the +H-GRS group was likely due to the increase in overall estimated CHD risk by at least 10% after including the GRS. Although in the +L-GRS subset the estimated CHD risk was lower, statin initiation was not significantly different than in the CRS group. One possibility is that clinicians may not be comfortable in downgrading risk estimated based on conventional risk factors. However, the shared medical decision process that was utilized in the trial ensured that the decision to start statins was made taking both physician and participant preferences into account.
We previously reported that +GRS participants in this trial had higher perceived personal control and genetic counseling satisfaction than those who received conventional risk factor information.26 However, disclosure of CHD genetic risk did not lead to changes in dietary fat intake and physical activity levels. McBride et al.27 demonstrated that disclosure of genetic risk for cancer predisposition did not affect smoking cessation rates. Similarly, in volunteers who underwent direct-to-consumer genome-wide testing for various medical conditions, there were no significant changes in participants’ dietary or physical activity behaviors.28 Our results highlight that prompting patients to adopt and sustain lifestyle changes remains challenging despite the provision of personalized disease risk estimates.
There is concern that disclosure of genetic risk for a disease may increase anxiety levels in patients with high genetic risk and induce a sense of invulnerability in those at low genetic risk. We found that disclosure of CHD genetic risk was not associated with greater anxiety levels consistent with prior studies of disclosing genetic risk of common disorders.29, 30 Also, we did not observe increased dietary fat intake or decreased physical activity levels in those at low genetic risk compared to the other study groups (Supplemental Tables 5 and 6).
To minimize potential confounding due to presence of family history of CHD, we randomized patients to study arms based on such history. The GRS tended to be higher in patients with family history but this difference was not statistically significant suggesting family history and GRS may provide additive information about CHD risk. Family history was not associated with post randomization LDL-C levels although it tended to be associated with greater statin use at the end of the study period.
Our study has implications for prevention of CHD which often manifests as sudden death or myocardial infarction. Several circulating biomarkers have been proposed for improving risk stratification for adverse CHD events but most are associated to a varying degree with known risk factors.31 Although the genetic susceptibility variants measured in this study have modest effects, these were not associated with established factors (GRS and CRS were not correlated in our study) and therefore provide an orthogonal means of risk assessment. As genome sequencing becomes more common, it will be possible to estimate a GRS for common diseases such as CHD and further refine risk estimates and inform targeted therapy. However, large clinical trials will be needed to investigate the effects of such an approach on reducing adverse CHD outcomes and on health care costs and utilization.
Our study demonstrates that genetic risk information for a common disease can be incorporated into the EHR to enable shared decision making regarding drug therapy. Blood draws and genotyping were done in a CLIA-environment and results were placed in the EHR. Several limitations deserve mention. We did not prospectively validate the GRS; however in a recent study,8 a GRS based on 27 genetic variants was independently associated with adverse cardiovascular outcomes and response to statin therapy.8 CHD risk scores are not static and newly discovered variants may need to be included.32 The study sample size was relatively small and the intervention was not blinded. We were not able to use the risk calculator and categories recommended in the latest ACC/AHA guidelines which appeared after the study began. Of note, the majority (82%) of participants were appropriately initiated on statins based on these guidelines i.e., they had a 10-year risk of ≥7.5% (Supplemental Table 7). The short term and modest reduction in LDL-C levels observed in this study may not ultimately translate to improved outcomes and large clinical trials will be needed to prove clinical utility of a GRS for CHD. Additional studies are needed to study the effects of disclosure of genetic risk for CHD in various ethnic groups and in the ‘real world’ setting of primary care.
Conclusions
We demonstrate that genetic risk information for a common disease can be effectively incorporated into the EHR and used at the point of care to guide therapy. Individuals who received a GRS in addition to a conventional risk estimate for CHD had lower LDL-C levels 6 months after disclosure than participants who received a conventional risk score alone. Shared decision making after CHD risk disclosure led to a greater proportion of patients who received CHD genetic risk being initiated on a statin medication. Disclosure of a GRS did not lead to significant changes in dietary fat intake, physical activity levels, or anxiety. The lowering of LDL-C was greatest in individuals with a high GRS for CHD compared to participants who did not receive GRS.
Supplementary Material
Clinical Perspectives.
Genome wide association studies have identified multiple genetic susceptibility loci for coronary heart disease (CHD) and several studies have reported that a genetic risk score (GRS) based on such loci is associated with adverse CHD events. However no prospective studies have investigated whether knowledge of genetic risk for CHD influences health-related outcomes. We conducted a randomized clinical trial to assess the effect of disclosure of a GRS for CHD on low-density lipoprotein cholesterol (LDL-C) levels. Risk disclosure (by a genetic counselor) and shared decision making regarding statin therapy (with a physician) were facilitated by a decision aid that was integrated in the electronic health record and modified to include genetic risk information. Disclosure of a CHD risk estimate that included GRS in addition to conventional risk factors led to lower LDL-C levels six months after disclosure of risk, compared to disclosure of a conventional risk estimate alone. The differences in LDL-C levels were due to higher proportion of participants in the GRS arm being started on a statin medication. Disclosure of a GRS did not lead to significant differences in dietary fat intake, physical activity or anxiety levels. Our study demonstrates that genetic risk information for CHD can be used at the point of care to enable shared decision making regarding statin therapy with subsequent change in LDL-C levels. The reduction in LDL-C levels observed in this study was modest and large clinical trials will be needed to prove the clinical utility of a GRS for CHD.
Acknowledgments
We would to acknowledge the staff of the Mayo Cardiovascular Health Clinic (Drs. Adelaide M. Arruda-Olson, Frank V. Brozovich, Salma Iftikhar, Stephen L. Kopecky, Francisco Lopez-Jimenez, Randal J. Thomas), Angela K. Dalenberg, Emma M. Heim, Dawn Reiss and Rachel Lorier for their help with the study.
Funding Sources: This study was funded as part of the NHGRI-supported eMERGE (Electronic Records and Genomics) Network (HG04599 and HG006379). Additional support was provided by the Mayo Center for Individualized Medicine. Dr. Green is supported by HG006500. Use of REDCap is funded by the Center for Clinical and Translational Science grant support (UL1TR000135).
Footnotes
Disclosures: None.
References
- 1.Manolio TA. Bringing genome-wide association findings into clinical use. Nat Rev Genet. 2013;14:549–558. doi: 10.1038/nrg3523. [DOI] [PubMed] [Google Scholar]
- 2.The Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011;43:339–344. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- 3.Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein BA, Stirrups K, Konig IR, Cazier JB, Johansson A, Hall AS, Lee JY, Willer CJ, Chambers JC, Esko T, Folkersen L, Goel A, Grundberg E, Havulinna AS, Ho WK, Hopewell JC, Eriksson N, Kleber ME, Kristiansson K, Lundmark P, Lyytikainen LP, Rafelt S, Shungin D, Strawbridge RJ, Thorleifsson G, Tikkanen E, Van Zuydam N, Voight BF, Waite LL, Zhang W, Ziegler A, Absher D, Altshuler D, Balmforth AJ, Barroso I, Braund PS, Burgdorf C, Claudi-Boehm S, Cox D, Dimitriou M, Do R, Doney AS, El Mokhtari N, Eriksson P, Fischer K, Fontanillas P, Franco-Cereceda A, Gigante B, Groop L, Gustafsson S, Hager J, Hallmans G, Han BG, Hunt SE, Kang HM, Illig T, Kessler T, Knowles JW, Kolovou G, Kuusisto J, Langenberg C, Langford C, Leander K, Lokki ML, Lundmark A, McCarthy MI, Meisinger C, Melander O, Mihailov E, Maouche S, Morris AD, Muller-Nurasyid M, Nikus K, Peden JF, Rayner NW, Rasheed A, Rosinger S, Rubin D, Rumpf MP, Schafer A, Sivananthan M, Song C, Stewart AF, Tan ST, Thorgeirsson G, van der Schoot CE, Wagner PJ, Wells GA, Wild PS, Yang TP, Amouyel P, Arveiler D, Basart H, Boehnke M, Boerwinkle E, Brambilla P, Cambien F, Cupples AL, de Faire U, Dehghan A, Diemert P, Epstein SE, Evans A, Ferrario MM, Ferrieres J, Gauguier D, Go AS, Goodall AH, Gudnason V, Hazen SL, Holm H, Iribarren C, Jang Y, Kahonen M, Kee F, Kim HS, Klopp N, Koenig W, Kratzer W, Kuulasmaa K, Laakso M, Laaksonen R, Lind L, Ouwehand WH, Parish S, Park JE, Pedersen NL, Peters A, Quertermous T, Rader DJ, Salomaa V, Schadt E, Shah SH, Sinisalo J, Stark K, Stefansson K, Tregouet DA, Virtamo J, Wallentin L, Wareham N, Zimmermann ME, Nieminen MS, Hengstenberg C, Sandhu MS, Pastinen T, Syvanen AC, Hovingh GK, Dedoussis G, Franks PW, Lehtimaki T, Metspalu A, Zalloua PA, Siegbahn A, Schreiber S, Ripatti S, Blankenberg SS, Perola M, Clarke R, Boehm BO, O’Donnell C, Reilly MP, Marz W, Collins R, Kathiresan S, Hamsten A, Kooner JS, Thorsteinsdottir U, Danesh J, Palmer CN, Roberts R, Watkins H, Schunkert H, Samani NJ. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thanassoulis G, Peloso GM, Pencina MJ, Hoffmann U, Fox CS, Cupples LA, Levy D, D’Agostino RB, Hwang SJ, O’Donnell CJ. A genetic risk score is associated with incident cardiovascular disease and coronary artery calcium: the Framingham Heart Study. Circ Cardiovasc Genet. 2012;5:113–121. doi: 10.1161/CIRCGENETICS.111.961342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paynter NP, Chasman DI, Pare G, Buring JE, Cook NR, Miletich JP, Ridker PM. Association between a literature-based genetic risk score and cardiovascular events in women. JAMA. 2010;303:631–637. doi: 10.1001/jama.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ganna A, Magnusson PK, Pedersen NL, de Faire U, Reilly M, Arnlov J, Sundstrom J, Hamsten A, Ingelsson E. Multilocus genetic risk scores for coronary heart disease prediction. Arterioscler Thromb Vasc Biol. 2013;33:2267–2272. doi: 10.1161/ATVBAHA.113.301218. [DOI] [PubMed] [Google Scholar]
- 7.Tikkanen E, Havulinna AS, Palotie A, Salomaa V, Ripatti S. Genetic risk prediction and a 2-stage risk screening strategy for coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33:2261–2266. doi: 10.1161/ATVBAHA.112.301120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield MJ, Devlin JJ, Nordio F, Hyde CL, Cannon CP, Sacks FM, Poulter NR, Sever PS, Ridker PM, Braunwald E, Melander O, Kathiresan S, Sabatine MS. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet. 2015;385:2264–2271. doi: 10.1016/S0140-6736(14)61730-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morrison AC, Bare LA, Chambless LE, Ellis SG, Malloy M, Kane JP, Pankow JS, Devlin JJ, Willerson JT, Boerwinkle E. Prediction of coronary heart disease risk using a genetic risk score: The Atherosclerosis Risk in Communities Study. Am J Epidemiol. 2007;166:28–35. doi: 10.1093/aje/kwm060. [DOI] [PubMed] [Google Scholar]
- 10.Ripatti S, Tikkanen E, Orho-Melander M, Havulinna AS, Silander K, Sharma A, Guiducci C, Perola M, Jula A, Sinisalo J, Lokki ML, Nieminen MS, Melander O, Salomaa V, Peltonen L, Kathiresan S. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet. 2010;376:1393–1400. doi: 10.1016/S0140-6736(10)61267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–795. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson PW, D’Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–1847. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 13.Kullo IJ, Jarvik GP, Manolio TA, Williams MS, Roden DM. Leveraging the electronic health record to implement genomic medicine. Genet Med. 2013;15:270–271. doi: 10.1038/gim.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weymiller AJ, Montori VM, Jones LA, Gafni A, Guyatt GH, Bryant SC, Christianson TJ, Mullan RJ, Smith SA. Helping patients with type 2 diabetes mellitus make treatment decisions: statin choice randomized trial. Arch Intern Med. 2007;167:1076–1082. doi: 10.1001/archinte.167.10.1076. [DOI] [PubMed] [Google Scholar]
- 15.Goff DC, Jr, Lloyd-Jones DM, Bennett G, Coady S, D’Agostino RB, Sr, Gibbons R, Greenland P, Lackland DT, Levy D, O’Donnell CJ, Robinson J, Schwartz JS, Shero ST, Smith SC, Jr, Sorlie P, Stone NJ, Wilson PW. 2013 ACC/AHA Guideline on the Assessment of Cardiovascular Risk: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129:S49–73. doi: 10.1161/01.cir.0000437741.48606.98. [DOI] [PubMed] [Google Scholar]
- 16.Olson JE, Ryu E, Johnson KJ, Koenig BA, Maschke KJ, Morrisette JA, Liebow M, Takahashi PY, Fredericksen ZS, Sharma RG, Anderson KS, Hathcock MA, Carnahan JA, Pathak J, Lindor NM, Beebe TJ, Thibodeau SN, Cerhan JR. The Mayo Clinic Biobank: a building block for individualized medicine. Mayo Clin Proc. 2013;88:952–962. doi: 10.1016/j.mayocp.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding K, Bailey KR, Kullo IJ. Genotype-informed calculation of risk of coronary heart disease based on genome-wide association data linked to the electronic medical record. BMC Cardiovasc Disord. 2011;11:66. doi: 10.1186/1471-2261-11-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kullo IJ, Jouni H, Olson JE, Montori VM, Bailey KR. Design of a randomized controlled trial of disclosing genomic risk of coronary heart disease: the Myocardial Infarction Genes (MI-GENES) study. BMC Med Genomics. 2015;8:51. doi: 10.1186/s12920-015-0122-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, McKillop JH, Packard CJ. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333:1301–1307. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- 20.Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, Beere PA, Langendorfer A, Stein EA, Kruyer W, Gotto AM., Jr Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279:1615–1622. doi: 10.1001/jama.279.20.1615. [DOI] [PubMed] [Google Scholar]
- 21.Pocock SJ, Simon R. Sequential treatment assignment with balancing for prognostic factors in the controlled clinical trial. Biometrics. 1975;31:103–115. [PubMed] [Google Scholar]
- 22.Thompson FE, Midthune D, Subar AF, Kipnis V, Kahle LL, Schatzkin A. Development and evaluation of a short instrument to estimate usual dietary intake of percentage energy from fat. J Am Diet Assoc. 2007;107:760–767. doi: 10.1016/j.jada.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Mayer CJ, Steinman L, Williams B, Topolski TD, LoGerfo J. Developing a Telephone Assessment of Physical Activity (TAPA) questionnaire for older adults. Prev Chronic Dis. 2008;5:A24. [PMC free article] [PubMed] [Google Scholar]
- 24.Spielberger C, Spielberger C. Manual for the State-Trait Anxiety Inventory, STAI (Form Y) 1983 [Google Scholar]
- 25.Paulos JA. Innumeracy: mathematical illiteracy and its consequences. New York: Hill and Wang; 1988. [Google Scholar]
- 26.Robinson CL, Jouni H, Kruisselbrink TM, Austin EE, Christensen KD, Green RC, Kullo IJ. Disclosing genetic risk for coronary heart disease: effects on perceived personal control and genetic counseling satisfaction. Clin Genet. 2015 Feb 23; doi: 10.1111/cge.12577. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McBride CM, Bepler G, Lipkus IM, Lyna P, Samsa G, Albright J, Datta S, Rimer BK. Incorporating genetic susceptibility feedback into a smoking cessation program for African-American smokers with low income. Cancer Epidemiol Biomarkers Prev. 2002;11:521–528. [PubMed] [Google Scholar]
- 28.Bloss CS, Schork NJ, Topol EJ. Effect of direct-to-consumer genomewide profiling to assess disease risk. N Engl J Med. 2011;364:524–534. doi: 10.1056/NEJMoa1011893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green RC, Roberts JS, Cupples LA, Relkin NR, Whitehouse PJ, Brown T, Eckert SL, Butson M, Sadovnick AD, Quaid KA, Chen C, Cook-Deegan R, Farrer LA. Disclosure of APOE genotype for risk of Alzheimer’s disease. N Engl J Med. 2009;361:245–254. doi: 10.1056/NEJMoa0809578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lautenbach DM, Christensen KD, Sparks JA, Green RC. Communicating genetic risk information for common disorders in the era of genomic medicine. Annu Rev Genomics Hum Genet. 2013;14:491–513. doi: 10.1146/annurev-genom-092010-110722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jonathan E, Derrick B, Emma L, Sarah P, John D, Jane A, Rory C. C-reactive protein concentration and the vascular benefits of statin therapy: an analysis of 20,536 patients in the Heart Protection Study. Lancet. 2011;377:469–476. doi: 10.1016/S0140-6736(10)62174-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, Webb TR, Zeng L, Dehghan A, Alver M, Armasu SM, Auro K, Bjonnes A, Chasman DI, Chen S, Ford I, Franceschini N, Gieger C, Grace C, Gustafsson S, Huang J, Hwang SJ, Kim YK, Kleber ME, Lau KW, Lu X, Lu Y, Lyytikainen LP, Mihailov E, Morrison AC, Pervjakova N, Qu L, Rose LM, Salfati E, Saxena R, Scholz M, Smith AV, Tikkanen E, Uitterlinden A, Yang X, Zhang W, Zhao W, de Andrade M, de Vries PS, van Zuydam NR, Anand SS, Bertram L, Beutner F, Dedoussis G, Frossard P, Gauguier D, Goodall AH, Gottesman O, Haber M, Han BG, Jalilzadeh S, Kessler T, Konig IR, Lannfelt L, Lieb W, Lind L, Lindgren CM, Lokki ML, Magnusson PK, Mallick NH, Mehra N, Meitinger T, Memon FU, Morris AP, Nieminen MS, Pedersen NL, Peters A, Rallidis LS, Rasheed A, Samuel M, Shah SH, Sinisalo J, Stirrups KE, Trompet S, Wang L, Zaman KS, Ardissino D, Boerwinkle E, Borecki IB, Bottinger EP, Buring JE, Chambers JC, Collins R, Cupples LA, Danesh J, Demuth I, Elosua R, Epstein SE, Esko T, Feitosa MF, Franco OH, Franzosi MG, Granger CB, Gu D, Gudnason V, Hall AS, Hamsten A, Harris TB, Hazen SL, Hengstenberg C, Hofman A, Ingelsson E, Iribarren C, Jukema JW, Karhunen PJ, Kim BJ, Kooner JS, Kullo IJ, Lehtimaki T, Loos RJ, Melander O, Metspalu A, Marz W, Palmer CN, Perola M, Quertermous T, Rader DJ, Ridker PM, Ripatti S, Roberts R, Salomaa V, Sanghera DK, Schwartz SM, Seedorf U, Stewart AF, Stott DJ, Thiery J, Zalloua PA, O’Donnell CJ, Reilly MP, Assimes TL, Thompson JR, Erdmann J, Clarke R, Watkins H, Kathiresan S, McPherson R, Deloukas P, Schunkert H, Samani NJ, Farrall M. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.