

Abstract
Better prediction of in vivo drug efficacy using in vitro models should greatly improve in vivo success. Here we utilize 3D highly porous chitosan-alginate complex scaffolds to probe how various components of the glioblastoma microenvironment including extracellular matrix and stromal cells affect tumor cell stem-like state.
The failure of new therapies in in vivo and clinical trials represents a costly bottleneck for new drug development. The vast majority of cancer drug development begins with in vitro testing using cell lines. However, many of these therapies that show promise in vitro fail during in vivo trials. Failure can be caused by various factors such as poor biodistribution, low serum stability, high off-target toxicity, and lower efficacy in cancer cells growing as part of the larger tumor microenvironment. The ability to better predict in vivo drug efficacy in vitro could rapidly accelerate novel drug development and reduce costs associated with these animal experiments.1
We have developed 3D highly porous scaffolds of polyelectrolyte chitosan-alginate (CA) complexes for in vitro culture of cancer cells that better mimic the behavior of cells growing as part of a tumor than cells culture on 2D well plates.2 We have shown that culturing cancer cells in these scaffolds drastically alters their growth, shape, and gene expression profile towards being more malignant. Indeed, we have shown enrichment of the cancer stem-like cell population indicating the more aggressive phenotype of cells cultured on these scaffolds, which was attributed to the 3D chemical extracellular matrix (ECM) environment afforded by the scaffolds.3 However, the tumor microenvironment comprises not only ECM but contains a variety of stromal cells that can both affect tumor growth, progression, and stem-like state.4 In fact, the tumor microenvironment has recently received significant attention in the development of new anti-cancer therapies.4b, 5 Therefore, the ability to model, in vitro, how the various parts of the tumor microenvironment affect tumor behavior could greatly accelerate the development of these next-generation therapies. Our CA scaffolds are well suited to systematically test how each stromal compartment affects cancer cell behavior.
Here we aim to utilize our CA scaffolds to probe how the tumor microenvironment of glioblastoma (GBM), the most common and deadly brain cancer, affects their growth and malignancy. We test how differences in extracellular composition and stromal cell type affect the stem-like properties of GBM cells. CA scaffolds were coated with hyaluronic acid (HA), a major ECM protein in GBM,6 or polycaprolactone (PCL) as a control to block the chemical structure of the CA scaffold so that only the 3D environment was present. GBM cells were also co-cultured in scaffolds with the primary stromal cells of GBM, astrocytes and endothelial cells.7 The effects of these changes in the tumor microenvironment on GBM cells were analyzed through protein and gene expression analyses.
CA scaffolds were coated with PCL as reported previously3 to generate PCL-CA scaffolds, and coated with HA following a similar procedure to generate HA-CA scaffolds. HA was dissolved in acetic acid at 0.5% and then diluted in methanol to 0.05%. Scaffolds were added to the HA solution and allowed to coat for 4 hrs before quickly drying with an air gun, sterilized with 70% ethanol overnight, and then washed with PBS before use. HA coating was confirmed with FTIR (Fig. 1b). A unique characteristic peak at 1380 cm−1 from HA was observed in HA-CA scaffolds (Fig. S1) indicating HA present on the scaffolds. The presence of PCL was observed from the unique characteristic peak at 1750 cm−1. To ensure pore structure was not altered, scaffolds were imaged using SEM (Fig. 1a). Pore structure remained similar to uncoated CA scaffolds as determined from low magnification images. High magnification images show the uniform coating of PCL or HA on the surface of the scaffold walls.
Figure 1.

Scaffold characterization. a) SEM images of scaffolds with different coatings showing pore structure at low magnification and wall structure at high magnification. Scale bars for low and high magnification correspond to 500 μm and 20 μm, respectively. b) FTIR characterization indicating the presence of HA or PCL on the scaffolds.
Cells (red fluorescent protein (RFP) expressing U-87 MG) were seeded (50,000 in 50 μL) on optimized coated scaffolds (uncoated, PCL, HA) and imaged over 8 days. Cells cultured in uncoated CA scaffolds showed similar growth as seen previously,2b, 3 generating ~40 μm diameter tumor spheres within the scaffold in 8 days (Figure 2). Cells cultured in HA-CA scaffolds showed similar growth trend, but the number of tumor spheres within the scaffold was much more numerous (Fig. S2). Cells cultured in PCL-CA scaffolds displayed a more elongated structure similar to that seen with 2D culture, and no tumor spheres were observed in the lower magnification images (Fig. S2).
Figure 2.
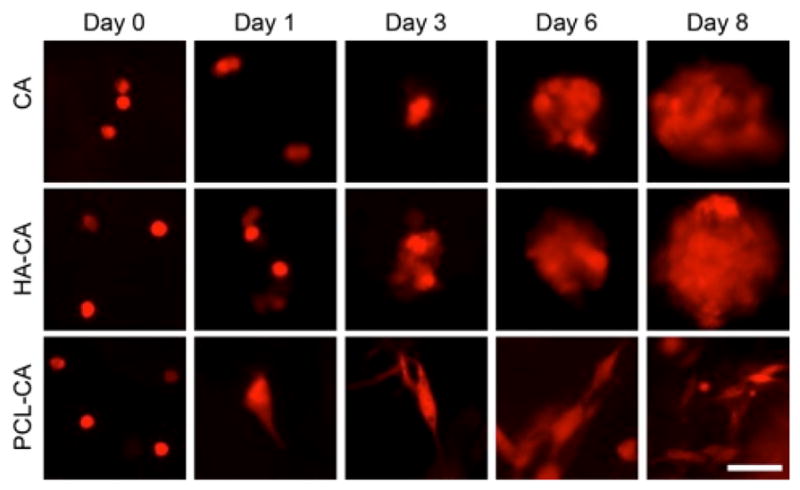
Fluorescence images of U-87 MG cells cultured on coated and uncoated CA scaffolds for 8 days. Scale bar corresponds to 20 μm.
On day 10, cells were imaged and collected for SEM, Western blot, and PCR analyses. SEM imaging confirmed that the cell structure observed with fluorescence imaging tumor spheroids in the CA and HA-CA scaffolds and elongated cells in the PCL-CA scaffolds (Fig. 3a–b). Western blot was used to detect expression of CD44 and Id1 (Fig. S3), whose increased expressions suggest an increase in the stem-like cell state of the cells.8 Fig. 3c shows an increase in expression of both CD44 and Id1 protein in cells cultured in HA-CA scaffolds suggesting cells on these scaffolds were more stem-like. CD44 is the cell surface receptor for HA so is likely upregulated in response to HA present on the scaffold. Overexpression of Id1, an inhibitor of DNA binding protein involved in regulation of self-renewal in neural stem cells9 and cancer metastases,10 in combination with CD44 is thought to represent a stem-like population of GBM cells.11 To further assess the stem-like characteristics of these cells, expression of the cancer stem-like cell gene CD13312 was assessed using PCR (Fig. 3d) to avoid confusing glycosylated and non-glycosylated forms of CD133 protein in Western blots.13 CD133+ GBM subpopulations are well characterized to be enriched in cancer stem-like cells.12b, 14 Cells cultured in HA-CA scaffolds showed a 3.5-fold increase in CD133 expression, nearly double that observed in cells cultured in CA scaffolds. This suggests that changing the chemical microenvironment altered the cancer stem-like properties of cultured GBM cells.
Figure 3.

U-87 MG cells cultured in different scaffolds for 10 days. a) Fluorescence images of cells cultured in different scaffolds indicating their morphology. The scale bar corresponds to 20 μm. b) Colorized SEM images confirming morphology. Cells were identified and pseudocolored in Photoshop. The scale bar corresponds to 20 μm. c) Differential expression of CD44 and Id1 protein in U-87 MG cells cultured in different scaffolds as determined by Western blot. Band densities were normalized to β-actin and 2D culture. d) Differential CD133 mRNA abundance in U-87 MG cells cultured in different scaffolds as determined by PCR. β-actin was used as the reference gene and mRNA abundance was normalized to 2D culture. **indicates a statistical difference from CA culture as determined by Student’s t test (p < 0.001).
The most common stromal cells present in GBM are endothelial cells and astrocytes.7 These cells have been shown to provide a niche for GBM stem-like cells to invade surrounding brain15 and promote drug resistance.7a, 16 For co-culture experiments, U-87 MG cells were mixed at various ratios with human astrocytes or endothelial cells (HUVEC) prior to seeded on scaffolds. Ratios between stromal:tumor cells were 1:5, 1:1, and 5:1 with 50,000 U-87 MG cells seeded in each scaffold. The growth behavior of the cells was visualized through live cell fluorescence imaging using RFP expressing U-87 MG cells and stromal cells labeled with green Vybrant CFDA SE Cell Tracer dye at 10 μM following the manufacturer’s protocol, which gave visible fluorescence over 10 days without observable toxicity to the cells (data not shown).
Co-culture with either astrocytes or HUVECs resulted in smaller tumor spheres forming (Fig. S4) suggesting a slower growth rate of the tumor cells (Fig. 4). Tumor spheres were observed to form around or on the stromal cells, which grew significantly slower, so final tumor sphere had only one to a few stromal cells associated with them. The lower ratio of stromal cell to tumor cell showed similar tumor sphere sizes to tumor cells grown without stromal cells likely because of the minimal number of tumor cells that had a stromal cell to incorporate with.
Figure 4.

Fluorescence images of U-87 MG cells cultured on CA scaffolds with human astrocytes or HUVEC cells for 10 days. Scale bar corresponds to 20 μm. U-87 MG cells are red and stromal cells are green.
Western blot revealed an increased expression of CD44 and Id1 (Fig. S5) with increased ratios of astrocytes or HUVECs to U-87 MG cells (Fig. 5a–b). Additionally, this increase was accompanied by increased CD133 mRNA abundance (Fig. 5c) suggesting co-culture with stromal cells increased the stem-like properties of U-87 MG cells. This is in accordance with the smaller tumor spheres observed at higher stroma:tumor cell radios, since the slower growth of tumor cells may suggest reversion to a more stem-like state rather than simply uncontrolled cell proliferation, which we’ve observed previously.3, 17 The slower growth of tumor spheres combined with the increased expression of stem-like cancer cell markers provides strong evidence that we can finely adjust the tumor microenvironment in vitro to better mimic in vivo conditions.
Figure 5.

Cancer stem-like cell (CSC) gene expression analysis after 10-day culture of U-87 MG cells in CA scaffolds with different stromal cells. a) Western blot analysis of CD44. b) Western blot analysis of Id1. c) PCR analysis of CD133. Data is normalized to cells cultured on 2D. The U87 label represents U-87 MG cells cultured on CA scaffolds without stromal cells. Statistical significance was determined by Student’s t test (* indicates p < 0.01).
In conclusion, we showed that altering the tumor microenvironment in vitro alters the expression of glioblastoma stem-like cell markers. This correlates well with the growing body of evidence that the stromal compartment plays a significant role in tumor aggressiveness and progression, and that this can be modeled in vitro. We found that both the chemical structure of the microenvironment and the presence of stromal cells impact glioblastoma stem-like cell state. We hope these modified scaffolds will be used to further probe how each stromal compartment affects tumor behavior in order to accelerate the development of more effective cancer therapies.
Supplementary Material
Acknowledgments
This work was supported by NIH grant R01CA172455. F.K. acknowledges support from an American Brain Tumor Association Fellowship in Honor of Susan Kramer, K.W. acknowledges support from the University of Washington College of Engineering Dean’s Fellowship, and A.E. acknowledges support from the National Science Foundation Graduate Research Fellowship. We acknowledge the use of the SEM at the University of Washington Center for Nanotechnology. We acknowledge laboratory assistance from Ms. Anna A. Thompson and Ms. Melanie J. Coyne for running Western blots.
Footnotes
Electronic Supplementary Information (ESI) available: Detailed methods and low magnification images of tumor spheres growing in scaffolds. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Hutmacher DW. Nature materials. 2010;9:90. doi: 10.1038/nmat2619. [DOI] [PubMed] [Google Scholar]
- 2.(a) Florczyk SJ, Liu G, Kievit FM, Lewis AM, Wu JD, Zhang M. Advanced healthcare materials. 2012;1:590. doi: 10.1002/adhm.201100054. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kievit FM, Florczyk SJ, Leung MC, Veiseh O, Park JO, Disis ML, Zhang M. Biomaterials. 2010;31:5903. doi: 10.1016/j.biomaterials.2010.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Leung M, Kievit FM, Florczyk SJ, Veiseh O, Wu J, Park JO, Zhang M. Pharm Res. 2010;27:1939. doi: 10.1007/s11095-010-0198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Florczyk SJ, Kim DJ, Wood DL, Zhang M. J Biomed Mater Res A. 2011;98:614. doi: 10.1002/jbm.a.33153. [DOI] [PubMed] [Google Scholar]
- 3.Kievit FM, Florczyk SJ, Leung MC, Wang K, Wu JD, Silber JR, Ellenbogen RG, Lee JS, Zhang M. Biomaterials. 2014;35:9137. doi: 10.1016/j.biomaterials.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Junttila MR, de Sauvage FJ. Nature. 2013;501:346. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]; (b) Hanahan D, Weinberg RA. Cell. 2011;144:646. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.(a) Quail DF, Joyce JA. Nat Med. 2013;19:1423. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jain RK. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31:2205. doi: 10.1200/JCO.2012.46.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Ananthanarayanan B, Kim Y, Kumar S. Biomaterials. 2011;32:7913. doi: 10.1016/j.biomaterials.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Toole BP. Nat Rev Cancer. 2004;4:528. doi: 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- 7.(a) Golebiewska A, Bougnaud S, Stieber D, Brons NHC, Vallar L, Hertel F, Klink B, Schrock E, Bjerkvig R, Niclou SP. Brain. 2013;136:1462. doi: 10.1093/brain/awt025. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rath BH, Fair JM, Jamal M, Camphausen K, Tofilon PJ. PLoS One. 2013;8:e54752. doi: 10.1371/journal.pone.0054752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pietras A, Katz AM, Ekstrom EJ, Wee B, Halliday JJ, Pitter KL, Werbeck JL, Amankulor NM, Huse JT, Holland EC. Cell Stem Cell. 2014;14:357. doi: 10.1016/j.stem.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nam HS, Benezra R. Cell Stem Cell. 2009;5:515. doi: 10.1016/j.stem.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupta GP, Perk J, Acharyya S, de Candia P, Mittal V, Todorova-Manova K, Gerald WL, Brogi E, Benezra R, Massague J. Proc Natl Acad Sci U S A. 2007;104:19506. doi: 10.1073/pnas.0709185104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anido J, Saez-Borderias A, Gonzalez-Junca A, Rodon L, Folch G, Carmona MA, Prieto-Sanchez RM, Barba I, Martinez-Saez E, Prudkin L, Cuartas I, Raventos C, Martinez-Ricarte F, Poca MA, Garcia-Dorado D, Lahn MM, Yingling JM, Rodon J, Sahuquillo J, Baselga J, Seoane J. Cancer Cell. 2010;18:655. doi: 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 12.(a) Lathia JD, Hitomi M, Gallagher J, Gadani SP, Adkins J, Vasanji A, Liu L, Eyler CE, Heddleston JM, Wu Q, Minhas S, Soeda A, Hoeppner DJ, Ravin R, McKay RD, McLendon RE, Corbeil D, Chenn A, Hjelmeland AB, Park DM, Rich JN. Cell Death Dis. 2011;2:e200. doi: 10.1038/cddis.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tirino V, Desiderio V, d’Aquino R, De Francesco F, Pirozzi G, Graziano A, Galderisi U, Cavaliere C, De Rosa A, Papaccio G, Giordano A. PLoS One. 2008;3:e3469. doi: 10.1371/journal.pone.0003469. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Nature. 2004;432:396. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 13.(a) Osmond TL, Broadley KW, McConnell MJ. Int J Mol Med. 2010;25:883. doi: 10.3892/ijmm_00000418. [DOI] [PubMed] [Google Scholar]; (b) Bidlingmaier S, Zhu X, Liu B. J Mol Med (Berl) 2008;86:1025. doi: 10.1007/s00109-008-0357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hermansen SK, Christensen KG, Jensen SS, Kristensen BW. J Histochem Cytochem. 2011;59:391. doi: 10.1369/0022155411400867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Nature. 2006;444:756. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 15.(a) Grodecki J, Short AR, Winter JO, Rao SS, Winter JO, Otero JJ, Lannutti JJ, Sarkar A. Biotechnology progress. 2015 doi: 10.1002/btpr.2123. [DOI] [PubMed] [Google Scholar]; (b) Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, Frank A, Bayazitov IT, Zakharenko SS, Gajjar A, Davidoff A, Gilbertson RJ. Cancer Cell. 2007;11:69. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 16.Yang N, Yan T, Zhu H, Liang X, Leiss L, Sakariassen PO, Skaftnesmo KO, Huang B, Costea DE, Enger PO, Li X, Wang J. Journal of translational medicine. 2014;12:278. doi: 10.1186/s12967-014-0278-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Florczyk SJ, Wang K, Jana S, Wood DL, Sytsma SK, Sham JG, Kievit FM, Zhang M. Biomaterials. 2013;34:10143. doi: 10.1016/j.biomaterials.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.