

Abstract
Lung injury associated with hyperoxia reflects in part the secondary effects of pulmonary inflammation and the associated production of reactive oxygen species due to activation of NADPH oxidase, type 2 (NOX2). Activation of NOX2 requires the phospholipase A2 (PLA2) activity of peroxiredoxin 6 (Prdx6). Therefore, we evaluated whether blocking Prdx6 PLA2 activity using the inhibitor MJ33 would be protective in a mouse model of acute lung injury resulting from hyperoxic exposure. Mice were treated with an intraperitoneal injection of MJ33 (2.5 nmol/g body weight) at the start of exposure (zero time) and at 48 h during continuous exposure to 100% O2 for 80 h. Treatment with MJ33 reduced the number of neutrophils and the protein content in the fluid obtained by bronchoalveolar lavage, inhibited the increase in lipid peroxidation products in lung tissue, decreased the number of apoptotic cells in the lung, and decreased the perivascular edema associated with the 80 h exposure to hyperoxia. Thus, blocking Prdx6 PLA2 activity by MJ33 significantly protected lungs against damage from hyperoxia, presumably by preventing the activation of NOX2 and the amplification of lung injury associated with inflammation. These findings demonstrate that MJ33, a potent inhibitor of Prdx6 PLA2 activity, can protect mouse lungs against the manifestations of acute lung injury due to oxidative stress.
Keywords: Acute lung injury, Oxidative stress, Lipid peroxidation, MJ33, Inflammation
Abbreviations: BALF, bronchoalveolar lavage fluid; FOX, ferrous oxidation-xylenol orange; ip, intraperitoneal; LOOH, lipid hydroperoxides; MJ33, 1-hexadecyl-3-(trifluoroethyl)-sn-glycero-2-phosphomethanol; NOX2, NADPH oxidase, type 2; PLA2, phospholipase A2; ROS, reactive oxygen species; TBARS, thiobarbituric acid reactive substances; TUNEL, terminal transferase dUTP nick-end-labeling
Graphical Abstract
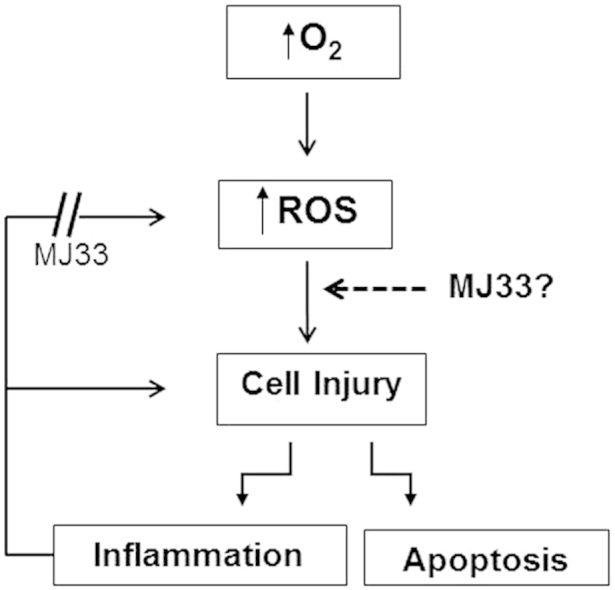
Highlights
-
•
Exposure of mice to sublethal O2 (100% O2 for 80 h) resulted in acute lung injury (ALI).
-
•
Manifestations of ALI included increased PMN, lung edema, tissue lipid peroxidation, and cellular apoptosis.
-
•
MJ33, an inhibitor of lung acidic, Ca2+-independent phospholipase A2 (aiPLA2) activity, markedly decreased manifestations of tissue injury with hyperoxia.
-
•
The proposed mechanism for lung protection is the prevention of NADPH oxidase activation associated with lung inflammation.
-
•
Possible additional roles for MJ33 have not yet been studied.
Introduction
Continuous exposure to O2 at concentrations greater than 60% in the inspired air can result in irreversible pulmonary toxicity and death [11], [12], [15], [16], [30], [38]. A major mechanism for lung injury associated with hyperoxia is the oxidation of tissue components, including lipids, proteins, and DNA, that results subsequent to the formation of reactive oxygen species (ROS) [15], [19], [39]. ROS can be generated directly from the interaction of O2 with tissue components or can result from a secondary inflammatory response that amplifies the primary lung injury.
A major source of secondary ROS in hyperoxic lung injury is the oxidative burst of inflammatory cells including polymorphonuclear leukocytes (PMN) and alveolar macrophages (AM). This burst results from activation of a membrane-bound NADPH oxidase (NOX), the enzyme that is primarily responsible for ROS generation associated with inflammation [3]. The generation of ROS is crucial for the bactericidal function of these phagocytic cells but an over-exuberant response can damage normal host cells [1], [37]. The NOX enzyme family comprises 7 members; NOX2 is considered as the prototypical NOX and is the family member associated with the respiratory burst in phagocytic cells [34]. NOX2 also is present in pulmonary endothelium and other cell types where it plays an important role in cell signaling, including the recruitment of phagocytic cells associated with inflammation [5], [50]. Thus, the activation of NOX2 is considered to be an important factor in the amplification of lung injury and preventing this activation could be beneficial in preventing tissue injury under these conditions [8], [17].
Currently available NOX inhibitors have relatively low selectivity, potency and bioavailability [20], [24]. Recently, 1-hexadecyl-3-(trifluoroethyl)-sn-glycero-2-phosphomethanol (MJ33) has been described as a more potent and non-toxic inhibitor of agonist-induced activation of NOX2 [9], [27]. The mechanism is through competitive inhibition of the phospholipase A2 (PLA2) activity of peroxiredoxin 6 that is required for NOX2 activation [9]. We have termed the PLA2 activity of Prdx6 as aiPLA2 [13]. MJ33 has been shown to markedly inhibit lung injury associated with lung inflammation in mice treated with endotoxin [26]. MJ33 is a fluorinated lipid analog that serves to inhibit the enzyme by acting as a mimic of the transition state of the substrate [18]. MJ33 has specificity for both pancreatic (type 1B) PLA2 and aiPLA2 activities, but does not inhibit cytosolic (type IV) PLA2, phospholipases C/D, or other lipases [13], [14], [18], [23]. For the present study, we proposed that blocking NOX2 activation using MJ33 would be protective in the mouse model of acute lung injury associated with exposure to hyperoxia.
Materials and methods
Reagents
MJ33 (as the lithium salt), 2-thiobarbituric acid (TBA), and butylated hydroxytoluene were purchased from Sigma-Aldrich (St. Louis, MO). 4′,6-Diamidino-2-phenylindole (DAPI) and Dulbecco's phosphate-buffered saline (DPBS) were obtained from Life Technologies (Grand Island, NY). Medical grade O2 was obtained from Air Products (Allentown, PA). Ferrous oxidation-xylenol orange (FOX) assay kit was from Northwest Life Science (Vancouver, WA). Trichloroacetic acid and formalin (10%) were purchased from Fisher Scientific (Fair Lawn, NJ). Coomassie blue protein assay reagent was purchased from Bio-Rad Laboratories (Hercules, CA). Proteinase K was purchased from DAKO (Carpinteria, CA). Terminal transferase dUTP nick-end-labeling (TUNEL) Label and TUNEL Enzyme were purchased from Roche (Indianapolis, IN).
Animals
Male C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and used at 8–10 weeks of age. Protocols for the use of mice for these studies were approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC).
Exposure to hyperoxia
Mice were exposed to 100% O2 at 1 atmosphere absolute in a Plexiglas chamber (Braintree Scientific Inc., Braintree, MA) as described previously [46]. Oxygen was passed through a bubble humidifier and introduced into the sealed chamber at 6–8 l/min to provide ~5 gas exchanges per hour. The oxygen concentration of the chamber was measured continuously with an oxygen analyzer (Pacifitech, Temecula, CA) and exceeded 98%; the exposure under these conditions is termed 100% O2. Chamber CO2 was absorbed with a soda lime filter and was maintained at <0.2%. Relative humidity in the chamber was 45–50%. Mice were allowed food and water ad libitum and maintained on a 12-h dark:light cycle. Cages were opened daily for 5 min for change of water, food, and bedding.
To determine the effect of the inhibitor, MJ33 in aqueous solution (50 nmol in 20 µl PBS) was injected intraperitoneally (ip) at 0 and 48 h of hyperoxic exposure. Control mice were injected with the same volume of PBS. Some mice were sacrificed at 48 h in order to determine PLA2 activity of the lung homogenate. Remaining mice were sacrificed after 80 h of O2 exposure. At the end of exposure, mice were anesthetized with a cocktail of ketamine/xylazine/acepromazine (100/15/2 mg/kg body wt) injected intraperitoneally, a midline laparotomy/thoracotomy was performed, the trachea was cannulated for continuous ventilation, and mice were exsanguinated by transection of the abdominal aorta.
Analytical procedures
Bronchoalveolar lavage
Immediately after sacrifice of mice, the lungs were lavaged three times by instillation followed by aspiration of 1 ml PBS containing 0.5 mmol EDTA, pH 8.0, to obtain the bronchoalveolar lavage fluid (BALF). These lungs were then discarded. BALF was analyzed for total nucleated cells using a hemocytometer and the protein concentration of the supernatant was assayed using the Bradford protocol (see below).
Lipid peroxidation
Tissue lipid peroxidation was evaluated in lungs that were not lavaged after mice were sacrificed. The pulmonary vasculature was flushed with PBS by cannulation of the pulmonary artery via the right ventricle, followed by en bloc removal of the heart and lungs. The heart and large airways were dissected away from the lungs and discarded. The lung tissue was then rapidly frozen in liquid nitrogen and stored at −80 °C for later analysis of lipid peroxidation using assays that have been described previously in detail [28]. For assay, an aliquot of frozen lung was homogenized under N2 in PBS (1:10) containing 0.01% butylated hydroxytoluene (BHT). To determine TBA-reactive substances (TBARS), the absorbance of the lung homogenate after addition of thiobarbituric acid was measured at 535 nm. For determination of lipid hydroperoxides (LOOH) by the FOX method, absorbance of the Fe3+-xylenol orange complex was measured at 550 nm. TBARS and lipid LOOH were normalized to lung homogenate protein. Protein concentration of lung extracts was measured by Bradford assay with Coomassie blue (Bio-Rad) and bovine γ-globulin as the standard; absorbance was read at 595 nm. Absorbance measurements utilized a Cary 50 Bio UV–visible spectrophotometer (Agilent Technologies, Foster City, CA).
PLA2 activity
For measurement of aiPLA2 activity, lungs were cleared of blood as described above and then homogenized in PBS. Mixed unilamellar liposomes containing tracer [3H-9, 10-palmitate]- DPPC (4400 dpm/nmol) were used as substrate and were incubated with lung homogenate at 37 °C for 1 h at pH 4 in Ca2+-free medium and then analyzed for 3H-palmitate as described previously [13], [35]. PLA2 activity was normalized to the lung homogenate protein content used for assay.
Histology and apoptosis
Lungs used for histological evaluation were removed from the thorax of the mouse after sacrifice and inflated via a cannula by gentle infusion of fixative (10% phosphate-buffered formalin, pH 7.0) over 5 min to reach a constant fluid pressure of 25 cm H2O. The trachea was tied with a ligature, and the lungs were placed in a glass vial containing 10% formalin and kept on ice for 24 h. All tissue samples were processed by the Pathology Core at the Children's Hospital of Philadelphia (Abramson Research Center, Philadelphia, PA). After fixation, individual lung lobes were embedded in paraffin and blocks were sectioned; some sections were stained with hematoxylin and eosin (H&E) while others were stained for TUNEL assay. For TUNEL assay, lung sections were hydrated and endogenous hydrogen peroxide was blocked by treatment with hydrogen peroxide-methanol followed by Proteinase K; sections were then treated with TUNEL label and TUNEL enzyme and counterstained with DAPI [28].
Whole lobe mounts stained with H&E were examined independently by 3 observers and randomly selected fields were chosen for comparison. Lung lobes from control mice (room air) and from mice that were exposed to oxygen for 80 h with or without MJ33 were evaluated (n=3 for each condition). For quantitation of perivascular edema and apoptosis, the results from 5 to 6 fields were averaged to obtain the mean result for each lung. Perivascular edema was quantitated in H&E stained sections by measuring the ratio of total area (vessel plus perivascular space) to luminal area in peripheral arterioles using Aperio ImageScope software as described previously [28]. TUNEL staining showed considerable variability in different areas of the O2-exposed lungs. Therefore, we selected fields that showed a high or a low degree of apoptosis for further quantitation, and these are reported separately. The percentage of apoptotic (TUNEL-positive) cells was determined in TUNEL-stained sections by Cyto-Nuclear FL Quantification algorithm software (Fluorescent Toolbox, Leica Biosystems Imaging, Buffalo Grove, Il).
Statistical analysis
Data are expressed as mean±SE. Statistical significance was assessed by analysis of variance using SigmaStat software (Jandel Scientific, San Jose, CA). Group differences were evaluated by Student's t-test as appropriate. Differences between mean values were considered statistically significant at P≤0.05.
Results
PLA2 activity was measured in lung homogenates of mice that were sacrificed at 48 or 80 h after treatment with MJ33 and kept in room air or exposed continuously to 100% O2. Mice sacrificed after 48 h of O2 received only a single dose of MJ33 at zero time while mice sacrificed at 80 h received a second dose of MJ33 at 48 h of O2 exposure. aiPLA2 activity was higher in lungs from hyperoxic mice compared to room air controls (Table 1) consistent with the induction of Prdx6 expression with O2 exposure as we have noted previously [22]. There was a marked decrease in lung aiPLA2 activity with MJ33 treatment in both room air control mice and mice at 48 and 80 h of hyperoxia indicating efficacy of the MJ33 treatment protocol (Table 1). The residual PLA2 activity after MJ33 (~20% of control) has been a consistent finding that appears to be unrelated to the dose of the inhibitor and may reflect the activity of a lung PLA2 enzyme that is not inhibited by MJ33.
Table 1.
Effect of MJ33 on aiPLA2 activity of lung homogenate.
|
aiPLA2 activity nmol/h/mg prot |
||
|---|---|---|
| 48 h⁎ | 80 h⁎ | |
| MJ33 | 2.5±0.3 | − |
| O2 | 12.5±0.1† | 13.2±0.2† |
| O2+MJ33 | 2.8±0.3 | 2.5±0.1 |
PLA2 activity of control (no treatment) lungs was 8.8±0.2 nmol/h/mg protein. Values are mean±SE (n=3).
Time after the initial dose of MJ33 (50 nmol ip) and start of O2 exposure; for 80 h experiments, a second dose of MJ33 was given at 48 h.
P<0.05 vs. MJ33±O2.
Analysis of BALF showed a significant increase in the number of nucleated cells in lungs after 80 h of hyperoxia; this increase was abolished by treatment of mice with MJ33 (Table 2). Likewise, the protein content of BALF was increased significantly by 80 h of hyperoxia and was decreased by MJ33 treatment (Table 2).
Table 2.
Nucleated cells and protein in broncho-alveolar lung lavage fluid (BALF) following O2 exposure for 80 h: effect of MJ33.
|
Total cells ×104 |
Protein mg |
|
|---|---|---|
| Control | 7.3±0.8 | 0.2±0.04 |
| O2 | 19.2±1.1⁎ | 15.9±1.3⁎ |
| O2+MJ33 | 5.2±0.14† | 5.7+0.6⁎† |
Values are mean±SE (n=5).
P≤0.05 vs. control.
P≤0.05 vs. O2.
Lipid peroxidation was measured in whole lung homogenates by two different assays (TBARS, FOX) that are sensitive to different lipid oxidation products. TBARS reflects breakdown products of lipid metabolism such as aldehydes while the FOX assay is sensitive to hydroperoxides. Both assays indicate lung lipid peroxidation after 80 h of hyperoxia that was significantly decreased by treatment with MJ33 (Table 3).
Table 3.
Lung lipid peroxidation following O2 exposure for 80 h: effect of MJ33.
|
TBARS pmol/mg prot |
Lipid OOH pmol/mg prot |
|
|---|---|---|
| Control | 63.0±9 | 19.9±3 |
| O2 | 173±13⁎ | 54.8±2.1⁎ |
| O2+MJ33 | 77.0±8† | 31.7±3† |
Values are mean±SE (n=5).
P≤0.05 vs. control.
P≤0.05 vs. O2.
Lung histology was evaluated with H&E stained tissue sections. Hyperoxia resulted in increased alveolar wall thickness, interstitial and alveolar edema, and entrapped red blood cells with significant lymphatic cuffing as indicated by enlargement of peribronchial and perivascular spaces (Fig. 1). Perivascular edema as quantitated from the ratio of the total area of vessel plus perivascular space to the luminal area was increased in hyperoxic lungs (Table 4). MJ33 treatment significantly improved lung morphology and reduced the perivascular edema associated with hyperoxia (Fig. 1, Table 4).
Fig. 1.

Lung histology. Hematoxylin and eosin stained sections of mouse lungs after exposure to room air (control) or 100% O2 for 80 h with or without MJ33 treatment. The green lines outline the luminal area and total vascular area (including the perivascular space) of a small vein. The enlarged perivascular space in the O2 exposed mice indicates alveolar edema. Upper row: scale bar 300 µm, original magnification ×100; lower row: scale bar 200 µm, original magnification ×200. A, Airway; V, vessel.
Table 4.
Quantitation of perivascular edema and cellular apoptosis following O2 exposure for 80 h: effect of MJ33.
| Total vessel area/luminal area |
Apoptosisa% of cells |
||
|---|---|---|---|
| Low | High | ||
| Control | 1.8±0.1 | 0.2±0.01 | 0.2±0.04 |
| O2 | 80±1.1† | 1.5±0.3† | 47±1.8† |
| O2+MJ33 | 3.8 ±0.3†⁎ | 0.4±0.1⁎ | 0.7±0.01⁎ |
Values are mean±SE for n=3; at least 5 fields were evaluated for each section.
Apoptosis varied considerably within different fields of the same section; we chose fields in each section that showed relatively low or high rates of apoptosis. The values indicate the percentage of total cells that are stained positively for TUNEL.
P≤0.05 vs. control.
P≤0.05 vs. O2.
Apoptotic cell death was evaluated by TUNEL staining of lung sections (Fig. 2). These were quantitated as the % of total cells that stained positively with the fluorescent probe (Table 4). Apoptosis was low in lungs of room air control mice but increased significantly although in a patchy fashion, in lungs of mice exposed to 80 h of hyperoxia; the percentage of TUNEL positive cells in fields with low levels as well as high levels of staining was significantly increased with hyperoxia (Fig. 2; Table 4). MJ33 treatment significantly reduced apoptotic cell death caused by hyperoxia (Fig. 2; Table 4).
Fig. 2.

Apoptosis of lung cells. Apoptosis was evaluated by TUNEL staining after exposure of mice for 80 h to room air (control), 100% O2, or 100% O2+MJ33. TUNEL-positive cells show as bright green dots. As the TUNEL staining was heterogeneous, we separately assessed regions with low and high levels of staining in each section. (A) Sections from control mice (room air) and mice exposed to 100% O2 for 80 h without or with MJ33 treatment. Upper row, low levels of staining; lower row, high levels of staining. Scale bar = 500 µm. (B) Areas enclosed in the box from upper row, low staining (left) or lower row; high staining (right).
Discussion
As a first step to evaluate the use of MJ33 in mice that were exposed to hyperoxia, we determined the appropriate dose of MJ33 to inhibit lung aiPLA2 activity. We elected to administer MJ33 by intraperitoneal injection in order to avoid the more invasive procedures that would be required for intratracheal or intravenous administration. MJ33 treatment at zero time inhibited PLA2 activity by ~80% at 48 h compared to control. Assay at earlier time points showed similar inhibition and no further inhibition was observed after the direct addition of MJ33 to the lung homogenate used for assay (not shown). Thus, this dose of MJ33 appeared to result in maximal inhibition of lung aiPLA2 activity. A second dose of MJ33 that was administered at 48 h resulted in maximal inhibition of lung aiPLA2 activity for the 80 h duration of exposure to hyperoxia.
The time course that we used to assess hyperoxia-induced lung injury in mice was based on previous studies that showed early signs of lung injury at 60 h of exposure and death of 50% of wild type mice (LT50) at 87 h of 100% O2 [28], [46]. We selected 80 h as a sub-lethal exposure that should result in significant lung injury and would provide a good test for the efficacy of MJ33.
Analysis of BALF obtained after 80 h of hyperoxia showed lung inflammation as indicated by increased recovery of PMN. An increase in BALF protein also was observed and reflects increased alveolar permeability with leakage of plasma proteins into the airspaces. This latter finding is compatible with the histological finding of perivascular cuffing that reflects lung edema. The presence of increased TBARS and lipid hydroperoxides in the lung homogenate indicates the oxidation of tissue lipid components. Lipid peroxidation leads to cellular membrane disruption and can result in cell death, compatible with results of the TUNEL assay. All of these abnormalities induced by exposure of mice to hyperoxia were markedly reduced by treatment with MJ33. Based on our previous studies, we propose that the marked effect of MJ33 in ameliorating lung injury reflects the inhibition of aiPLA2 activity and, consequently, the inhibition of NOX2 activation in pulmonary endothelium and lung inflammatory cells. Thus, We propose that MJ33 inhibits the amplification of lung injury associated with lung inflammation by preventing the activation of NOX2.
There are several important questions that require consideration with respect to this proposed mechanism for protection against lung injury by MJ33. First, are there other effects of MJ33 that could contribute to protection? To date, the only effect described for MJ33 has been inhibition of several PLA2 activities. MJ33 is a non-reactive compound that does not interact chemically with its target; indeed, it inhibits PLA2 activity by serving as a mimic of the enzymatic transition state that binds tightly to the protein preventing catalytic processing. This lack of reactivity also is the basic for the non-toxicity of the compound as described previously [27]. The only PLA2 enzymes that are presently known to be inhibited by MJ33 are aiPLA2 and pancreatic secreted (Type IB) PLA2. It is unlikely that inhibition of pancreatic PLA2 has a significant effect on pulmonary injury with O2 since this protein has not been observed in the lung. Thus, at this time possible off-target effects of MJ33 would not seem to explain its role in preventing acute lung injury with hyperoxia.
A second possible confounding factor is whether inhibition of aiPLA2 activity by MJ33 has effects other than preventing NOX2 activation; for example, is aiPLA2 activity required for the activation of other enzymes besides NOX2? With respect to the other NOX proteins, there is evidence, albeit limited, indicating that NOX1 activation is independent of Prdx6 activity [27]. NOX3 has recently been identified in the lung, but regulation of its activity is not understood [49]. NOX4 is constitutively active (i.e. increased activity requires increased transcription) so that inhibition of NOX4 activation by MJ33 is extremely unlikely. NOX5 is not present in mouse lungs. Possible activation of DUOX enzymes has not been studied but they are not known to be expressed at significant levels in the lung alveolar cells. Thus, based on current knowledge, altered activation of NOX enzymes other then NOX2 cannot be considered as an explanation for the effect of MJ33. A possible role for aiPLA2 in the activation of other (non-NOX) enzymes by aiPLA2 has neither been proposed nor studied.
As indicated above, we have proposed that a block in NOX2 activation is the primary mechanism for the effectiveness of MJ33 in preventing lung injury during hyperoxia. Is there evidence from other studies to support this hypothesis? MJ33 inhibits the PLA2 activity of Prdx6, but does not inhibit its peroxidase activity [10]. Thus, it is not possible to study the effects of the loss of aiPLA2 activity using Prdx6 null mice. Indeed, these latter mice show increased sensitivity to the toxic effects of oxygen and other oxidants indicating an important anti-oxidant role for the peroxidase function of Prdx6 [42], [43], [44], [45]. The availability of mice expressing only the peroxidase and not the PLA2 activity of Prdx6 could provide important insights into the mechanism for the effect of MJ33, but those studies have not yet been done.
Another possibility to gain insight into the mechanism for the effectiveness of MJ33 would be to evaluate the effect of elevated O2 on lungs of NOX2 null mice. Those experiments have been reported in several publications but there does not appear to be a consensus concerning the role of NOX2 in hyperoxia; one study showed an important role for NOX2 in lung injury with hyperoxia [32] while the second study did not [7]. Numerous studies related to the role of PMN (a major source of NOX2) in hyperoxia as well as other lung disease models have suggested that ROS from this source contribute significantly to lung injury, although here again the results have been variable [2], [4], [21], [25], [29], [33], [36], [41], [47], [48]. Pulmonary endothelium is another source of NOX2 that could play an important role in lung injury. The role of ROS produced by lung microvascular endothelial cells in the pathophysiology of hyperoxic lung injury has been demonstrated but the source of ROS has not been adequately evaluated [6], [31], [40]; NOX2 appears to be important but other pathways may also play a role [6], [32]. So, the question of whether MJ33 may have have effects through mechanisms in addition to its inhibition of NOX2 activation remain unsettled.
Conclusions
Our data indicate that inhibition of the PLA2 activity of the Prdx6 by MJ33 inhibits the oxidative stress that plays an important role in lung injury during hyperoxia. Thus, our novel findings regarding the inhibition of Prdx6 PLA2 activity by MJ33 may constitute a promising new therapeutic approach for the prevention of lung injury associated with exposure to elevated concentrations of O2.
Acknowledgements
We thank Dr. Daniel Martinez from the Pathology Core at Children's Hospital of Philadelphia (Abramson Research Center, Philadelphia, PA) for assistance with lung histology and TUNEL quantitation and Ms. Dawn Williams for typing the manuscript. The results of these studies have presented in part at the 2014 Experimental Biology (EB) meeting in San Diego, Ca. Supported by HL-R-01-105509 from the National Institutes of Health of the United States.
References
- 1.Babior B.M. The leukocyte NADPH oxidase. The Israel Medical Association Journal. 2002;4(11):1023–1024. [PubMed] [Google Scholar]
- 2.Barry B.E., Crapo J.D. Patterns of accumulation of platelets and neutrophils in rat lungs during exposure to 100% and 85% oxygen. The American Review of Respiratory Disease. 1985;132(3):548–555. doi: 10.1164/arrd.1985.132.3.548. 4037528 [DOI] [PubMed] [Google Scholar]
- 3.Bellavite P. The superoxide-forming enzymatic system of phagocytes. Free Radical Biology and Medicine. 1988;4(4):225–261. doi: 10.1016/0891-5849(88)90044-5. 2834275 [DOI] [PubMed] [Google Scholar]
- 4.Boyce N.W., Campbell D., Holdsworth S.R. Granulocyte independence of pulmonary oxygen toxicity in the rat. Experimental Lung Research. 1989;15(3):491–498. doi: 10.3109/01902148909087873. 2743954 [DOI] [PubMed] [Google Scholar]
- 5.Browning E.A., Chatterjee S., Fisher A.B. Stop the flow: a paradigm for cell signaling mediated by reactive oxygen species in the pulmonary endothelium. Annual Review of Physiology. 2012;74:403–424. doi: 10.1146/annurev-physiol-020911-153324. 22077215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brueckl C., Kaestle S., Kerem A., Habazettl H., Krombach F., Kuppe H., Kuebler W.M. Hyperoxia-induced reactive oxygen species formation in pulmonary capillary endothelial cells in situ. American Journal of Respiratory Cell and Molecular Biology. 2006;34(4):453–463. doi: 10.1165/rcmb.2005-0223OC. 16357365 [DOI] [PubMed] [Google Scholar]
- 7.Carnesecchi S., Deffert C., Pagano A., Garrido-Urbani S., Métrailler-Ruchonnet I., Schäppi M., Donati Y., Matthay M.A., Krause K.H., Barazzone Argiroffo C. NADPH oxidase-1 plays a crucial role in hyperoxia-induced acute lung injury in mice. American Journal of Respiratory and Critical Care Medicine. 2009;180(10):972–981. doi: 10.1164/rccm.200902-0296OC. 19661248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carnesecchi S., Pache J.C., Barazzone-Argiroffo C. NOX enzymes: potential target for the treatment of acute lung injury. Cellular and Molecular Life Sciences. 2012;69(14):2373–2385. doi: 10.1007/s00018-012-1013-6. 22581364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chatterjee S., Feinstein S.I., Dodia C., Sorokina E., Lien Y.C., Nguyen S., Debolt K., Speicher D., Fisher A.B. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. Journal of Biological Chemistry. 2011;286(13):11696–11706. doi: 10.1074/jbc.M110.206623. 21262967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J.W., Dodia C., Feinstein S.I., Jain M.K., Fisher A.B. 1-Cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. Journal of Biological Chemistry. 2000;275(37):28421–28427. doi: 10.1074/jbc.M005073200. 10893423 [DOI] [PubMed] [Google Scholar]
- 11.Clark J.M., Lambertsen C.J. Pulmonary oxygen toxicity: a review. Pharmacological Reviews. 1971;23(2):37–133. 4948324 [PubMed] [Google Scholar]
- 12.Deneke S.M., Fanburg B.L. Normobaric oxygen toxicity of the lung. New England Journal of Medicine. 1980;303(2):76–86. doi: 10.1056/NEJM198007103030204. 6247652 [DOI] [PubMed] [Google Scholar]
- 13.Fisher A.B., Dodia C. Role of phospholipase A2 enzymes in degradation of dipalmitoylphosphatidylcholine by granular pneumocytes. The Journal of Lipid Research. 1996;37(5):1057–1064. 8725157 [PubMed] [Google Scholar]
- 14.Fisher A.B., Dodia C., Chander A., Jain M. A competitive inhibitor of phospholipase A2 decreases surfactant phosphatidylcholine degradation by the rat lung. Biochemical Journal. 1992;288(2):407–411. doi: 10.1042/bj2880407. 1463444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fisher A.B., Forman H.J., Glass M. Mechanisms of pulmonary oxygen toxicity. Lung. 1984;162(5):255–259. doi: 10.1007/BF02715655. 6392761 [DOI] [PubMed] [Google Scholar]
- 16.Gore A., Muralidhar M., Espey M.G., Degenhardt K., Mantell L.L. Hyperoxia sensing: from molecular mechanisms to significance in disease. Journal of Immunotoxicology. 2010;7(4):239–254. doi: 10.3109/1547691X.2010.492254. [DOI] [PubMed] [Google Scholar]
- 17.Griffith B., Pendyala S., Hecker L., Lee P.J., Natarajan V., Thannickal V.J. NOX enzymes and pulmonary disease. Antioxidants and Redox Signaling. 2009;11(10):2505–2516. doi: 10.1089/ars.2009.2599. 19331546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jain M.K., Tao W.J., Rogers J., Arenson C., Eibl H., Yu B.Z. Active-site-directed specific competitive inhibitors of phospholipase A2: novel transition-state analogues. Biochemistry. 1991;30(42):10256–10268. doi: 10.1021/bi00106a025. 1931954 [DOI] [PubMed] [Google Scholar]
- 19.Jamieson D., Chance B., Cadenas E., Boveris A. The relation of free radical production to hyperoxia. Annual Review of Physiology. 1986;48:703–719. doi: 10.1146/annurev.ph.48.030186.003415. 3010832 [DOI] [PubMed] [Google Scholar]
- 20.Jaquet V., Scapozza L., Clark R.A., Krause K.H., Lambeth J.D. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxidants and Redox Signaling. 2009;11(10):2535–2552. doi: 10.1089/ars.2009.2585. 19309261 [DOI] [PubMed] [Google Scholar]
- 21.Keeney S.E., Mathews M.J., Haque A.K., Rudloff H.E., Schmalstieg F.C. Oxygen-induced lung injury in the guinea pig proceeds through CD18-independent mechanisms. American Journal of Respiratory and Critical Care Medicine. 1994;149(2 1):311–319. doi: 10.1164/ajrccm.149.2.7905767. 7905767 [DOI] [PubMed] [Google Scholar]
- 22.Kim H.S., Manevich Y., Feinstein S.I., Pak J.H., Ho Y.S., Fisher A.B. Induction of 1-cys peroxiredoxin expression by oxidative stress in lung epithelial cells. The American Journal of Physiology – Lung Cellular and Molecular Physiology. 2003;285(2):L363–L369. doi: 10.1152/ajplung.00078.2003. 12851211 [DOI] [PubMed] [Google Scholar]
- 23.Kim T.S., Dodia C., Chen X., Hennigan B.B., Jain M., Feinstein S.I., Fisher A.B. Cloning and expression of rat lung acidic Ca(2+)-independent PLA2 and its organ distribution. American Journal of Physiology. 1998;274(5 1):L750–L761. doi: 10.1152/ajplung.1998.274.5.L750. 9612290 [DOI] [PubMed] [Google Scholar]
- 24.Krause K.H., Lambeth D., Krönke M. NOX enzymes as drug targets. Cellular and Molecular Life Sciences. 2012;69(14):2279–2282. doi: 10.1007/s00018-012-1006-5. 22585058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krieger B.P., Loomis W.H., Spragg R.G. Granulocytes and hyperoxia act synergistically in causing acute lung injury. Experimental Lung Research. 1984;7(2):77–83. doi: 10.3109/01902148409069668. 6525983 [DOI] [PubMed] [Google Scholar]
- 26.Lee I., Dodia C., Chatterjee S., Feinstein S.I., Fisher A.B. Protection against LPS-induced acute lung injury by a mechanism-based inhibitor of NADPH oxidase (type 2) The American Journal of Physiology – Lung Cellular and Molecular Physiology. 2014;306(7):L635–L644. doi: 10.1152/ajplung.00374.2013. 24487388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee I., Dodia C., Chatterjee S., Zagorski J., Mesaros C., Blair I.A., Feinstein S.I., Jain M., Fisher A.B. A novel nontoxic inhibitor of the activation of NADPH oxidase reduces reactive oxygen species production in mouse lung. Journal of Pharmacology and Experimental Therapeutics. 2013;345(2):284–296. doi: 10.1124/jpet.112.201079. 23475902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu G., Feinstein S.I., Wang Y., Dodia C., Fisher D., Yu K., Ho Y.S., Fisher A.B. Comparison of glutathione peroxidase 1 and peroxiredoxin 6 in protection against oxidative stress in the mouse lung. Free Radical Biology and Medicine. 2010;49(7):1172–1181. doi: 10.1016/j.freeradbiomed.2010.07.002. 20627125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Min J.H., Codipilly C.N., Nasim S., Miller E.J., Ahmed M.N. Synergistic protection against hyperoxia-induced lung injury by neutrophils blockade and EC-SOD overexpression. Respiratory Research. 2012;13:58. doi: 10.1186/1465-9921-13-58. 22816678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pagano A., Barazzone-Argiroffo C. Alveolar cell death in hyperoxia-induced lung injury. Annals of the New York Academy of Sciences. 2003;1010:405–416. doi: 10.1196/annals.1299.074. 15033761 [DOI] [PubMed] [Google Scholar]
- 31.Parinandi N.L., Kleinberg M.A., Usatyuk P.V., Cummings R.J., Pennathur A., Cardounel A.J., Zweier J.L., Garcia J.G., Natarajan V. Hyperoxia-induced NAD(P)H oxidase activation and regulation by MAP kinases in human lung endothelial cells. The American Journal of Physiology – Lung Cellular and Molecular Physiology. 2003;284(1):L26–L38. doi: 10.1152/ajplung.00123.2002. 12388366 [DOI] [PubMed] [Google Scholar]
- 32.Pendyala S., Gorshkova I.A., Usatyuk P.V., He D., Pennathur A., Lambeth J.D., Thannickal V.J., Natarajan V. Role of Nox4 and Nox2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxidants and Redox Signaling. 2009;11(4):747–764. doi: 10.1089/ars.2008.2203. 18783311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perkowski S., Scherpereel A., Murciano J.C., Arguiri E., Solomides C.C., Albelda S.M., Muzykantov V., Christofidou-Solomidou M. Dissociation between alveolar transmigration of neutrophils and lung injury in hyperoxia. The American Journal of Physiology – Lung Cellular and Molecular Physiology. 2006;291(5):L1050–L1058. doi: 10.1152/ajplung.00067.2006. 16815892 [DOI] [PubMed] [Google Scholar]
- 34.Petry A., Weitnauer M., Görlach A. Receptor activation of NADPH oxidases. Antioxidants and Redox Signaling. 2010;13(4):467–487. doi: 10.1089/ars.2009.3026. 20001746 [DOI] [PubMed] [Google Scholar]
- 35.Rahaman H., Zhou S., Dodia C., Feinstein S.I., Huang S., Speicher D., Fisher A.B. Increased phospholipase A2 activity with phosphorylation of peroxiredoxin 6 requires a conformational change in the protein. Biochemistry. 2012;51(27):5521–5530. doi: 10.1021/bi300380h. 22663767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shasby D.M., Fox R.B., Harada R.N., Repine J.E. Reduction of the edema of acute hyperoxic lung injury by granulocyte depletion. Journal of Applied Physiology: Respiratory, Environmental and Exercise Physiology. 1982;52:1237–1244. doi: 10.1152/jappl.1982.52.5.1237. [DOI] [PubMed] [Google Scholar]
- 37.Shepherd V.L. The role of the respiratory burst of phagocytes in host defense. Seminars in Respiratory Infections. 1986;1(2):99–106. 2825313 [PubMed] [Google Scholar]
- 38.Tinits P. Oxygen therapy and oxygen toxicity. Annals of Emergency Medicine. 1983;12(5):321–328. doi: 10.1016/s0196-0644(83)80520-4. 6414343 [DOI] [PubMed] [Google Scholar]
- 39.Tyurina Y.Y., Tyurin V.A., Kaynar A.M., Kapralova V.I., Wasserloos K., Li J., Mosher M., Wright L., Wipf P., Watkins S., Pitt B.R., Kagan V.E. Oxidative lipidomics of hyperoxic acute lung injury: mass spectrometric characterization of cardiolipin and phosphatidylserine peroxidation. The American Journal of Physiology – Lung Cellular and Molecular Physiology. 2010;299(1):L73–L85. doi: 10.1152/ajplung.00035.2010. 20418384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Usatyuk P.V., Singleton P.A., Pendyala S., Kalari S.K., He D., Gorshkova I.A., Camp S.M., Moitra J., Dudek S.M., Garcia J.G., Natarajan V. Novel role for non-muscle myosin light chain kinase (MLCK) in hyperoxia-induced recruitment of cytoskeletal proteins, NADPH oxidase activation, and reactive oxygen species generation in lung endothelium. Journal of Biological Chemistry. 2012;287(12):9360–9375. doi: 10.1074/jbc.M111.294546. 22219181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang W., Suzuki Y., Tanigaki T., Rank D.R., Raffin T.A. Effect of the NADPH oxidase inhibitor apocynin on septic lung injury in guinea pigs. American Journal of Respiratory and Critical Care Medicine. 1994;150(5 1):1449–1452. doi: 10.1164/ajrccm.150.5.7952574. 7952574 [DOI] [PubMed] [Google Scholar]
- 42.Wang X., Phelan S.A., Forsman-Semb K., Taylor E.F., Petros C., Brown A., Lerner C.P., Paigen B. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. Journal of Biological Chemistry. 2003;278(27):25179–25190. doi: 10.1074/jbc.M302706200. 12732627 [DOI] [PubMed] [Google Scholar]
- 43.Wang Y., Feinstein S.I., Fisher A.B. Peroxiredoxin 6 as an antioxidant enzyme: protection of lung alveolar epithelial type II cells from H2O2-induced oxidative stress. Journal of Cellular Biochemistry. 2008;104(4):1274–1285. doi: 10.1002/jcb.21703. 18260127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y., Feinstein S.I., Manevich Y., Ho Y.S., Fisher A.B. Lung injury and mortality with hyperoxia are increased in peroxiredoxin 6 gene-targeted mice. Free Radical Biology and Medicine. 2004;37(11):1736–1743. doi: 10.1016/j.freeradbiomed.2004.09.006. 15528033 [DOI] [PubMed] [Google Scholar]
- 45.Wang Y., Feinstein S.I., Manevich Y., Ho Y.S., Fisher A.B. Peroxiredoxin 6 gene-targeted mice show increased lung injury with paraquat-induced oxidative stress. Antioxidants and Redox Signaling. 2006;8(1–2):229–237. doi: 10.1089/ars.2006.8.229. 16487056 [DOI] [PubMed] [Google Scholar]
- 46.Wang Y., Manevich Y., Feinstein S.I., Fisher A.B. Adenovirus-mediated transfer of the 1-cys peroxiredoxin gene to mouse lung protects against hyperoxic injury. The American Journal of Physiology – Lung Cellular and Molecular Physiology. 2004;286(6):L1188–L1193. doi: 10.1152/ajplung.00288.2003. 15136296 [DOI] [PubMed] [Google Scholar]
- 47.Wegner C.D., Wolyniec W.W., LaPlante A.M., Marschman K., Lubbe K., Haynes N., Rothlein R., Letts L.G. Intercellular adhesion molecule-1 contributes to pulmonary oxygen toxicity in mice: role of leukocytes revised. Lung. 1992;170(5):267–279. doi: 10.1007/BF00566679. 1355574 [DOI] [PubMed] [Google Scholar]
- 48.Welty S.E., Rivera J.L., Elliston J.F., Smith C.V., Zeb T., Ballantyne C.M., Montgomery C.A., Hansen T.N. Increases in lung tissue expression of intercellular adhesion molecule-1 are associated with hyperoxic lung injury and inflammation in mice. American Journal of Respiratory Cell and Molecular Biology. 1993;9(4):393–400. doi: 10.1165/ajrcmb/9.4.393. 8104435 [DOI] [PubMed] [Google Scholar]
- 49.Zhang X., Shan P., Jiang G., Cohn L., Lee P.J. Toll-like receptor 4 deficiency causes pulmonary emphysema. Journal of Clinical Investigation. 2006;116(11):3050–3059. doi: 10.1172/JCI28139. 17053835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zimmerman B.J., Granger D.N. Mechanisms of reperfusion injury. The American Journal of the Medical Sciences. 1994;307(4):284–292. doi: 10.1097/00000441-199404000-00009. 8160724 [DOI] [PubMed] [Google Scholar]