

Abstract
Astrocytes tile the entire CNS, but their functions within neural circuits in health and disease remain incompletely understood. We used genetically encoded Ca2+ and glutamate indicators to explore the rules for astrocyte engagement in the corticostriatal circuit of adult wild-type (WT) and Huntington's disease (HD) model mice at ages not accompanied by overt astrogliosis (at approximately postnatal days 70–80). WT striatal astrocytes displayed extensive spontaneous Ca2+ signals, but did not respond to cortical stimulation, implying that astrocytes were largely disengaged from cortical input in healthy tissue. In contrast, in HD model mice, spontaneous Ca2+ signals were significantly reduced in frequency, duration, and amplitude, but astrocytes responded robustly to cortical stimulation with evoked Ca2+ signals. These action-potential-dependent astrocyte Ca2+ signals were mediated by neuronal glutamate release during cortical stimulation, accompanied by prolonged extracellular glutamate levels near astrocytes and tightly gated by Glt1 glutamate transporters. Moreover, dysfunctional Ca2+ and glutamate signaling that was observed in HD model mice was largely, but not completely, rescued by astrocyte specific restoration of Kir4.1, emphasizing the important contributions of K+ homeostatic mechanisms that are known to be reduced in HD model mice. Overall, our data show that astrocyte engagement in the corticostriatal circuit is markedly altered in HD. Such prodromal astrocyte dysfunctions may represent novel therapeutic targets in HD and other brain disorders.
SIGNIFICANCE STATEMENT We report how early-onset astrocyte dysfunction without detectable astrogliosis drives disease-related processes in a mouse model of Huntington's disease (HD). The cellular mechanisms involve astrocyte homeostasis and signaling mediated by Kir4.1, Glt1, and Ca2+. The data show that the rules for astrocyte engagement in a neuronal circuit are fundamentally altered in a brain disease caused by a known molecular defect and that fixing early homeostasis dysfunction remedies additional cellular deficits. Overall, our data suggest that key aspects of altered striatal function associated with HD may be triggered, at least in part, by dysfunctional astrocytes, thereby providing details of an emerging striatal microcircuit mechanism in HD. Such prodromal changes in astrocytes may represent novel therapeutic targets.
Keywords: astrocyte, calcium, GCaMP, Huntington's disease
Introduction
Astrocytes tile the entire brain, where they serve trophic, active, and homeostatic roles (Barres, 2008; Khakh and Sofroniew, 2015). In addition, evidence indicates that astrocytes contribute to neurological and psychiatric disorders (Maragakis and Rothstein, 2006; Barres, 2008; McGann et al., 2012). However, key questions concerning astrocyte engagement in neural circuits, such as whether active or homeostatic roles dominate and if astrogliosis or astrocyte dysfunctions are relevant in the context of brain disorders, remain unresolved.
Huntington's disease (HD) is characterized by motor, cognitive, and psychiatric disturbances associated with neuronal dysfunction and atrophy of the striatum and other brain areas (Ghosh and Tabrizi, 2015). HD is caused by an expanded polyglutamine repeat localized to the N-terminal region of the huntingtin protein (HTT) that causes intracellular accumulation and aggregation of mutant huntingtin (mHTT) (Mangiarini et al., 1996). The molecular, cellular, and circuit mechanisms that produce disease phenotypes remain incompletely understood (Waldvogel et al., 2015), although important progress has been made by focusing on mHTT expression within neurons of the corticostriatal pathway (Plotkin and Surmeier, 2015). In addition, several recent studies suggest that astrocytes are also involved in HD (Shin et al., 2005; Bradford et al., 2009; Faideau et al., 2010), which supports data showing that brains from HD patients and mouse models display accumulation of mHTT in striatal astrocytes (Shin et al., 2005; Faideau et al., 2010; Tong et al., 2014). However, much remains unknown about how astrocytes contribute to HD pathogenesis.
Recently, Kir4.1 potassium ion channel expression was shown to be decreased in astrocytes that express mHTT at early stages in transgenic R6/2 and knock-in Q175 mouse models of HD (Tong et al., 2014). Kir4.1 channels are astrocyte enriched and involved in K+ homeostasis (Kofuji and Newman, 2004; Sibille et al., 2015), along with other molecules (Larsen et al., 2014). Alterations in HD model mice occurred at stages with little or no evidence of accompanying astrogliosis (Mangiarini et al., 1996; Tong et al., 2014; Ben Haim et al., 2015), which is similar to observations on the lack of astrogliosis during early stages of the human disease (Faideau et al., 2010). Also consistent with the human disease, astrogliosis increased dramatically at late stages of pathology in R6/2 and Q175 mice at time points associated with overt neurodegeneration and striatal tissue loss (Mangiarini et al., 1996; Faideau et al., 2010; Tong et al., 2014). It is well established that HD patients display prodromal symptoms reflecting altered neurophysiology before marked striatal tissue atrophy (Tabrizi et al., 2009). From this perspective, the discovery of early astrocyte Kir4.1 deficits in HD mice at ages not accompanied by astrogliosis hints at causative astrocyte dysfunctions without attendant astrogliosis.
In the present study, we sought to explore astrocyte signaling in the corticostriatal circuit in wild-type (WT) and HD model mice. We used genetically encoded indicators of Ca2+ and glutamate to explore the dynamics of astrocyte spontaneous signaling, as well as astrocyte signaling during cortical input. Our goal was to evaluate systematically the rules, conditions, and constraints for astrocyte engagement in the corticostriatal circuit in healthy mice and in a mouse model of HD.
Materials and Methods
Molecular biology and adeno-associated virus generation.
Protocols used were described previously (Shigetomi et al., 2013). Briefly, to generate an adeno-associated virus (AAV2/5) capable of expressing GCaMP3, iGluSnFR, tdTomato, or Kir4.1-GFP (herein called Kir4.1) in astrocytes, we modified plasmid “pZac2.1final” (Penn Vector Core). We removed the CMV promoter flanked by BglII and HindIII sites and replaced it with the minimal (∼700 bp) GfaABC1D astrocyte-specific promoter, which was amplified by PCR from Addgene plasmid 19974. We then cloned GCaMP3 into this modified pZac2.1 vector between EcoRI and XbaI sites to generate plasmids that we called pZac2.1 gfaABC1D GCaMP3. pZac2.1 gfaABC1D iGluSnFR and tdTomato were made identically. The fully sequenced “pZac2.1” plasmids were sent to the Penn Vector Core, which used them to generate AAV 2/5 for each construct at a concentration of ∼2 × 1013 genome copies/ml (gc/ml). Our virus constructs have been deposited at Addgene in the Khakh lab repository, and the AAVs are available from the UPenn Vector Core Catalog.
Mouse models.
R6/2 and noncarrier control WT mice were obtained from Jackson Laboratories (strain B6CBA-Tg(HDexon1)62Gpb/1J; stock #006494). Whenever R6/2 mice were used, the control was always the noncarrier WT, which we refer to simply as WT in the text and figures. In other cases when R6/2 mice were not used (e.g., much of the initial validation work), the WT mice were C57BL/6N (Taconic) and these are referred to in the text as C57BL/6N. These were generated from an in-house breeding colony. R6/2 mice were genotyped by PCR and CAG repeat length determined by Laragen. The CAG repeat length of mutant offspring was between 118 and 140. Ip3r2 knock-out mice were obtained from Dr. Ju Chen at University of California–San Diego and maintained as a heterozygous line (Srinivasan et al., 2015). Homozygotes and WT littermates were used for experiments when they reached the age of postnatal day 56 (P56) to P80 (Srinivasan et al., 2015). Aldh1l1-eGFP mice were from an in-house colony and have been characterized previously (Cahoy et al., 2008). Breeders were obtained from the Mutant Mouse Resource and Regional Centers [MMRC strain name STOCK Tg(Aldh1l1-EGFP)OFC789Gsat/Mmucd].
Surgery and in vivo microinjections of AAV 2/5.
Protocols used were described previously (Shigetomi et al., 2013; Haustein et al., 2014; Jiang et al., 2014), with minor modifications (Jiang et al., 2014). Male and female P49–P56 C57BL/6N, R6/2, or noncarrier WT mice were used in all experiments, in accordance with institutional guidelines. All surgical procedures were conducted under general anesthesia using continuous isoflurane (induction at 5%, maintenance at 1–2.5% v/v). After induction of anesthesia, the mice were fitted into a stereotaxic frame with their heads secured by blunt ear bars and their noses placed into an anesthesia and ventilation system (David Kopf Instruments). Mice were administered 0.05 ml of buprenorphine (Buprenex, 0.1 mg/ml) subcutaneously before surgery. The surgical incision site was then cleaned three times with 10% povidone iodine and 70% ethanol. Skin incisions were made, followed by craniotomies 2–3 mm in diameter above the left parietal cortex using a small steel burr (Fine Science Tools) powered by a high-speed drill (K.1070; Foredom). Saline (0.9%) was applied onto the skull to reduce heating caused by drilling. Unilateral viral injections were performed by using a stereotaxic apparatus (David Kopf Instruments) to guide the placement of bevelled glass pipettes (1B100–4; World Precision Instruments) into the left striatum (coordinates from the bregma were as follows: anterior–posterior +0.8 mm, medial–lateral +2 mm, and dorsal–ventral −2.4 mm from the pial surface). Either 2 μl of AAV2/5 gfaABC1D iGluSnFR (4.2 × 1012 gc/ml) or 1.5 μl of AAV2/5 gfaABC1D GCaMP3 (1.5 × 1013 gc/ml) was injected by using a syringe pump at ∼200 nl/min (Pump11 PicoPlus Elite; Harvard Apparatus). Glass pipettes were left in place for at least 10 min. Surgical wounds were closed with single external 6–0 nylon sutures. After surgery, animals were allowed to recover overnight in cages placed partially on a low-voltage heating pad. Buprenorphine was administered 2 times/d for up to 2 d after surgery. In addition, trimethoprim/sulfamethoxazole (40 and 200 mg, respectively per 500 ml of water) was dispensed in the drinking water for 1 week. Mice were killed 14–20 d after surgery for imaging.
Preparation of brain slices for imaging and electrophysiology.
Striatal slices were prepared from ∼P70 WT and R6/2 mice. Briefly, animals were deeply anesthetized and decapitated. The brains were placed in ice-cold modified artificial CSF (aCSF) containing the following (in mm): 194 sucrose, 30 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 d-glucose and cut into 300-μm-thick coronal or parasagittal slices containing the striatum and cortex. Brain slices were allowed to equilibrate for at least 30 min at 32–34°C in normal aCSF containing (in mm); 124 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 d-glucose continuously bubbled with a mixture of 95% O2/5% CO2, stored at room temperature in the same buffer, and used for experiments within about 6 h of slicing.
Electrophysiological recording from medium spiny neurons in brain slices.
Methods were as described previously (Adermark and Lovinger, 2008; Tong et al., 2014). Cells were visualized with infrared optics on an upright microscope (BX51WI, Olympus). pCLAMP10 software and a MultiClamp 700B amplifier was used for electrophysiology (Molecular Devices). For striatal medium spiny neuron (MSN) recordings, the aCSF contained the following (in mm): 124 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 d-glucose continuously bubbled with a mixture of 95% O2/5% CO2. For EPSC recordings, the intracellular solution in the patch pipette contained the following (in mm): 135 potassium gluconate, 3 KCl, 0.1 CaCl2, 10 HEPES, 1 EGTA, 8 Na2-phosphocreatine, 4 Mg-ATP, 0.3 Na2-GTP, and 1 QX314 bromide, pH 7.3 adjusted with KOH. For cortical axonal stimulation, electrical field stimulation (EFS) were achieved using a bipolar matrix electrode (FHC) that was placed on the dorsolateral corpus callosum to evoke glutamate release (Vergara et al., 2003). The astrocytes or neurons to be assessed were typically located ∼250–300 μm away from the stimulation site. For GCaMP3 or iGluSnFR imaging of Ca2+ and glutamate, individual pulses were 0.2 ms in duration and stimuli were delivered at 5 mA.
Tissue dissociation, astrocyte sorting and qPCR.
Mice expressing eGFP under the astrocyte specific 10-formyltetrahydrofolate dehydrogenase (Aldh1l1) promoter (Cahoy et al., 2008) were used to purify astrocytes by fluorescence-activated cell sorting (FACS). The striata from heterozygous 30-d-old mice were dissociated following published guidelines (Foo, 2013) with slight modifications. Briefly, the striata from 4 mice were dissected and digested together for 90 min at 36°C in a 35 mm Petri dish with 2.5 ml of papain solution (1 × EBSS, 0.46% d-glucose, 26 mm NaHCO3, 50 mm EDTA, 75 U/ml DNase 1, 300 units of papain, and 2 mm l-cysteine) while bubbling with 5% CO2 and 95% O2. After digestion, the tissue was washed 4 times with ovomucoid solution (1× EBSS, 0.46% d-glucose, 26 mm NaHCO3, 1 mg/ml ovomucoid, 1 mg/ml BSA, and 60 U/ml DNase 1) and mechanically dissociated with 2 fire-polished borosilicate glass pipettes with different bore sizes. A bottom layer of concentrated ovomucoid solution (1× EBSS, 0.46% d-glucose, 26 mm NaHCO3, 5.5 mg/ml ovomucoid, 5.5 mg/ml BSA, and 25 U/ml DNase 1) was added to the cell suspension. The tubes were centrifuged at room temperature at 300 × g for 10 min and the resultant pellet was resuspended in d-PBS with 0.02% BSA and 13 U/ml of DNase 1 and filtered with a 20 μm mesh. FACS was performed in a FACSAria II (BD Bioscience) with a 70 μm nozzle using standard methods at the University of California–Los Angeles (UCLA) Cell Sorting Core. Sorted cells, in d-PBS with 0.1% BSA, were centrifuged for 10 min at 4°C and 2000 × g. The RNA was extracted from the pelleted cells using RNeasy Plus Micro Kit (QIAGEN). Amplified cDNA from the RNA samples was generated using Ovation PicoSL WTA System V2 (Nugen). The cDNA was then purified with a QIAquick PCR Purification Kit (QIAGEN) and quantified with a Nanodrop 2000. qPCR was performed in a LightCycler 96 Real-Time PCR System (Roche). Amplified cDNA from both eGFP-negative and eGFP-positive cell populations from three separate sorts was used. One nanogram of each sample was loaded per well and the expression of Grm3, Grm5, and Arbp was analyzed using the primers shown below. To calculate the expression of Grm3 and Grm5, their Ct values were measured relative to Arbp using the following formulas: 2−ΔCt (Grm3-Arbp) and 2−ΔCt (Grm5-Arbp), respectively.
Data analyses.
Slow drifts in astrocyte position (∼2–5 μm) were corrected with the TurboReg Plugin in ImageJ. Astrocyte territory sizes for all experiments were estimated by measuring the area of a region of interest (ROI) that surrounded the largest fluorescence projection profile of bushy astrocytes gathered from a series of confocal images. We performed Ca2+-imaging experiments in a single optical plane. Ca2+ transients were measured by plotting the intensity of ROIs over time after the intensity of a background ROI had been subtracted. ROIs were selected based on the appearance of Ca2+ signals in the time series images, which were processed by the 3d Hybrid Median Filter Plugin in ImageJ. A signal was declared as a Ca2+ transient if it exceeded the baseline by greater than twice the baseline noise (SD). For line scan analysis, ROIs were selected and data extracted via Image2Data software written by Raul Serrano (UCLA) before being analyzed in the same way as frame scan data. GECIquant software was used as described previously (Srinivasan et al., 2015) to measure the areas covered by Ca2+ signals (as in Fig. 1). However, the properties of Ca2+ signals were analyzed with manually selected ROIs as described previously (Shigetomi et al., 2013; Haustein et al., 2014). Spontaneous synaptic currents were analyzed using MiniAnalysis Program version 6.0.7 (Synaptosoft) and evoked EPSCs were analyzed using Clampfit version 10.2 (Molecular Devices).
Figure 1.

Three types of striatal astrocyte Ca2+ signals observed in C57BL/6N mice differ in the areas that they cover. A, Representative images and tracings for the areas of global waves, local waves, and microdomains. B, Average data from six mice for experiments and analyses such as those shown in A. Scatter graph shows average area covered by the three types of Ca2+ signals. Statistical comparisons were made using one-way ANOVA. For the box-and-whisker plots in B, the circle represents the mean, the box the SEM, and the whisker the SD. C, Distinct cumulative probability plots for areas covered by global waves, local waves, and microdomains.
All statistical tests were run in GraphPad InStat 3 or Origin 9. The graphs were created in Origin 9 and assembled in CorelDraw12. Data are presented as mean ± SEM. Note that, in some of the graphs, the SEM bars are smaller than the symbols used to represent the mean. For each set of data to be compared, we determined within GraphPad Instat whether the data were normally distributed. If they were normally distributed, we used parametric tests. If the data were not normally distributed, we used nonparametric tests. For qPCR experiments, we assumed a normal distribution and statistical significance was calculated using a t test with Welch correction. Paired and unpaired Student's two-tailed t tests (as appropriate) and two-tailed Mann–Whitney tests were used for most statistical analyses with significance declared at p < 0.05, but stated in each case with a precise a p-value. When the p-value was <0.00001, it is stated as p < 0.00001 to save space on the figure panels and tables. Herein, the results of statistical tests (p-values and n numbers) are reported in the figure panels. Numbers (n) are defined as the numbers of cells or mice throughout.
Drugs and chemicals.
All chemicals were purchased from Sigma-Aldrich, Tocris Bioscience, or VWR.
Results
GCaMP3 to monitor striatal astrocyte Ca2+ signals in C57BL/6N mice
Because intracellular Ca2+ signaling is a prevalent feature of astrocytes that is proposed to be related to neuronal function (Volterra et al., 2014; Khakh and McCarthy, 2015; Khakh and Sofroniew, 2015), we set out to measure Ca2+ signals within striatal astrocytes as a metric of their functional engagement in the corticostriatal circuit. We expressed GCaMP3 within entire striatal astrocytes using in vivo AAV2/5 microinjections in adult mice. These procedures were reliable; astrocyte specific; did not cause astrogliosis in the striatum (Jiang et al., 2014; Tong et al., 2014), hippocampus (Shigetomi et al., 2013; Haustein et al., 2014), or cortex (Bonder and McCarthy, 2014); and did not alter astrocyte properties (Shigetomi et al., 2013; Haustein et al., 2014). Striatal astrocyte Ca2+ signals have not been described previously in detail. We started by quantifying spontaneous Ca2+ signals within astrocyte somata and branches (Figs. 1, 2).
Figure 2.
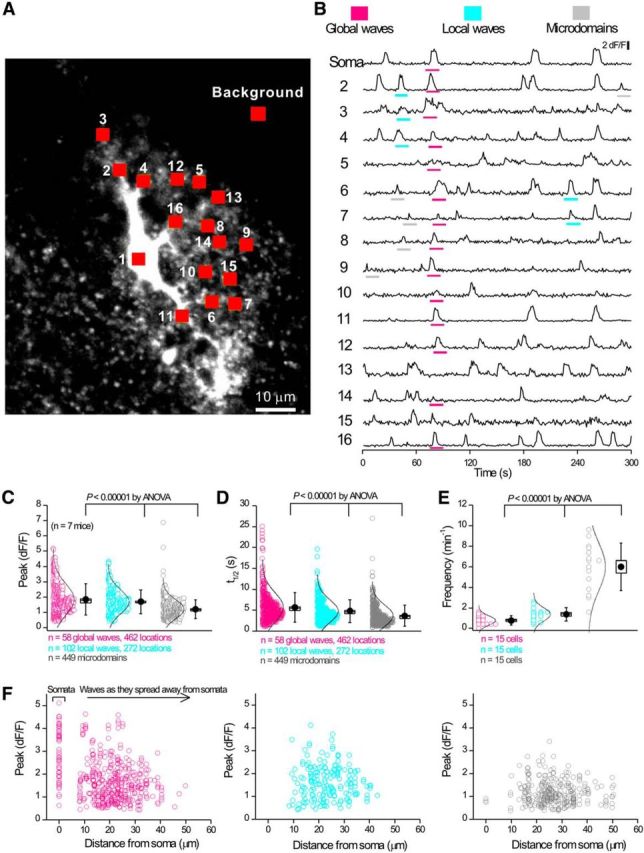
Striatal astrocytes in C57BL/6N mice display three types of intracellular Ca2+ signals. A, Image of a striatal astrocyte expressing GCaMP3 with 16 ROIs indicated. B, Representative traces from the 16 ROIs shown in A. Global waves, local waves, and microdomains are highlighted in separate colors. C–E, Scatter graphs for average data for traces such as those in B show that the three types of Ca2+ signals differ in amplitude, half-width, and frequency. Statistical tests in C–E were done using one-way ANOVA. F, Scatter graph of amplitude against distance (from the soma) for the three types of intracellular Ca2+ signals. Local waves and microdomains largely occurred in branches. Statistical comparisons were made using one-way ANOVA. For the box-and-whisker plots in C–E, the circle represents the mean, the box the SEM, and the whisker the SD.
Using GCaMP3, three types of spontaneous Ca2+ signals were readily observed and could be designated descriptively as global waves, local waves, and microdomains. Global waves were spontaneously occurring and encompassed the soma and large parts of astrocyte branches (∼50% of their length). Local waves were spontaneously occurring, but were restricted and covered smaller parts of branches. Microdomains occurred spontaneously, were frequent, and covered only small, micrometer-scale areas of branches and somata (n = 15 cells, n = 7 mice). Representative data are shown in Figure 1, A–C. Figure 1A shows image frames that display global waves, local waves, and microdomains and the tracing of their areas using GECIquant software (Srinivasan et al., 2015). The astrocyte territory area could not be traced with GECIquant and was drawn by hand, as described previously (Srinivasan et al., 2015). The tracings in Figure 1A clearly show that global waves cover the largest areas. Local waves represent the next largest areas and microdomains cover the smallest areas. This trend was supported by analyses across multiple cells and mice (n = 6 cells, n = 6 mice) and the differences were statistically significant (Fig. 1B). Therefore, analyses of astrocyte Ca2+ signal areas (Fig. 1B) showed that global waves covered ∼881 ± 93 μm2, local waves covered 183 ± 14 μm2, and microdomains covered 14 ± 1 μm2 (Fig. 1B; n = 6 cells, n = 6 mice). In comparison, the maximum projected approximate territory area for these six astrocytes was 1847 ± 113 μm2. Moreover, the distributions representing areas for global waves, local waves, and microdomains were clearly distinct (Fig. 1C). Together, these data provide compelling evidence for the existence of three types of discernible Ca2+ signals in striatal astrocytes, as we have shown previously in hippocampus and cortex (Haustein et al., 2014; Srinivasan et al., 2015).
Having identified global waves, local waves, and microdomains based on their areas, we next determined whether the cognate Ca2+ signals differed significantly in their peak amplitude (Fig. 2A–C), duration (Fig. 2A,B,D), and frequency (Fig. 2A,B,E). We found that the global waves, local waves, and microdomains differed significantly in these parameters; the graphs in Figure 2, C and D, show the distributions of the raw data, as well as box-and-whisker plots of mean, SE, and SD. Note, however, that the designations into global waves, local waves, and microdomains were first made based on the areas of the signals (Fig. 1). Nonetheless, three separable peaks were clearly observed for frequency (Fig. 2E). Although the amplitude and duration were significantly different for global waves, local waves, and microdomains, the underlying distributions did overlap (Fig. 2C,D). Moreover, whereas local waves and microdomains were restricted to the branches of astrocytes, the global waves encompassed the somata and branches, frequently starting in the somata and spreading out into the branches (Fig. 2F). Global Ca2+ waves spread in either direction to and from somata at a velocity of 14 ± 4 μm/s (n = 11 cells, n = 5 mice) and dissipated with distance.
Astrocyte “fast” Ca2+ signals lasting hundreds of milliseconds have been reported in some fields of the hippocampus (Di Castro et al., 2011; Panatier et al., 2011), but not in others (Haustein et al., 2014). To determine whether fast spontaneous Ca2+ signals existed in striatal astrocytes, we performed a specific set of experiments with 200 Hz line scan imaging along the somata and main branches of striatal astrocytes (Fig. 3A,B; n = 23 cells, n = 4 mice). On average, the signals lasted ∼6–10 s in duration in branches and somata (Fig. 3C,D), which was similar to the frame scan data shown in Figure 2. Therefore, we found no compelling evidence for predominantly fast Ca2+ signals lasting hundreds of milliseconds in striatal astrocytes. This is similar to our past findings with astrocytes from the hippocampal CA3 region and visual cortex (Haustein et al., 2014; Srinivasan et al., 2015).
Figure 3.
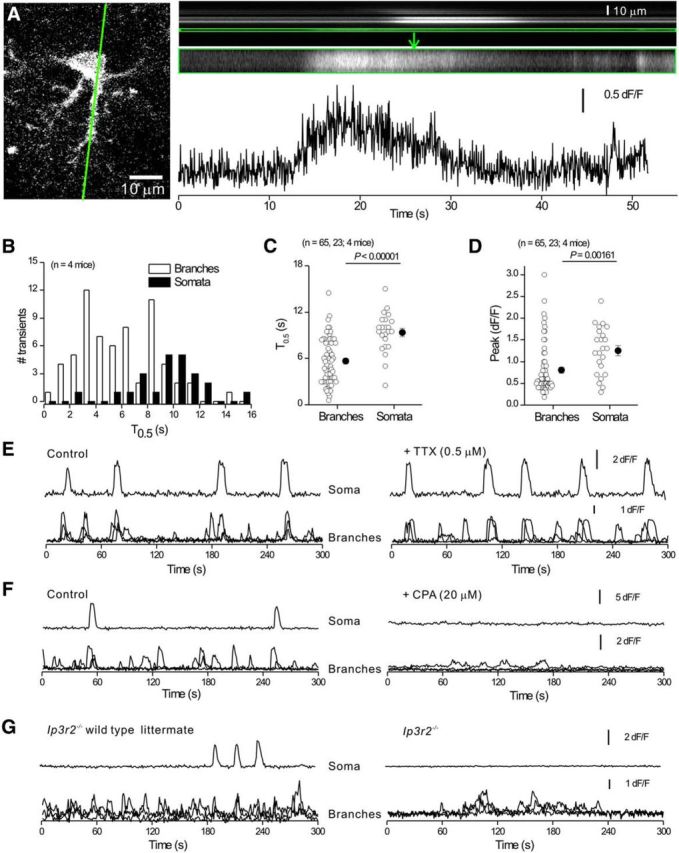
Properties of intracellular Ca2+ signals in striatal astrocytes in C57BL/6N mice. A, Confocal image of a striatal astrocyte with a green line indicating the approximate position of the region chosen for 200 Hz line scan imaging. Top right, Line scan imaging data for the green line, with an expanded region corresponding to a branch shown below. In these images, the x-axis is time and the y-axis is distance along the scanned line. The trace is plotted for the selected region (framed in green) shown in the recorded image above. B, Distributions showing Ca2+ signal half-widths from line scan experiments for somatic and branch regions. C, D, Scatter plots summarizing Ca2+ signal properties such as amplitude and half-width for line scan data. Statistical tests in C and D were using unpaired Student's t test. E, F, Representative single trace for the soma and three superimposed traces for branches under control conditions and then in the presence of TTX (E) or CPA (F). G, Representative single trace for the soma and three superimposed traces for branches for spontaneous Ca2+ signals recorded from Ip3r2−/− mice and their WT littermates. Average data for TTX, CPA, and Ip3r2−/− experiments (and WT controls) are shown in Table 1. In some cases, the error bars signifying SEM are smaller than the symbols used.
We next investigated whether astrocyte spontaneous Ca2+ signals were driven by action potential firing, as is proposed in some brain areas (Khakh and McCarthy, 2015). Application of 0.5 μm tetrodotoxin (TTX) produced no effect on Ca2+ signal dF/F or frequency in somata or branches (Fig. 3E, n = 6 cells, n = 4 mice, Table 1). However, depletion of intracellular Ca2+ stores with cyclopiazonic acid (CPA; 20 μm, n = 6 cells, n = 3 mice) almost completely (>95%) abolished the Ca2+ signals in astrocyte somata, but residual Ca2+ signals persisted in branches (Fig. 3F, Table 1). We also assessed Ca2+ signals in WT and inositol triphosphate type 2 receptor knock-out (Ip3r2−/−) mice (Agulhon et al., 2010) and found few Ca2+ signals in astrocyte somata from Ip3r2−/− mice, but detected significant numbers of Ca2+ signals in astrocyte branches (Fig. 3G, n = 11 cells, n = 4 mice, Table 1). Similar data have been reported for hippocampus and cortex (Haustein et al., 2014; Srinivasan et al., 2015).
Table 1.
Properties of Ca2+ global waves, local waves, and microdomains measured in striatal astrocytes in situ with GCaMP3
| Peak δF/F | T0.5 (s) | Frequency (min−1) | No. of mice | |
|---|---|---|---|---|
| Basic properties | ||||
| Global waves | ||||
| Control | 1.81 ± 0.08 (n = 26 waves, 189 locations) | 4.94 ± 0.16 (n = 26 waves, 189 locations) | 0.87 ± 0.22 (n = 6 cells) | 4 |
| +TTX | 1.66 ± 0.07 (n = 34 waves, 244 locations) | 4.47 ± 0.13 (n = 34 waves, 244 locations) | 1.13 ± 0.31 (n = 6 cells) | 4 |
| p | 0.07 | 0.02 | 0.136 | |
| Local waves | ||||
| Control | 1.72 ± 0.09 (n = 34 waves, 103 locations) | 4.22 ± 0.19 (n = 34 waves, 103 locations) | 1.13 ± 0.30 (n = 6 cells) | 4 |
| +TTX | 1.64 ± 0.08 (n = 39 waves, 106 locations) | 4.14 ± 0.22 (n = 39 waves, 106 locations) | 1.17 ± 0.08 (n = 6 cells) | 4 |
| p | 0.558 | 0.5 | 0.58 | |
| Microdomains | ||||
| Control | 1.29 ± 0.07 (n = 145 events) | 3.01 ± 0.16 (n = 145 events) | 4.83 ± 0.99 (n = 6 cells) | 4 |
| +TTX | 1.20 ± 0.05 (n = 151 events) | 2.98 ± 0.11 (n = 151 events) | 5.03 ± 1.04 (n = 6 cells) | 4 |
| p | 0.40 | 0.16 | 0.713 | |
| Store depletion | ||||
| Global waves | ||||
| Control | 1.77 ± 0.07 (n = 23 waves, 161 locations) | 5.77 ± 0.37 (n = 23 waves, 161 locations) | 0.77 ± 0.18 (n = 6 cells) | 3 |
| +CPA | – (n = 0 waves) | – (n = 0 waves) | 0 ± 0 | 3 |
| p | ND | ND | 0.0084 | |
| Local waves | ||||
| Control | 1.60 ± 0.06 (n = 50 waves, 103 locations) | 5.15 ± 0.34 (n = 50 waves, 103 locations) | 1.67 ± 0.22 (n = 6 cells) | 3 |
| +CPA | 1.60 ± 0.09 (n = 3 waves, 4 locations) | 6.78 ± 1.14 (n = 3 waves, 4 locations) | 0.1 ± 0.07 (n = 6 cells) | 3 |
| p | 0.98 | 0.10 | 0.0011 | |
| Microdomains | ||||
| Control | 1.14 ± 0.03 (n = 189 events) | 4.0 ± 0.21 (n = 189 events) | 6.3 ± 0.83 (n = 6 cells) | 3 |
| +CPA | 1.19 ± 0.09 (n = 55 events) | 4.0 ± 0.28 (n = 55 events) | 1.83 ± 0.22 (n = 6 cells) | 3 |
| p | 0.55 | 0.58 | 0.0033 | |
| Ip3r2 involvement | ||||
| Global waves | ||||
| WT littermate | 2.84 ± 0.13 (n = 25 waves, 273 locations) | 6.15 ± 0.2 (n = 25 waves, 273 locations) | 0.46 ± 0.15 (n = 11 cells) | 4 |
| Ip3r2−/− | – (n = 0 waves) | – (n = 0 waves) | 0 ± 0 (n = 15 cells) | 4 |
| p | ND | ND | 0.00016 | |
| Local waves | ||||
| WT littermate | 2.10 ± 0.08 (n = 122 waves, 241 locations) | 4.11 ± 0.13 (n = 122 waves, 241 locations) | 2.27 ± 0.34 (n = 11 cells) | 4 |
| Ip3r2−/− | 1.35 ± 0.21 (n = 8 waves, 20 locations) | 5.53 ± 0.99 (n = 8 waves, 20 locations) | 0.02 ± 0.02 (n = 15 cells) | 4 |
| p | 0.003 | 0.19 | 0.00001 | |
| Microdomains | ||||
| WT littermate | 1.36 ± 0.04 (n = 394 events) | 3.21 ± 0.09 (n = 394 events) | 7.20 ± 1.0 (n = 11 cells) | 4 |
| Ip3r2−/− | 1.07 ± 0.04 (n = 310 events) | 3.63 ± 0.15 (n = 310 events) | 4.0 ± 0.6 (n = 15 cells) | 4 |
| p | <0.00001 | 0.25 | 0.0003 |
CPA was used at 20 μm. TTX was used at 0.5 μm. Statistical tests were paired Student's t test for the CPA and TTX experiments and unpaired Student's t test or nonparametric Mann–Whitney tests as appropriate for the experiments with Ip3r2−/− mice. ND means not determined because there was not enough data to permit the comparison. Precise p-values are indicated. Significance was declared at p < 0.05. The experiments are described in the text.
Overall, the data reported in Figures 1, 2, and 3 provide validation of GCaMP3 to explore astrocyte Ca2+ signaling in HD mice (reported in later sections) and also provide important details of basic striatal astrocyte Ca2+ signal properties.
Spontaneous Ca2+ signals are reduced in striatal astrocytes from HD model mice
In the present study, we explored astrocytes in the widely used early onset exon 1 human mHTT transgenic R6/2 model (Mangiarini et al., 1996) because of past findings (Tong et al., 2014), where we observed similar astrocyte deficits in R6/2 and the more slowly developing knock-in Q175 model (Heikkinen et al., 2012; Menalled et al., 2012). The R6/2 mouse model was attractive for the current study, because symptom onset occurs before detectable astrogliosis (Mangiarini et al., 1996; Tong et al., 2014). In addition, the use of P50–P70 mice was necessary for using AAV microinjections to express GCaMP3, Kir4.1, tdTomato, and iGluSnFR (as reported in later sections). These approaches have only been tested and deployed for astrocytes from mice at these ages (Shigetomi et al., 2013; Haustein et al., 2014; Jiang et al., 2014; Tong et al., 2014; Srinivasan et al., 2015). In contrast, Q175 mice display HD phenotypes at 8–12 months of age (Tong et al., 2014); that is, at time points when the genetic tools to explore astrocytes have not been validated to be specific or innocuous. For a combination of these reasons, we studied R6/2 mice at ∼P70.
Using GCaMP3, we monitored spontaneous Ca2+ signals in striatal astrocytes from R6/2 and noncarrier WT mice (Fig. 4A–C). In WT mice, we observed global waves, local waves, and microdomains (Fig. 4A, n = 20–29 cells, n = 7 mice each) that were indiscernible from those measured in C57BL/6N mice (Fig. 2). The same three types of Ca2+ signals were also observed in R6/2 mice (Fig. 4B) but, relative to WT, they were significantly reduced in amplitude, duration, and frequency across the board for global waves, local waves, and microdomains (Fig. 4C; n = 20–29 cells, n = 7 mice each). The most dramatic effects were reductions in the frequency of local waves and microdomains within astrocyte branches (Fig. 4C). The results were highly significant and readily visible from the raw traces (Fig. 4A,B). Therefore, in contrast to past work with Alzheimer's disease, cerebral ischemia, and status epilepticus, which are accompanied by elevated astrocyte Ca2+ signals (Ding et al., 2007; Takano et al., 2007; Ding et al., 2009; Kuchibhotla et al., 2009; Delekate et al., 2014), we found that astrocytes from R6/2 mice display significantly reduced spontaneous Ca2+ signals, which reflects the specific nature of the molecular defect associated with HD.
Figure 4.

Striatal astrocytes in R6/2 mice displayed reduced spontaneous intracellular Ca2+ signals. A, B, Representative traces from eight selected ROIs for spontaneous Ca2+ signals in astrocytes from a WT (A) or from a R6/2 mouse (B). C, Quantification of peak amplitude, duration, and frequency for three types of Ca2+ signals in striatal astrocytes in WT and R6/2 mice (global waves, local waves, and microdomains; see text for further details). D, Schematic showing that CPA blocks SERCA and leads to depletion of intracellular Ca2+ stores due to Ca2+ release from the endoplasmic reticulum (ER). E, Individual traces (gray) and averages (black or red) showing the effect of CPA on astrocyte Ca2+ in WT and R6/2 mice. F, Average data for CPA-evoked Ca2+ increases such as those shown in E. The CPA-mobilized Ca2+ store was smaller in R6/2 mice. In some cases, the error bars signifying SEM are smaller than the symbols used.
A substantial proportion of spontaneous Ca2+ signals in striatal astrocytes are due to store-mediated Ca2+ release via Ip3r2 (Fig. 3, Table 1), prompting us to evaluate whether intracellular Ca2+ store capacity was lower in R6/2 mice relative to WT. To explore this possibility, we applied CPA (20 μm), a blocker of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), and measured the change in fluorescence of GCaMP3 expressing astrocytes from WT and R6/2 mice (Fig. 4D–F). We detected significantly smaller CPA-evoked changes in fluorescence from R6/2 mice compared with WT (Fig. 4E,F; n = 29–37 cells, n = 4 mice each). These data imply that CPA-mobilized Ca2+, which includes both intracellular Ca2+ store capacity and Ca2+ entry due to store depletion, was lower in R6/2 mice than WT. In the most straightforward interpretation, the loss of spontaneous Ca2+ signals observed in astrocytes from R6/2 mice (Fig. 4A–C) may reflect lower Ca2+ store capacity.
Evoked Ca2+ signals are increased in striatal astrocytes from HD model mice
The striatum receives strong glutamatergic input from the cortex that can be targeted for selective stimulation in parasagittal neostriatal brain slices (Vergara et al., 2003). We used this approach (Fig. 5A) to stimulate cortical inputs and began by recording from MSNs. Based on past work (Haustein et al., 2014), we applied brief trains of 4 EFS in 1 s and recorded large-amplitude, fast, evoked EPSCs onto MSNs that were completely blocked by 0.5 μm TTX (Fig. 5B; n = 7 cells, n = 4 mice). Having established a reliable protocol to stimulate cortical inputs, we next imaged glutamate signals using iGluSnFR (Marvin et al., 2013), which was expressed on the surface of striatal astrocytes (Haustein et al., 2014) in C57BL/6N mice (Fig. 5C,D). Each round of EFS evoked robust and short-lived iGluSnFR signals (Fig. 5C,D, n = 12 cells, n = 4 mice) that were completely blocked by TTX (Fig. 5E, n = 15 cells, n = 4 mice). Therefore, striatal astrocytes “see” glutamate released from cortical axons. Using this protocol (Fig. 5A–E), we assessed EFS-evoked iGluSnFR and Ca2+ signals onto striatal astrocytes in WT and R6/2 mice.
Figure 5.

Striatal astrocytes in R6/2 mice displayed robust Ca2+ signals in response to cortical axon EFS, but those in WT mice did not. A, Photomicrograph of a parasagittal slice of mouse brain showing the position of the stimulating electrode and the imaging site. B, Confocal image (left) and diagram (middle) showing GCaMP3-expressing astrocytes (green) and a MSN filled through the patch-pipette with Alexa Fluor 546. To the right are evoked EPSCs recorded from a MSN in response to 3 EFS; these were abolished in the presence of TTX (0.5 μm). C, Representative traces for a 2-pulse protocol in which 4 EFS were applied twice (first and second stimulation) to cortical axons 10 min apart. Individual traces (gray) and averages (black) for iGluSnFR imaging are shown in response to 4 EFS (arrows). The lines on top of the traces illustrate the time windows in which the peak of the three phases including the baseline, the peak, and later epochs were measured. D, Average data for peak amplitude measured at the three epochs shown in C. E, As in D, with two pulses of 4 EFS at an interval of 10 min, but with the second pulse in the presence of TTX. Note that 2 rounds of 4 EFS produced similar peak responses, whereas interpulse application of TTX abolished the second response. F, Individual traces (gray) and averages (black) for iGluSnFR imaging in response to EFS (arrows) for astrocytes from WT and R6/2 mice. G–I, Average data for peak amplitude, area, and kinetics for traces such as those shown in F. J, K, Top left, Individual traces (gray) and averages (black) for GCaMP3 imaging within somata (J) or branches (K) of astrocytes in WT and R6/2 mice, with arrows indicating the time points for EFS and peak response. Note that a delayed slow response was present in both WT and R6/2, but this slow response was not abolished by TTX (see main text). The graphs next to the traces show the percentage of cells responding to EFS. The bar graphs below the traces show average data for peak responses in response to EFS under various conditions for somata and branches. In some cases, the error bars signifying SEM are smaller than the symbols used.
We recorded short latency (<2 s), robust, and equally sized EFS-evoked iGluSnFR signals onto astrocytes in WT and R6/2 mice (Fig. 5F,G; n = 21, n = 25 cells, n = 4 mice). However, the decay times of the iGluSnFR signals were significantly longer in R6/2 compared with WT mice (Fig. 5H). Therefore, EFS-evoked release of glutamate persisted in the extracellular space near astrocytes for longer in R6/2 mice than in the WT, which was reflected in the area of the iGluSnFR signals (Fig. 5I). In the simplest interpretation, these data imply that astrocytes from WT and R6/2 mice received approximately equivalent glutamate from cortical afferents, but that the glutamate was less efficiently cleared in R6/2 mice.
We next used EFS to determine whether evoked astrocyte Ca2+ signaling differed between R6/2 mice and WT (Fig. 5J,K). We measured these evoked signals within ∼10 s of EFS (arrowheads in Fig. 5J,K) because these time points correspond to when glutamate was detected on astrocytes (Fig. 5C,F). For both astrocyte somata and branches, we found that significantly greater proportions of astrocytes responded to EFS in R6/2 mice compared with their WT controls (Fig. 5J,K). This elevated evoked astrocyte Ca2+ signaling was apparent when we measured the peak dF/F across all cells for astrocyte somata and branches (bar graphs in Fig. 5J,K). Evoked Ca2+ signals from astrocyte somata and branches in R6/2 mice were blocked by TTX (0.5 μm) and by the mGluR2/3 antagonist LY341495 (10 μm; Fig. 5J,K), which is known to mediate evoked astrocyte Ca2+ responses in the hippocampus (Haustein et al., 2014). Therefore, in R6/2 mice, astrocytes display action potential-dependent corticostriatal glutamate (Fig. 5F–I) and Ca2+ signaling (Fig. 5J,K), whereas action-potential-independent spontaneous Ca2+ signals were reduced in R6/2 mice (Fig. 4). Conversely, WT mice do not display evoked astrocyte Ca2+ signals during cortical stimulation, but do display extensive spontaneous Ca2+ signals.
Very slow astrocyte Ca2+ responses that occurred with a latency of >20 s after EFS and lasted ∼60 s were also observed. These can be seen in the traces shown in Figure 5, J and K, but they were not blocked by TTX (n = 12 cells, n = 4 mice). We interpret this to indicate that they were not the result of neuronal action potential firing and thus these peculiar slow events were not studied further, but may reflect mechanical stimulation at the EFS site and gap junctional intracellular spread.
We further evaluated the functional expression of mGluR3 receptors in striatal astrocytes from adult WT and R6/2 mice by applying the mGluR2/3 agonist LY354740 (3 μm). We measured equivalent LY354740-evoked Ca2+ signals within astrocyte somata and branches from WT and R6/2 mice, providing strong evidence that striatal astrocytes express functional mGluR3 receptors that elevate Ca2+ to similar levels in both genotypes (Fig. 6A,B). To further explore mGluR3 expression, we used FACS to isolate a pure population of striatal astrocytes from adult Aldh1l1-eGFP mice (Cahoy et al., 2008) and performed qPCR for Grm3 and Grm5, the genes that encode mGluR3 and mGluR5, respectively (Sun et al., 2013). These experiments showed significantly higher Grm3 expression in striatal astrocytes compared with Grm5 (Fig. 6C–E), which is consistent with past work on adult cortical astrocytes (Sun et al., 2013) and hippocampal astrocytes (Haustein et al., 2014). Our use of a selective agonist (Fig. 6A,B), antagonist (Fig. 5J,K) and qPCR from striatal astrocytes (Fig. 6C–E) provide compelling evidence for the expression of mGluR3 within the striatum. These observations are consistent with detailed immunohistochemistry reporting mGluR3 protein in striatal glia (Petralia et al., 1996; Tamaru et al., 2001).
Figure 6.

Functional and molecular evidence for mGluR3 expression in striatal astrocytes. A, Individual traces (gray) and averages (black) for LY354740-evoked somatic Ca2+ signals within astrocytes for WT and R6/2 mice. The scatter plot to the right shows the average data. B, As in A, but for measurements in branches. C, Schematic for purification of striatal astrocytes from Aldh1l1-eGFP mice at ∼P30 using FACS (see Materials and Methods). D, GFP-negative population (Neg) was defined by the background emission on the FITC channel of GFP-positive cells, and the GFP-positive population was defined by high eGFP fluorescence not overlapping with the GFP-negative cells. E, Bar graph for qPCR analysis of Grm3 and Grm5 from the two purified fractions. The gene expression levels were normalized to the reference gene Arbp using the following formula: 2−ΔCt (Gene of interest-Arbp). In some cases, the error bars signifying SEM are smaller than the symbols used.
Glutamate uptake gates striatal astrocyte Ca2+ and glutamate signaling
Astrocytes in the striatum express Glt1, the expression of which is reduced in HD model mice (Liévens et al., 2001), leading us to evaluate whether altered astrocyte Ca2+ and glutamate signaling observed in R6/2 mice could be triggered simply as a consequence of the functional loss of Glt1-mediated glutamate uptake. To explore this possibility, we used WT C57BL/6N mice and applied the specific Glt1/GLAST blocker TBOA (1 μm) and found that it significantly increased astrocyte Ca2+ signals (Fig. 7A; n = 7 cells, n = 4 mice), increased basal iGluSnFR signals (Fig. 7B; n = 28 cells, n = 4 mice), and increased the peak, decay time, and integrated area of EFS-evoked astrocyte iGluSnFR signals (Fig. 7C–E; 21–25 cells, n = 4 mice). Moreover, blockade of Glt1 resulted in robust EFS-evoked Ca2+ signals in astrocyte somata and branches (Fig. 7F–I; n = 24 cells, n = 7 mice) that covered large parts of the GCaMP3-observable astrocyte area (Fig. 7J; n = 26 cells, n = 7 mice). For both somata and branches, the EFS-evoked signals in the presence of TBOA were completely blocked by TTX (0.5 μm) and LY341495 (10 μm), but were resistant to the mGluR5 antagonist MPEP (Fig. 7G,I; 50 μm). These data further indicate that striatal astrocytes express functional mGluR3 rather than mGluR5 receptors and suggest that the EFS-evoked astrocyte Ca2+ signals observed in R6/2 mice (Fig. 5) are not caused by increased expression of mGluR3 in HD model mice because similar mGluR3-mediated responses could be unmasked in C57BL/6N mice when glutamate transport was blocked. The findings suggest that glutamate uptake acts as a key regulator of astrocyte engagement in the corticostriatal circuit (Haustein et al., 2014).
Figure 7.

Blockade of Glt1 in C57BL/6N astrocytes leads to enhanced astrocyte iGluSnFR signals and enhanced astrocyte Ca2+ signals evoked by EFS of cortical axons. A, Individual traces (gray) and averages (black) for GCaMP3 imaging showing increased spontaneous Ca2+ signals in astrocytes from C57BL/6N mice in the presence of TBOA (1 μm). Bar graph to the right shows average data for the peak area before and during applications of TBOA. B, As in A, but for iGluSnFR imaging showing elevated basal glutamate levels in the presence of TBOA. C, Individual traces (gray) and averages (black) for 4 EFS-evoked iGluSnFR signals in the absence of TBOA as well as in its presence. D, E, Average data for peak amplitude and decay time for traces such as those shown in C. F, Individual traces (gray) and averages (black) for 4 EFS-evoked GCaMP3 Ca2+ signals in the presence of TBOA or TBOA/TTX measured in somata of astrocytes in C57BL/6N mice. Tilted arrows indicate the expected time for the EFS-evoked Ca2+ signals. G, Summary bar graph for experiments such as those shown in F in the presence of different drugs. H, I, As in F and G, but for measurements in branches instead of somata. J, Representative confocal images of a GCaMP3-expressing astrocyte taken before, during, and after 4 EFS in the presence of TBOA. Bar graph to the right shows the area of the astrocyte territory and the area of the 4 EFS-evoked Ca2+ signals. In some cases, the error bars signifying SEM are smaller than the symbols used.
A prediction from our findings with TBOA in C57BL/6N mice (Fig. 7) is that the effect of TBOA on astrocyte iGluSnFR and Ca2+ signals in R6/2 mice would be smaller than in noncarrier WT controls. This is because R6/2 mice have reduced Glt1 levels, so the observable consequence of blocking Glt1 with TBOA should be smaller. We tested for this systematically across all of the key experiments in both R6/2 and noncarrier WT mice and report the data in Tables 2, 3, and 4. We found that TBOA had a significantly greater effect on basal iGluSnFR signals in WT mice compared with R6/2 mice (Table 2) for experiments such as those shown in Figure 7B. We also found that TBOA had a significantly greater effect on EFS-evoked iGluSnFR signals in WT mice compared with R6/2 mice for experiments such as those shown in Figure 7 C–E. Finally, TBOA had a greater effect on EFS-evoked astrocyte Ca2+ signals in WT mice compared with R6/2 for experiments such as those shown in Figure 7 F–I. Together, these experiments provide strong evidence that the deficits observed in astrocyte glutamate and Ca2+ signaling in R6/2 mice are largely driven by dysfunction of Glt1.
Table 2.
TBOA has a greater effect on astrocyte basal iGluSnFR signals in WT mice than in R6/2 mice
| Baseline dF/F | + TBOA dF/F | p | Fold-change from baseline to TBOA | n (cells, mice) | |
|---|---|---|---|---|---|
| WT | 0.01 ± 0.01 | 0.39 ± 0.04 | <0.00001 | 39.8 ± 10.2 | 19, 4 |
| R6/2 | 0.03 ± 0.01 | 0.16 ± 0.03 | <0.00001 | 7.7 ± 2.5 | 24, 4 |
Data are from experiments such as those illustrated in Figure 7B, but summarized here for R6/2 and noncarrier controls (WT). TBOA significantly increased the iGluSnFR signals in WT and R6/2 mice, but the fold changes were significantly greater in WT (p < 0.00001). The mean values for fold changes calculated from individual experiments are shown.
Table 3.
TBOA has a greater effect on astrocyte EFS-evoked iGluSnFR signals in WT mice than in R6/2 mice
| Control | + TBOA | p | Fold-change from control to TBOA | n (cells, mice) | |
|---|---|---|---|---|---|
| WT | |||||
| Peak dF/F | 0.41 ± 0.09 | 1.07 ± 0.09 | <0.00001 | 7.0 ± 1.4 | 21, 4 |
| Decay (s) | 2.79 ± 0.55 | 10.85 ± 1.24 | <0.00001 | 7.2 ± 2.1 | 21, 4 |
| Area (dF/F · s) | 0.73 ± 0.14 | 5.18 ± 0.56 | <0.00001 | 10.3 ± 2.2 | 21, 4 |
| R6/2 | |||||
| Peak dF/F | 0.42 ± 0.05 | 0.54 ± 0.05 | 0.00025 | 1.8 ± 0.2 | 25, 4 |
| Decay (s) | 5.55 ± 0.40 | 12.77 ± 1.88 | 0.00037 | 2.5 ± 0.4 | 25, 4 |
| Area (dF/F · s) | 1.20 ± 0.09 | 3.13 ± 0.53 | 0.00146 | 2.5 ± 0.4 | 25, 4 |
Data are from experiments such as those illustrated in Figure 7C, but summarized here for R6/2 and noncarrier controls (WT). TBOA significantly increased the EFS-evoked iGluSnFR signals in WT and R6/2 mice, but the fold changes were significantly greater in WT mice for all three comparisons of peak δF/F (p = 0.00132), decay time (p = 0.01696), and area (p = 0.00296) between WT and R6/2 mice. The mean values for fold changes calculated from individual experiments are shown.
Table 4.
TBOA has a greater effect on astrocyte EFS-evoked Ca2+ signals in WT mice than in R6/2 mice
| Control | +TBOA | p | Fold change from control to TBOA | n (cells, mice) | |
|---|---|---|---|---|---|
| WT somata | |||||
| Peak dF/F | 0.18 ± 0.05 | 2.36 ± 0.60 | 0.00489 | 13.1 | 10–13, 4 |
| R6/2 somata | |||||
| Peak dF/F | 0.98 ± 0.40 | 3.13 ± 0.57 | 0.01795 | 3.2 | 12–15, 4 |
| WT branches | |||||
| Peak dF/F | 0.30 ± 0.04 | 1.83 ± 0.14 | <0.00001 | 6.1 | 10–13, 4 |
| R6/2 branches | |||||
| Peak dF/F | 0.70 ± 0.10 | 0.82 ± 0.07 | 0.00845 | 1.2 | 12–15, 4 |
Data are from experiments such as those illustrated in Figure 7, F and H, but summarized here for R6/2 and noncarrier controls (WT). TBOA significantly increased the EFS-evoked Ca2+ signals in WT and R6/2 mice somata, but the fold changes were greater in WT. For the experiments shown in this table, the fold changes were calculated from the average data because the control and +TBOA experiments were not always from the same cells. Nonetheless, the trend for greater effects of TBOA in WT mice was very clear and consistent with the data shown in Tables 2 and 3.
No evidence for increased pulsatile glutamate release onto astrocytes in HD model mice
A recent study using astrocyte cultures indicated that astrocytes from HD model mice display elevated glutamate exocytosis (Lee et al., 2013). Might this explain enhanced EFS-evoked mGluR3 mediated Ca2+ signals and the prolonged glutamate levels observed as a result of EFS in R6/2 mice (Fig. 5)? We considered this unlikely given our evidence implicating Glt1 (Fig. 7), but nonetheless we sought to measure iGluSnFR-detected glutamate “flashes” that might indicate elevated glutamate levels near astrocytes from any cellular source in R6/2 mice (Marvin et al., 2013). We measured infrequent iGluSnFR flashes in WT and R6/2 astrocytes at a rate of ∼1 every 5 min in fields of view containing ∼10 astrocytes (Fig. 8A–D; n = 6 mice). These signals typically covered a large astrocyte area of ∼400 μm2 and seemed unlikely to represent point-source release events and more likely to represent ambient glutamate fluctuations in the extracellular space due to neuronal release. Regardless of their origin, the iGluSnFR flashes were significantly reduced in frequency in R6/2 relative to WT mice (Fig. 8D; n = 6 mice) consistent with decreased corticostriatal neuronal glutamate release probability in R6/2 mice (Fig. 8E; n = 4 mice), which has been well documented previously (Cepeda et al., 2010). We also found no difference in the basal iGluSnFR intensity between WT and R6/2 mice (Fig. 7D), although this metric is less reliable than the quantification of the iGluSnFR flashes because the basal intensity also depends on expression levels. These experiments provided little evidence for increased glutamate levels (pulsatile or basal) in the absence of EFS (Fig. 5) near astrocytes in HD model mice.
Figure 8.
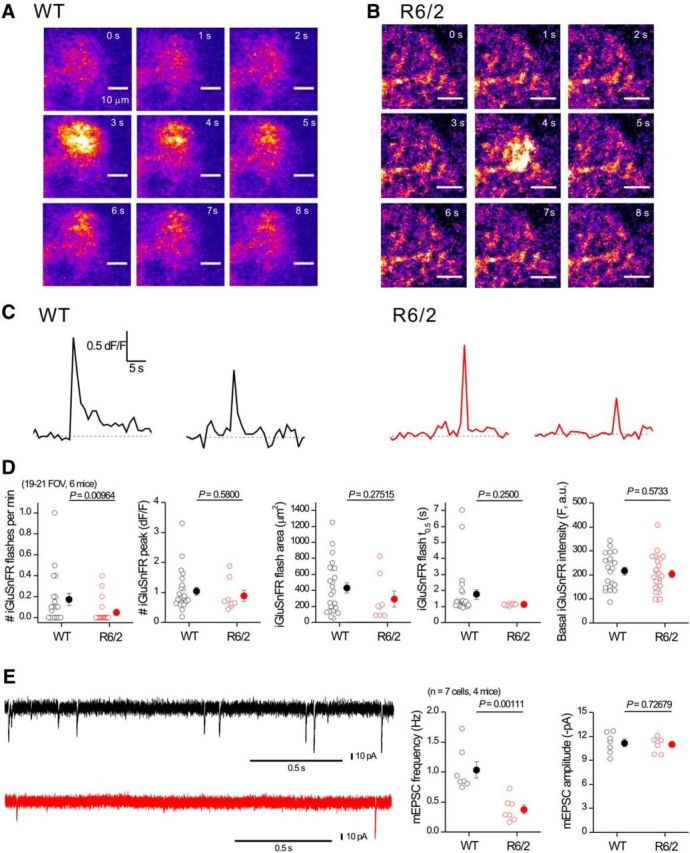
iGluSnFR flashes are not increased in striatal astrocytes from R6/2 mice. A, Representative time series of confocal images from a small region of an astrocyte expressing iGluSnFR from a WT mouse; an iGluSnFR flash occurred at ∼3 s. B, As in A, but for an astrocyte from a R6/2 mouse. C, Representative traces for transients such as those shown in A and B. D, Bar graphs showing average data for traces such as those shown in C. Note: the only significant difference in iGluSnFR signals between WT and R6/2 mice was in the frequency; no other parameter was changed in R6/2 compared with WT astrocytes. E, Representative traces and average data for mEPSCs recorded from MSNs at −70 mV from WT and R6/2 mice. In some cases, the error bars signifying SEM are smaller than the symbols used.
Restoring Kir4.1 function rescues astrocyte dysfunctions associated with HD
Astrocyte Kir4.1 channels are components of the machinery that regulate extracellular K+ levels and neuronal excitability (Kofuji and Newman, 2004; Larsen et al., 2014; Sibille et al., 2015). Based on the finding that HD-associated reductions in Glt1 can be remedied by rescuing Kir4.1 (Tong et al., 2014), we used AAVs to express GCaMP3 with either Kir4.1 or tdTomato in striatal astrocytes and investigated whether Ca2+ and glutamate signal dysfunctions observed in R6/2 mice were rescued in astrocytes that received Kir4.1 relative to those that received tdTomato as a control (Fig. 9A). Coinjecting two AAVs resulted in the coexpression of both fluorescent proteins in ∼90% of astrocytes (Fig. 9B; n = 38 cells, n = 4 mice). Moreover, our past work shows that AAV2/5 mediated delivery of Kir4.1 in R6/2 mice restores channel expression and function to levels equivalent to those in WT mice (Tong et al., 2014). In astrocytes from R6/2 mice expressing tdTomato, we observed small and infrequent spontaneous Ca2+ signals (Fig. 9C; n = 28 cells, n = 5 mice), as expected for R6/2 mice (Fig. 4). However, in astrocytes from R6/2 mice expressing Kir4.1, we observed significantly larger and more frequent spontaneous Ca2+ signals (Fig. 9C–E; n = 24 cells, n = 5 mice). When quantified, we measured significant recovery of astrocyte global waves, local waves, and Ca2+ microdomains in mice that had received Kir4.1 relative to those that received tdTomato (Fig. 9E).
Figure 9.
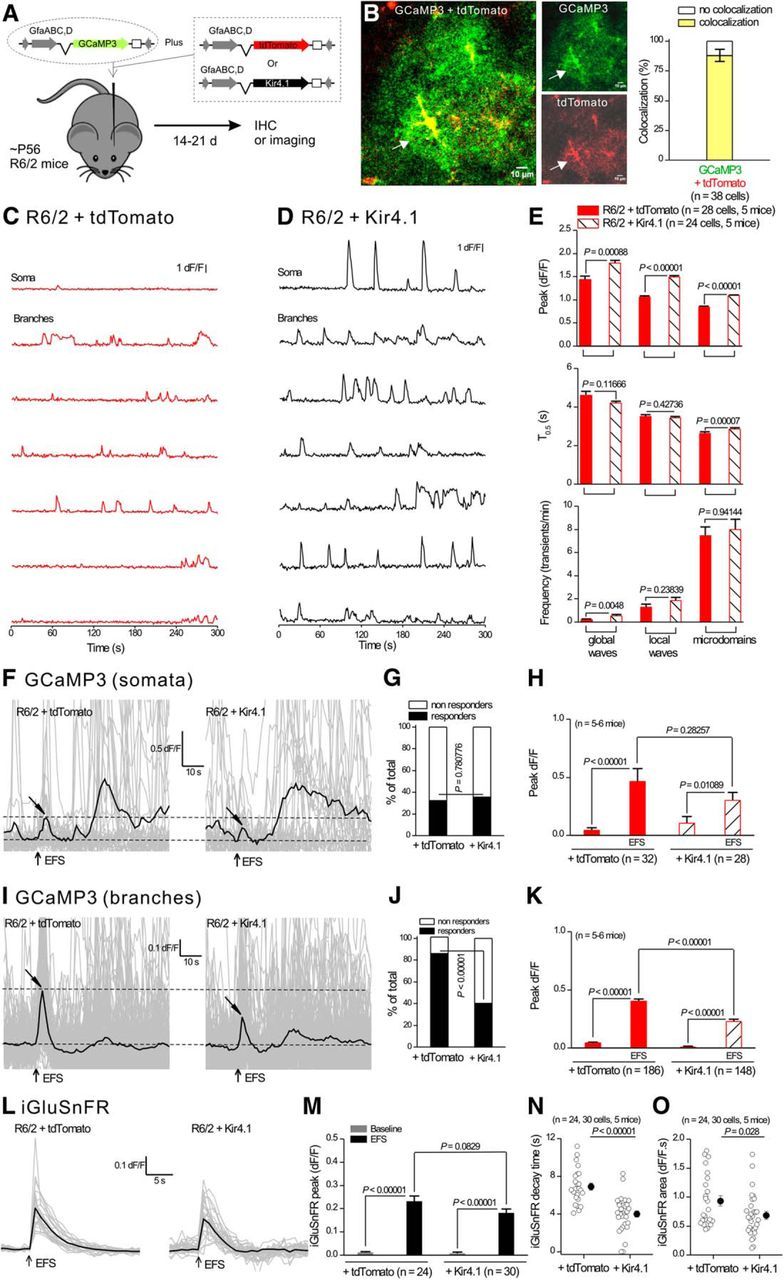
AAV2/5-mediated Kir4.1 expression within astrocytes rescued dysfunctional astrocyte Ca2+ and glutamate signaling in R6/2 mice. A, The diagram illustrates the viral constructs used, and that the mice were microinjected at ∼P50–P56 and studied at P70–P80. B, Confocal imaging for GCaMP3 and tdTomato shows colocalization when two viral constructs were coexpressed in striatal astrocytes. C, D, Representative traces from seven selected ROIs showing enhanced spontaneous Ca2+ signals in astrocytes from R6/2 mice that had received AAV2/5 Kir4.1 (D) compared with R6/2 mice receiving AAV2/5 tdTomato (C). E, Quantification of peak amplitude, duration, and frequency for the three types of Ca2+ signals in striatal astrocytes in R6/2 mice receiving AAV2/5 Kir4.1 or AAV2/5 tdTomato. F, Individual traces (gray) and averages (black) for eight EFS-evoked Ca2+ signals within somata of astrocytes in R6/2 mice receiving AAV2/5 Kir4.1 or AAV2/5 tdTomato. Tilted arrows indicate the peak of the response. G, Bar graph showing the percentage of cells responding to EFS. H, Average data for peak amplitude from traces such as those shown in F. I–K, As in F–H, but for measurements from branches instead of somata. L, Individual traces (gray) and averages (black) for eight EFS-evoked iGluSnFR signals within astrocyte territories from R6/2 mice receiving AAV2/5 Kir4.1 or AAV2/5 tdTomato. M–O, Quantification of peak amplitude, area, and kinetics for traces such as those shown in L for iGluSnFR. In some cases, the error bars signifying SEM are smaller than the symbols used.
Next, we evaluated EFS-evoked astrocyte Ca2+ signals in astrocytes from R6/2 mice that had received Kir4.1, relative to those that received tdTomato (Fig. 9F–H; n = 5–6 mice). This rescue, although partial relative to WT mice, was clear for astrocyte branches and was apparent as a reduction in the proportion of astrocytes responding to EFS and in the amplitude of EFS-evoked astrocyte Ca2+ signals (Fig. 9I–K). Moreover, expression of Kir4.1 in astrocytes from R6/2 mice also shortened the decay time and reduced the integrated area of the EFS-evoked iGluSnFR signals (Fig. 9L–O). Therefore, Ca2+ and glutamate signaling in astrocytes from R6/2 mice was rescued toward WT levels by the expression of exogenous Kir4.1 in astrocytes, but not by expression of control tdTomato.
Discussion
There are several key findings from this study, some of which are schematized in Figure 10. First, striatal astrocytes from symptomatic R6/2 mice, known to contain mHTT nuclear inclusions at ∼P70 before detectable astrogliosis, displayed Kir4.1 (Tong et al., 2014) and Glt1 dysfunctions that were related to each other. Second, astrocyte spontaneous Ca2+ signals were reduced in R6/2 mice, but evoked Ca2+ signals were increased in a manner that was dependent on Glt1. The action-potential-dependent evoked Ca2+ signals were mediated by astrocyte mGluR2/3 receptors. Third, the lifetime of EFS-evoked glutamate release near the surface of astrocytes was prolonged in R6/2 relative to WT mice, reflecting a loss of Glt1 function. Fourth, prolonged astrocyte iGluSnFR signals and elevated mGluR2/3-mediated evoked Ca2+ signals observed in R6/2 mice could be mimicked in C57BL/6N mice by blocking Glt1, implying that reduced glutamate uptake is the likely cause of these astrocyte dysfunctions in R6/2 mice. Fifth, many of the iGluSnFR and Ca2+ signal alterations observed in R6/2 mice could be rescued by astrocyte delivery of Kir4.1. However, restoration of Kir4.1 did not significantly increase the frequency of microdomain Ca2+ signals even though these were significantly attenuated in R6/2 mice. Therefore, the rescue by Kir4.1 was partial.
Figure 10.
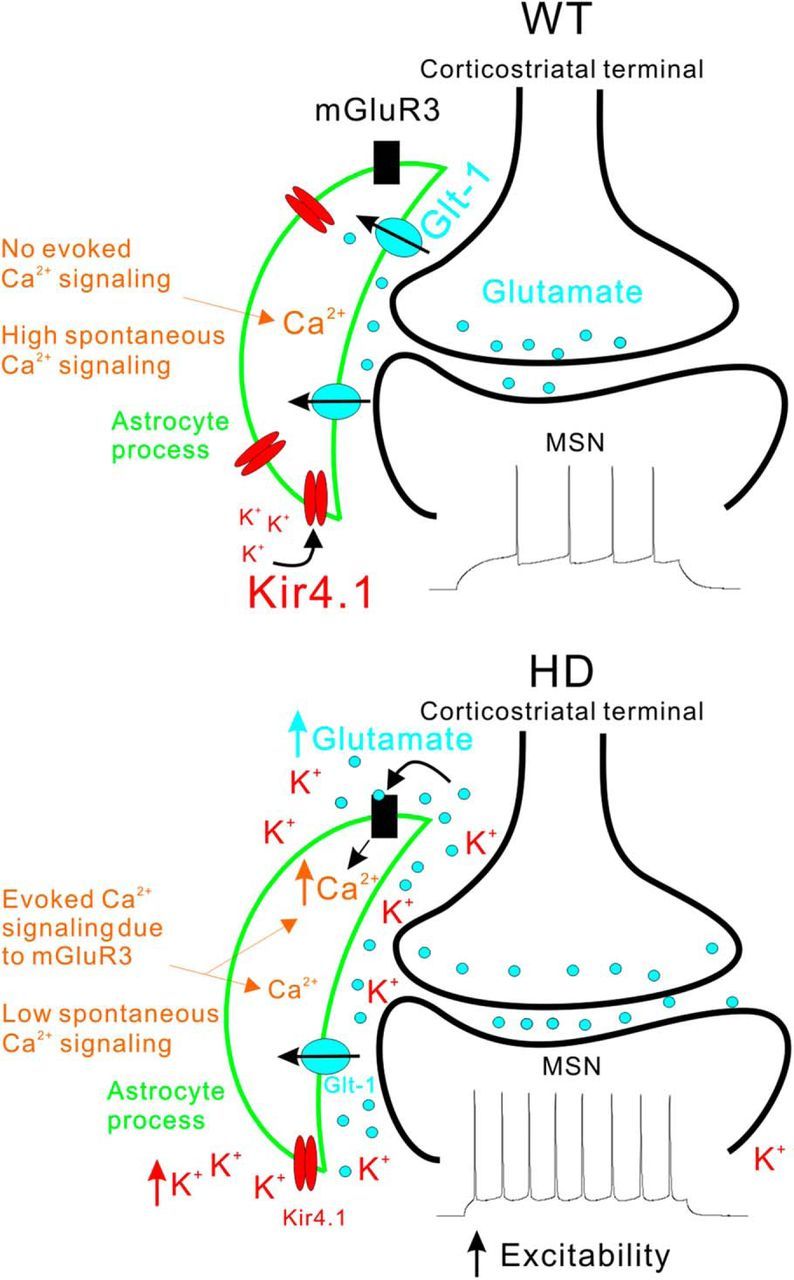
Schematic summary of the major astrocyte changes observed in R6/2 mice at ∼P70. The top and bottom diagrams summarize key aspects of the changes that occur in astrocytes in HD model mice and how they affect MSNs in the dorsolateral striatum (Tong et al., 2014). Only the key features of our findings are schematized and other differences between WT and R6/2 mice are discussed in the text. In the WT striatum, normal expression levels of Kir4.1 and Glt1 maintain low levels of extracellular K+ and glutamate near synapses. As a result, electrical stimulation of corticostriatal axons does not evoke robust mGluR3-mediated astrocyte Ca2+ signals. However, there are abundant spontaneous Ca2+ signals in striatal astrocytes. In HD model mice, expression levels of Kir4.1 and Glt1 are reduced and this has several observable consequences for astrocytes. Reduced Glt1 levels mean that astrocytes display robust mGluR3-mediated Ca2+ signals during stimulation of corticostriatal axons. This gain of evoked Ca2+ signals is accompanied by a loss of spontaneous Ca2+ signals. The elevated glutamate and K+ levels in the extracellular space increase the excitability of MSNs. Although the observations shown in the diagram were robust and highly significant (see text), at this stage, it is not possible to extend our cellular observations and propose a satisfying circuit based mechanism for how astrocytes cause or contribute to HD symptoms, largely because there is no widely accepted circuit based model for neuronal alterations that accompany HD (Waldvogel et al., 2015). Such circuit-based mechanisms are worthy of further investigation, but will require further detailed evaluations of astrocytes and neurons in WT and HD model mice.
Although several mouse models of HD exist, we explored astrocytes in the extensively studied transgenic R6/2 model (Mangiarini et al., 1996) based on past work (Tong et al., 2014). In addition, the early onset of symptoms in R6/2 mice permitted the use of AAVs to express fluorescent proteins as well as Ca2+ and glutamate indicators, which have not been validated for astrocyte studies in mice >∼P80 (Shigetomi et al., 2013; Haustein et al., 2014; Jiang et al., 2014; Tong et al., 2014; Srinivasan et al., 2015). At the ages used in this study, R6/2 mice display symptom onset before detectable astrogliosis (Mangiarini et al., 1996; Tong et al., 2014), which recalls the early stages of the human disease (Faideau et al., 2010) and may be portentous of symptoms in humans that occur before overt striatal tissue loss (Tabrizi et al., 2009). Overall, R6/2 mice present a number of advantages for exploring astrocytes. Nonetheless, in future work, additional evaluations in humans and perhaps other mouse models will be needed.
The present study was motivated by the discovery that correcting the loss of astrocyte Kir4.1 rescued the loss of Glt1 as assessed by Western blot (Tong et al., 2014) and sought to explore the functional implications of this observation within the striatal microcircuit. Previous studies have reported disruptions of astrocyte Glt1 function in HD mouse models (Behrens et al., 2002; Miller et al., 2008; Bradford et al., 2009; Bradford et al., 2010; Tong et al., 2014). Our aim was to determine how astrocyte physiology was altered in HD mouse models and shed light on the consequences with a focus on Kir4.1 and Glt1. More generally, past studies of several brain disorders suggested that they are accompanied by reduced Kir4.1 expression, including Alzheimer's disease (Wilcock et al., 2009), amyotrophic lateral sclerosis (Kaiser et al., 2006), spinocerebellar ataxia (Magaña et al., 2013), and epilepsy (Scholl et al., 2009). Furthermore, the genetic deletion of Kir4.1 causes severe pathology in mice (Neusch et al., 2001; Djukic et al., 2007; Chever et al., 2010). Therefore, studies of Kir4.1 in the context of HD may yield mechanistic information of general relevance.
In relation to Kir4.1, we explored Ca2+ and glutamate signaling, which are involved in multiple aspects of astrocyte biology (Volterra et al., 2014; Khakh and McCarthy, 2015; Khakh and Sofroniew, 2015) and implicated in disease (Nedergaard et al., 2010; Agulhon et al., 2012). Astrocyte Ca2+ signaling was increased in mouse models of Alzheimer's disease (Takano et al., 2007; Kuchibhotla et al., 2009; Delekate et al., 2014), after cerebral ischemia (Ding et al., 2009), and during status epilepticus (Ding et al., 2007). In all three of these settings, it is proposed that elevated astrocyte Ca2+ signaling may contribute to disease. In contrast, there was hitherto no available information for striatal astrocyte Ca2+ signaling in WT or HD mice. Consistent with recent studies in the hippocampus, we found that striatal astrocytes from C57BL/6N mice displayed three types of seconds-time-scale spontaneous Ca2+ signals that were action-potential independent and occurred mainly in astrocyte branches (Haustein et al., 2014; Srinivasan et al., 2015). Using selective stimulation of corticostriatal axons, we found that striatal astrocytes did not respond reliably with Ca2+ elevations when glutamate was released onto them; that is, they were largely disengaged from the corticostriatal circuit in C57BL/6N mice (Haustein et al., 2014).
By focusing on early stages in R6/2 mice, we found that astrocyte Ca2+ signaling in the striatum was altered dramatically. Specifically, we measured a loss of spontaneous Ca2+ signals and a gain of evoked action potential-dependent Ca2+ signals in R6/2 mice. The loss of spontaneous Ca2+ signals likely reflects lower store capacity, whereas the increase in evoked Ca2+ signals likely reflects the loss of Glt1, increased lifetime of glutamate in the extracellular space and subsequent activation of mGluR2/3 receptors. The endogenous activation of astrocyte mGluR2/3 receptors may represent an endogenous protective response because a mixed mGluR2/3 agonist is neuroprotective in R6/2 mice (Schiefer et al., 2004). A role for mGluR2/3 receptors is consistent with the expression of mGluR3 receptors in astrocytes from adult mice (Sun et al., 2013; Haustein et al., 2014). Recent studies (Rosenegger et al., 2015) also suggest that loss of astrocyte spontaneous Ca2+ signals may contribute to reduced cerebral blood flow in HD. We also speculate that reduced spontaneous Ca2+ signals alter astrocyte gene expression and thus alter function of astrocytes and neurons. Together, our data emphasize the critical role of glutamate clearance in determining the conditions and constraints for astrocyte Ca2+ signaling in the striatum. Additional studies are needed to explore how astrocyte Ca2+ signaling contributes to striatal microcircuit function in health and dysfunction in HD model mice.
We measured robust mGluR2/3-dependent Ca2+ signals in striatal astrocytes from C57BL/6N mice when Glt1 was blocked. Moreover, the loss of Kir4.1 and Glt1 appear to be related: both are reduced in HD mouse models and genetically restoring Kir4.1 also rescues Glt1. Glt1 is Na+ dependent and thus electrogenic: it functions most efficiently at negative resting membrane potentials. Therefore, the restoration of Kir4.1 is expected to restore function of expressed Glt1 and has been shown to lead to restored protein expression (Tong et al., 2014). Over activation of NMDA receptors has been shown to lead to decreased Kir4.1 expression (Obara-Michlewska et al., 2015), suggesting a possible feedback loop between Kir4.1 and the consequences of Glt1 loss in HD. A critical role for Kir4.1 in these phenomena is supported by past cell biological studies (Härtel et al., 2007; Kucheryavykh et al., 2007) and suggests that correcting Kir4.1-dependent homeostatic functions in astrocytes has downstream beneficial effects via Glt1 and Ca2+. Disentangling cause and effect and revealing potential convergent molecular mechanisms among Kir4.1, Glt1, and Ca2+ will require further detailed work, but could include gene and/or protein regulation, the influence of electrochemical driving force, altered ion selectivity (Härtel et al., 2007), or possibly all four. Moreover, the available data do not suggest a simple all-encompassing, circuit-based mechanism for how astrocytes cause or contribute to HD symptoms, largely because there is no accepted neural circuit based model for HD per se (Waldvogel et al., 2015; Plotkin and Surmeier, 2015). Such models are beyond the scope of the current work.
MSN phenotypes that occur in HD (Cepeda et al., 2010) involve multiple cell types because mHTT is expressed widely in neurons and glia. Our recent studies reported herein and in Tong et al., 2014 suggest that astrocytes play causative roles in neural circuit level dysfunctions. This suggestion is consistent with past work demonstrating that mHTT expression only in astrocytes can lead to HD-like phenotypes in vivo (Bradford et al., 2009) and the finding that restoring astrocyte Kir4.1 levels can rescue some in vivo HD phenotypes (Tong et al., 2014). HTT is known to associate with several transcriptional activators, including Sp1, in GFAP-HD model mice and mHTT was found to more readily associate with Sp1, leading to decreased occupancy of the conserved Sp1-binding site in the Glt1 promoter and concomitant reduced Glt1 expression (Bradford et al., 2009). The promoters of many Kir channels also have conserved Sp1-binding sites and these are crucial in regulating the expression of Kir2.1 (Redell and Tempel, 1998) and Kir3.1 (Schoots et al., 1997). Kir4.1 has a highly conserved Sp1-binding site in its promoter, implying that mHTT-Sp1 association may be a common mechanism for downregulation of Kir4.1 and Glt1 in HD.
In the future, astrocyte-specific inducible Cre lines may allow for the specific expression or deletion of mHTT in striatal astrocytes and thus provide opportunities to further explore non-cell-autonomous mechanisms in HD (McGann et al., 2012). Suitable Cre lines do not yet exist and striatal astrocytes are notable for their low expression of GFAP (Mangiarini et al., 1996; Tong et al., 2014). Therefore, available GFAP Cre lines, which have been used with aplomb in other brain disorders (McGann et al., 2012), may have limited utility for the striatum.
In summary, our data show that the loss of astrocyte Kir4.1- and Glt1-mediated homeostatic functions in R6/2 mice caused dysfunctional astrocyte glutamate and Ca2+ signaling, which we propose may in turn contribute to altered MSNs in the striatum. We suggest that striatal circuit defects in HD, and perhaps other brain disorders, may be remedied by correcting key astrocyte homeostatic dysfunctions that precede overt astrogliosis and neurodegeneration.
Footnotes
This work was supported by the CHDI Foundation and by the National Institutes of Health (Grants NS060677 and MH104069 to B.S.K.). We thank H. Chai, V. Beaumont, I. Munoz-Sanjuan, and R. Cachope for valuable comments and discussions during the course of this study; M. Sofroniew for sharing equipment; and Xiaoping Tong for comments on an earlier version of this manuscript.
The authors declare no competing financial interests.
References
- Adermark L, Lovinger DM. Electrophysiological properties and gap junction coupling of striatal astrocytes. Neurochem Int. 2008;52:1365–1372. doi: 10.1016/j.neuint.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agulhon C, Fiacco TA, McCarthy KD. Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science. 2010;327:1250–1254. doi: 10.1126/science.1184821. [DOI] [PubMed] [Google Scholar]
- Agulhon C, Sun MY, Murphy T, Myers T, Lauderdale K, Fiacco TA. Calcium signaling and gliotransmission in normal vs. reactive astrocytes. Front Pharmacol. 2012;3:139. doi: 10.3389/fphar.2012.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB. Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain. 2002;125:1908–1922. doi: 10.1093/brain/awf180. [DOI] [PubMed] [Google Scholar]
- Ben Haim L, Ceyzériat K, Carrillo-de Sauvage MA, Aubry F, Auregan G, Guillermier M, Ruiz M, Petit F, Houitte D, Faivre E, Vandesquille M, Aron-Badin R, Dhenain M, Déglon N, Hantraye P, Brouillet E, Bonvento G, Escartin C. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer's and Huntington's diseases. J Neurosci. 2015;35:2817–2829. doi: 10.1523/JNEUROSCI.3516-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonder DE, McCarthy KD. Astrocytic Gq-GPCR-linked IP3R-dependent Ca2+ signaling does not mediate neurovascular coupling in mouse visual cortex in vivo. J Neurosci. 2014;34:13139–13150. doi: 10.1523/JNEUROSCI.2591-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, Li S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci U S A. 2009;106:22480–22485. doi: 10.1073/pnas.0911503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford J, Shin JY, Roberts M, Wang CE, Sheng G, Li S, Li XJ. Mutant huntingtin in glial cells exacerbates neurological symptoms of Huntington disease mice. J Biol Chem. 2010;285:10653–10661. doi: 10.1074/jbc.M109.083287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Cummings DM, André VM, Holley SM, Levine MS. Genetic mouse models of Huntington's disease: focus on electrophysiological mechanisms. ASN Neuro. 2010;2:e00033. doi: 10.1042/AN20090058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chever O, Djukic B, McCarthy KD, Amzica F. Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J Neurosci. 2010;30:15769–15777. doi: 10.1523/JNEUROSCI.2078-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delekate A, Füchtemeier M, Schumacher T, Ulbrich C, Foddis M, Petzold GC. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer's disease mouse model. Nat Commun. 2014;5:5422. doi: 10.1038/ncomms6422. [DOI] [PubMed] [Google Scholar]
- Di Castro MA, Chuquet J, Liaudet N, Bhaukaurally K, Santello M, Bouvier D, Tiret P, Volterra A. Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat Neurosci. 2011;14:1276–1284. doi: 10.1038/nn.2929. [DOI] [PubMed] [Google Scholar]
- Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, Coulter DA, Carmignoto G, Haydon PG. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci. 2007;27:10674–10684. doi: 10.1523/JNEUROSCI.2001-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Wang T, Cui W, Haydon PG. Photothrombosis ischemia stimulates a sustained astrocytic Ca2+ signaling in vivo. Glia. 2009;57:767–776. doi: 10.1002/glia.20804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. 2007;27:11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G, Dufour N, Guillermier M, Brouillet E, Hantraye P, Déglon N, Ferrante RJ, Bonvento G. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington's disease subjects. Hum Mol Genet. 2010;19:3053–3067. doi: 10.1093/hmg/ddq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo LC. Purification of astrocytes from transgenic rodents by fluorescence-activated cell sorting. Cold Spring Harb Protoc. 2013;2013:551–560. doi: 10.1101/pdb.prot074229. [DOI] [PubMed] [Google Scholar]
- Ghosh R, Tabrizi SJ. Clinical aspects of Huntington's disease. Curr Top Behav Neurosci. 2015;22:3–31. doi: 10.1007/7854_2013_238. [DOI] [PubMed] [Google Scholar]
- Härtel K, Singaravelu K, Kaiser M, Neusch C, Hülsmann S, Deitmer JW. Calcium influx mediated by the inwardly rectifying K+ channel Kir4.1 (KCNJ10) at low external K+ concentration. Cell Calcium. 2007;42:271–280. doi: 10.1016/j.ceca.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Haustein MD, Kracun S, Lu XH, Shih T, Jackson-Weaver O, Tong X, Xu J, Yang XW, O'Dell TJ, Marvin JS, Ellisman MH, Bushong EA, Looger LL, Khakh BS. Conditions and constraints for astrocyte calcium signaling in the hippocampal mossy fiber pathway. Neuron. 2014;82:413–429. doi: 10.1016/j.neuron.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkinen T, Lehtimäki K, Vartiainen N, Puoliväli J, Hendricks SJ, Glaser JR, Bradaia A, Wadel K, Touller C, Kontkanen O, Yrjänheikki JM, Buisson B, Howland D, Beaumont V, Munoz-Sanjuan I, Park LC. Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington's disease. PLoS One. 2012;7:e50717. doi: 10.1371/journal.pone.0050717. doi: 10.1371/journal.pone.0050717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Haustein MD, Sofroniew MV, Khakh BS. Imaging intracellular Ca2+ signals in striatal astrocytes from adult mice using genetically-encoded calcium indicators. J Vis Exp. 2014;93:e51972. doi: 10.3791/51972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser M, Maletzki I, Hülsmann S, Holtmann B, Schulz-Schaeffer W, Kirchhoff F, Bähr M, Neusch C. Progressive loss of a glial potassium channel (KCNJ10) in the spinal cord of the SOD1 (G93A) transgenic mouse model of amyotrophic lateral sclerosis. J Neurochem. 2006;99:900–912. doi: 10.1111/j.1471-4159.2006.04131.x. [DOI] [PubMed] [Google Scholar]
- Khakh BS, McCarthy KD. Astrocyte calcium signals: from observations to functions and the challenges therein. Cold Spring Harb Perspect Biol. 2015;7:a020404. doi: 10.1101/cshperspect.a020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Sofroniew MV. Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci. 2015;18:942–952. doi: 10.1038/nn.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129:1045–1056. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, Eaton MJ. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia. 2007;55:274–281. doi: 10.1002/glia.20455. [DOI] [PubMed] [Google Scholar]
- Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science. 2009;323:1211–1215. doi: 10.1126/science.1169096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, Kaila K, Voipio J, MacAulay N. Contributions of the Na+/K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia. 2014;62:608–622. doi: 10.1002/glia.22629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Reyes RC, Gottipati MK, Lewis K, Lesort M, Parpura V, Gray M. Enhanced Ca(2+)-dependent glutamate release from astrocytes of the BACHD Huntington's disease mouse model. Neurobiol Dis. 2013;58:192–199. doi: 10.1016/j.nbd.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liévens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le Goff L, Bates GP. Impaired glutamate uptake in the R6 Huntington's disease transgenic mice. Neurobiol Dis. 2001;8:807–821. doi: 10.1006/nbdi.2001.0430. [DOI] [PubMed] [Google Scholar]
- Magaña JJ, Velázquez-Pérez L, Cisneros B. Spinocerebellar ataxia type 2: clinical presentation, molecular mechanisms, and therapeutic perspectives. Mol Neurobiol. 2013;47:90–104. doi: 10.1007/s12035-012-8348-8. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/S0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006;2:679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- Marvin JS, Borghuis BG, Tian L, Cichon J, Harnett MT, Akerboom J, Gordus A, Renninger SL, Chen TW, Bargmann CI, Orger MB, Schreiter ER, Demb JB, Gan WB, Hires SA, Looger LL. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat Methods. 2013;10:162–170. doi: 10.1038/nmeth.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGann JC, Lioy DT, Mandel G. Astrocytes conspire with neurons during progression of neurological disease. Curr Opin Neurobiol. 2012;22:850–858. doi: 10.1016/j.conb.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Kudwa AE, Miller S, Fitzpatrick J, Watson-Johnson J, Keating N, Ruiz M, Mushlin R, Alosio W, McConnell K, Connor D, Murphy C, Oakeshott S, Kwan M, Beltran J, Ghavami A, Brunner D, Park LC, Ramboz S, Howland D. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington's disease: zQ175. PLoS One. 2012;7:e49838. doi: 10.1371/journal.pone.0049838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Dorner JL, Shou M, Sari Y, Barton SJ, Sengelaub DR, Kennedy RT, Rebec GV. Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington's disease phenotype in the R6/2 mouse. Neuroscience. 2008;153:329–337. doi: 10.1016/j.neuroscience.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Rodríguez JJ, Verkhratsky A. Glial calcium and diseases of the nervous system. Cell Calcium. 2010;47:140–149. doi: 10.1016/j.ceca.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci. 2001;21:5429–5438. doi: 10.1523/JNEUROSCI.21-15-05429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara-Michlewska M, Ruszkiewicz J, Zielińska M, Verkhratsky A, Albrecht J. Astroglial NMDA receptors inhibit expression of Kir4.1 channels in glutamate-overexposed astrocytes in vitro and in the brain of rats with acute liver failure. Neurochem Int. 2015;88:20–25. doi: 10.1016/j.neuint.2014.10.006. [DOI] [PubMed] [Google Scholar]
- Panatier A, Vallée J, Haber M, Murai KK, Lacaille JC, Robitaille R. Astrocytes are endogenous regulators of basal transmission at central synapses. Cell. 2011;146:785–798. doi: 10.1016/j.cell.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Niedzielski AS, Wenthold RJ. The metabotropic glutamate receptors, mGluR2 and mGluR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience. 1996;71:949–976. doi: 10.1016/0306-4522(95)00533-1. [DOI] [PubMed] [Google Scholar]
- Plotkin JL, Surmeier DJ. Corticostriatal synaptic adaptations in Huntington's disease. Curr Opin Neurobiol. 2015;33:53–62. doi: 10.1016/j.conb.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redell JB, Tempel BL. Multiple promoter elements interact to control the transcription of the potassium channel gene, KCNJ2. J Biol Chem. 1998;273:22807–22818. doi: 10.1074/jbc.273.35.22807. [DOI] [PubMed] [Google Scholar]
- Rosenegger DG, Tran CH, Wamsteeker Cusulin JI, Gordon GR. Tonic local brain blood flow control by astrocytes independent of phasic neurovascular coupling. J Neurosci. 35:13463–13474. doi: 10.1523/JNEUROSCI.1780-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiefer J, Sprünken A, Puls C, Lüesse HG, Milkereit A, Milkereit E, Johann V, Kosinski CM. The metabotropic glutamate receptor 5 antagonist MPEP and the mGluR2 agonist LY379268 modify disease progression in a transgenic mouse model of Huntington's disease. Brain Res. 2004;1019:246–254. doi: 10.1016/j.brainres.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Scholl UI, Choi M, Liu T, Ramaekers VT, Häusler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A. 2009;106:5842–5847. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoots O, Voskoglou T, Van Tol HH. Genomic organization and promoter analysis of the human G-protein-coupled K+ channel Kir3.1 (KCNJ3/HGIRK1) Genomics. 1997;39:279–288. doi: 10.1006/geno.1996.4495. [DOI] [PubMed] [Google Scholar]
- Shigetomi E, Bushong EA, Haustein MD, Tong X, Jackson-Weaver O, Kracun S, Xu J, Sofroniew MV, MHE, Khakh BS. Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J Gen Physiol. 2013;141:633–647. doi: 10.1085/jgp.201210949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171:1001–1012. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibille J, Dao Duc K, Holcman D, Rouach N. The neuroglial potassium cycle during neurotransmission: role of Kir4.1 channels. PLoS Comput Biol Mar. 2015;11:e1004137. doi: 10.1371/journal.pcbi.1004137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Huang BS, Venugopal S, Johnston AD, Chai H, Zeng H, Golshani P, Khakh BS. Ca(2+) signaling in astrocytes from Ip3r2(-/-) mice in brain slices and during startle responses in vivo. Nat Neurosci. 2015;18:708–717. doi: 10.1038/nn.4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W, Lovatt D, Han X, Smith Y, Nedergaard M. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science. 2013;339:197–200. doi: 10.1126/science.1226740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Langbehn DR, Leavitt BR, Roos RA, Durr A, Craufurd D, Kennard C, Hicks SL, Fox NC, Scahill RI, Borowsky B, Tobin AJ, Rosas HD, Johnson H, Reilmann R, Landwehrmeyer B, Stout JC, Stout JC. Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol. 2009;8:791–801. doi: 10.1016/S1474-4422(09)70170-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Han X, Deane R, Zlokovic B, Nedergaard M. Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer's disease. Ann N Y Acad Sci. 2007;1097:40–50. doi: 10.1196/annals.1379.004. [DOI] [PubMed] [Google Scholar]
- Tamaru Y, Nomura S, Mizuno N, Shigemoto R. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience. 2001;106:481–503. doi: 10.1016/S0306-4522(01)00305-0. [DOI] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, Khakh BS. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat Neurosci. 2014;17:694–703. doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara R, Rick C, Hernández-López S, Laville JA, Guzman JN, Galarraga E, Surmeier DJ, Bargas J. Spontaneous voltage oscillations in striatal projection neurons in a rat corticostriatal slice. J Physiol. 2003;553:169–182. doi: 10.1113/jphysiol.2003.050799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volterra A, Liaudet N, Savtchouk I. Astrocyte Ca2+ signalling: an unexpected complexity. Nat Rev Neurosci. 2014;15:327–335. doi: 10.1038/nrn3725. [DOI] [PubMed] [Google Scholar]
- Waldvogel HJ, Kim EH, Tippett LJ, Vonsattel JP, Faull RL. The neuropathology of Huntington's disease. Curr Top Behav Neurosci. 2015;22:33–80. doi: 10.1007/7854_2014_354. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Vitek MP, Colton CA. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer's disease. Neuroscience. 2009;159:1055–1069. doi: 10.1016/j.neuroscience.2009.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]