

Abstract
Liver fibrosis is the pathological consequence of chronic liver diseases, where an excessive deposition of extracellular matrix (ECM) proteins occurs, concomitantly with the processes of repair and regeneration. It is characterized by increased production of matrix proteins, in particular collagens, and decreased matrix remodelling. The principal source of ECM accumulation is myofibroblasts (MFB). Most fibrogenic MFB are endogenous to the liver, coming from hepatic stellate cells (HSC) and portal fibroblasts. Dysregulated inflammatory responses have been associated with most (if not all) hepatotoxic insults and chronic oxidative stress play a role during the initial liver inflammatory phase and its progression to fibrosis. Redox-regulated processes are responsible for activation of HSC to MFB, as well as maintenance of the MFB function. Increased oxidative stress also induces hepatocyte apoptosis, which contributes to increase the liver injury and to transdifferentiate HSC to MFB, favouring the fibrogenic process. Mitochondria and other redox-active enzymes can generate superoxide and hydrogen peroxide as a by-product in liver cells. Moreover, accumulating evidence indicates that NADPH oxidases (NOXs), which play a critical role in the inflammatory response, may contribute to reactive oxygen species (ROS) production during liver fibrosis, being important players in HSC activation and hepatocyte apoptosis. Based on the knowledge of the pathogenic role of ROS, different strategies to prevent or reverse the oxidative damage have been developed to be used as therapeutic tools in liver fibrosis. This review will update all these concepts, highlighting the relevance of redox biology in chronic fibrogenic liver pathologies.
Abbreviations: 4-HNE, 4-hydroxi-2′-nonenal; CTGF, Connective Tissue Growth Factor; COX, cyclooxygenase; EMT, epithelial-to-mesenchymal transition; ECM, extracellular matrix; GSH, glutathione; HCC, hepatocellular carcinoma; IR, ischaemia/reperfusion; MSCs, mesenchymal stem cells; MMP, metalloproteases; MGSH, mitochondrial reduced glutathione; MFB, myofibroblasts; NOX, NADPH oxidase; NO, nitric oxide; NOS, nitric oxide synthase; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatiti; NFκB, Nuclear Factor kappa-B; PDGF, Platelet-Derived Growth Factor; RNS, reactive nitrogen species; ROS, reactive oxygen species; HSC, stellate cell; SOD, superoxide dismutase; TIMP, tissue inhibitors of metalloproteases; TLR4, toll-like receptor 4; TGF-β, Transforming Growth Factor-beta; TNF-α, Tumour Necrosis Factor-alpha; UPR, unfolding protein response; UDCA, ursodeoxycholic acid
Keywords: Hepatocyte, Stellate cell, Myofibroblast, TGF-beta, Inflammation, NOX
Graphical abstract
Highlights
Oxidative stress is a major cause for initiation/progression of liver fibrosis.
Redox-regulated processes activate hepatic stellate cells to myofibroblasts.
Increased oxidative stress induces hepatocyte apoptosis.
NOX inhibitors are considered as a new strategy to prevent/reverse liver fibrosis.
NADPH oxidases (NOX) have been involved in liver fibrogenic responses.
1. Introduction
In all organ systems, the normal mammalian response to injury occurs in three overlapping but distinct stages: inflammation, new tissue formation, and tissue remodelling. A critical step is when repair and regeneration take place, because excessive deposition of extracellular matrix (ECM) proteins leads to hypertrophic scars, which provoke tissue dysfunction [1]. Liver fibrosis is the pathological consequence of chronic liver diseases, resulting from the progressive accumulation of ECM, which is mainly enriched in types I and III fibrillar collagens. Scar deposition is a consequence of an altered wound healing response to prolonged parenchymal cell injury and/or inflammation. It is characterized by increased production of matrix proteins and decreased matrix remodelling. In advanced stages, fibrosis leads to cirrhosis, a condition defined by an abnormal liver architecture, failure in liver function, portal hypertension and high susceptibility to infection and to develop HCC [2], [3].
The principal source of ECM accumulation and prominent mediators of fibrogenesis are “activated fibroblasts” or myofibroblasts (MFB). Different origins for activated fibroblasts have been proposed: resident fibroblasts, bone marrow-derived fibrocytes, epithelial cells that undergo epithelial-to-mesenchymal transition (EMT), vascular smooth muscle cells and pericytes. However, recent knowledge indicates that most fibrogenic MFB are endogenous to the liver, coming from hepatic stellate cells (HSC) and portal fibroblasts. HSC are considered to be the major source of fibrogenic cells in response to chronic liver injury, while portal fibroblasts play an important role during cholestatic liver diseases [4]. A complex network of autocrine/paracrine fibrogenic signals promotes the activation, usually called transdifferentiation, of quiescent HSC to a myofibroblastic phenotype. This fibrogenic inputs include cytokines, chemokines, growth factors, lipid mediators and reactive oxygen species (ROS) that are produced by epithelial cells (hepatocytes and cholangiocytes), endothelial cells and cells of the immune system (macrophages, dendritic cells, and B and T lymphocytes). Apoptotic bodies derived from damaged hepatocytes can also transdifferentiate HSC to MFB favouring the fibrogenic process [2], [5]. A number of growth factors are pro-fibrotic in the liver, including Platelet-Derived Growth Factor (PDGF), angiotensin II, Connective Tissue Growth Factor (CTGF), and Transforming Growth Factor-beta (TGF-β) [6]. TGF-β has a pivotal role in fibrogenesis, and some of the other growth factors involved exert their effects by directly stimulating TGF-β production [7].
2. Inflammation, oxidative stress and liver fibrosis
Inflammation plays an essential role in the development of liver fibrosis. When a chronic injury takes place, a large infiltration of mononuclear cells, which include macrophages, lymphocytes and eosinophils, occur. Mobilization of lymphocytes produces lymphokines that activate macrophages, which, in turn, stimulate lymphocytes, fibroblasts, and other inflammatory cells, thus setting the stage for persistence of an inflammatory response. Dysregulated inflammatory responses have been associated with most (if not all) hepatotoxic insults, including ischaemia/reperfusion (IR) injuries; alcohol overconsumption; intoxications by xenobiotics or heavy metals; bacterial, viral and parasitic infections; as well as systemic metabolic conditions, such as non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), obesity, diabetes, and the metabolic syndrome [8] (Fig. 1). Macrophages produce pro-fibrotic mediators, including TGF-β and PDGF, and control ECM turnover by regulating the balance of various matrix metalloproteases (MMP) and tissue inhibitors of metalloproteases (TIMP). Examples of knock-out mice that are resistant to fibrosis because they have less inflammation include those with gene deletions of Tumour Necrosis Factor-alpha (TNF-α) or Toll-like receptor 4 (TLR4), among others [9], [10].
Fig. 1.

Different causes of chronic liver injury concur with inflammatory processes and oxidative stress.
ROS are short-lived, highly electrophilic molecules, generated by the partial reduction of oxygen; reactive nitrogen species (RNS) such as nitric oxide (NO) are sometimes included [11]. Different types of ROS have different intrinsic chemical properties, which dictate their reactivity, subcellular localization and preferred biological targets. ROS are critical intermediates in both the normal physiology and pathological conditions of liver cells. Overproduced ROS may directly deplete antioxidant molecules, such as glutathione (GSH) and inhibit the activities of antioxidant enzymes, such as superoxide dismutase (SOD), but they may also induce the expression of antioxidant genes to counteract oxidative stress effects [12]. When the balance between ROS generation and the antioxidant defence of cells is disrupted, it results in oxidative stress. Importantly, it has been recently reported that activated HSC have increased ROS-detoxifying capacity compared to quiescent HSC that protects them from ROS-induced apoptosis and necrosis [13].
High concentrations of ROS induce HSC death, however, non-toxic levels of ROS stimulate the activation, proliferation, and collagen production of HSC [14]. Furthermore, intracellular generation of ROS, through activation of specific signalling pathways, is a critical event for directional migration of HSC/MFB, as well as mesenchymal stem cells (MSCs) [15]. Indeed, in the liver, HSC transdifferentiation was shown to be inhibited by antioxidants [16], [17]. Oxidative stress markers have been detected in the serum and in biopsy samples from liver cirrhosis patients, as well as in experimental liver fibrosis/cirrhosis animals [18], [19]. In liver biopsies, areas of fibrosis were localized to areas with increased 4-hydroxi-2′-nonenal (4-HNE), a marker of lipid peroxidation [20], [21]. ROS production associated to steatosis affects nuclear redox state and induces modifications of nuclear proteins [22]. Furthermore, recent results indicate that hepatic stromal cells promote leucocyte migration through catalytic generation of ROS, which indicates that a clear cross-talk exists among liver inflammation, oxidative stress and fibrogenesis [23]. Indeed, chronic oxidative stress plays a role during both the initial inflammatory phase and its progression to fibrosis.
There are numerous potential sources of ROS within the cell. ROS can be produced as by-products of enzymatic processes, like in the case of mitochondrial ROS. Mitochondria generate ATP in an oxygen-dependent manner during which the flow of electrons down the respiratory chain eventually culminates at complex IV with the reduction of molecular oxygen to water. Throughout this process, superoxide is generated, predominantly at complex I and complex III of the cytochrome chain, when electrons initially derived from NADH or FADH2 react with oxygen. Normal mitochondria provide a low basal level of ROS in most of cells, but this level may increase during aging or mitochondrial damage. Mitochondrial oxidants have been historically viewed as purely toxic, however, recent evidence suggests that they can be also regulators of intracellular signalling pathways [11]. In addition to mitochondria, other redox-active enzymes can generate superoxide and hydrogen peroxide as a by-product. These include xanthine oxidase, cytochrome p450, cyclooxygenase, lipoxygenase, and nitric oxide synthase (NOS) [11], [24]. Moreover, accumulating evidence indicates that NADPH oxidases (NOXs), which play a critical role in the inflammatory response, may contribute to reactive oxygen species (ROS) production during liver fibrosis, being important players in HSC activation and hepatocyte apoptosis [14]. Recent findings also implicate the tumour suppressor p53 as a critical co-factor for several key fibrotic and cell cycle effectors. Increased oxidative stress associated with the fibrotic process is both a likely initiator and an upstream mediator of p53 signalling in the injured tissue [25].
3. NOXs and liver fibrosis
NOXs are a family of enzymes that generate ROS (either superoxide or hydrogen peroxide) as the primary species during the catalytic metabolism of oxygen for a range of host defence and signalling functions. Seven isoforms of NOX are expressed in mammalian cells (NOX1-5, DUOX1 and DUOX2). All NOX isoforms are membrane bound enzymes that rely on NADPH for their activity and the major source of ROS is generated when the flavin- and haem-containing protein complex transfer electrons from cytosolic NADPH to molecular oxygen to produce O2·− or H2O2 [11], [26]. Both parenchymal and non-parenchymal hepatic cells express different members of the NOX family. Hepatocytes and HSC express NOX1, NOX2, NOX4, DUOX1 and DUOX2; endothelial cells express mainly NOX1, NOX2 and NOX4; and Kupffer cells, which are hepatic-resident macrophages, express the phagocyte NOX2 [14], [27].
NOX-derived ROS have been previously related to fibrosis in several organs such as lung [28], pancreas [29], kidney [30], [31] and heart [32]. Recent evidence also suggests a key role for NOX proteins in the progression of hepatic fibrosis [33], [34], [35]. NOX mediates liver fibrogenic responses induced by different agonists [14], as well as phagocytosis of apoptotic bodies [36]. But, of relevance, NOX mediates TGF-β-induced MFB activation in different organs [28], [31], [32], [37], [38], [39]. HSC responds to TGF-β inducing NOX4-derived ROS [40], which play a key role in hepatic MFB activation both in vivo and in vitro [41], [42]. Indeed, TGF-β-induced HSC activation is attenuated either by NOX4 down regulation or in a Nox4−/− genetic background, and, interestingly, the MFB activated state could also be reversed by NOX4 down regulation [42]. Levels of NOX4 are elevated in patients with hepatitis C virus (HCV)-derived liver fibrosis, increasing along the fibrosis degree, as well as in patients with NASH [43]. Hepatocyte specific deletion of NOX4 reduced oxidative stress, lipid peroxidation and liver fibrosis in mice with diet-induced steatohepatitis [43]. All these data strongly suggest the essential role played by NOX4 in the development of liver fibrosis. However, studies performed in Nox1−/−, Nox2−/−, p47phox−/− have demonstrated also the importance of other NOX proteins, concretely NOX1 and NOX2 in fibrogenesis [35], [44], [45]. NOX1 seems to mediate the pro-fibrogenic effects in endogenous liver cells, through PTEN inactivation and positive regulation of the AKT/FOXO4/p27 signalling pathway [34] and it may further contribute to the inflammatory process, promoting cyclooxygenase (COX)-2 expression and prostaglandin synthesis in hepatocytes [46]. NOX2 could be implicated in both endogenous liver cells and bone marrow-derived cells [35], possibly acting in the process of phagocytosis of dead hepatocytes [47] (Fig. 2).
Fig. 2.

Role of NOXs on HSC activation and regulation of hepatocyte apoptosis. Role of TGF-β, growth and inflammatory factors in this process. [See text for further details.]
Hepatocyte apoptosis is another crucial event during fibrogenesis since it triggers Kupffer cells and HSC activation by secreting cytokines, chemokines and microparticles. TGF-β induces hepatocyte apoptosis through ROS that are derived from both mitochondria and NOX activity [48]. In addition, NOX4 also mediates apoptosis induced by other stimuli since NOX4−/− hepatocytes are resistant to apoptosis induced by CD95L and TNF-α/actinomycin D [41]. Hepatocyte apoptosis during fibrosis might be relevant to blunt regeneration and create a pro-fibrogenic microenvironment. In agreement with these results, it has been proposed a role for NOX4 in epithelial cell death during development of bleomycin-induced lung fibrosis. Using a model of NOX4 deficient mice, authors demonstrated that these animals were resistant to fibrosis due to the abrogation of TGF-β-induced apoptosis in epithelial cells [49]. Of note, hepatocytes express not only NOX4 but also other NOX proteins, and they play opposite roles in the control of hepatocyte survival and death. Indeed, whereas NOX4 is necessary to mediate apoptosis induced by TGF-β [50], [51], this pro-apoptotic effect of the cytokine can be attenuated after NOX1 activation by EGF or other growth factors [52], [53]. Phagocytosis of hepatocyte apoptotic bodies by HSC induces their activation to MFB, a process that involves NOX2 activation, thereby contributing to exacerbate the fibrotic response [36], [47] (Fig. 2).
4. Targeting liver fibrosis by hampering ROS/NOX and/or modulating redox-related intracellular signals
Regression of fibrosis can be achieved by the successful control of chronic liver injury, with the consequent termination of the fibrogenic reaction followed by the clearance of hepatic MFB and restoration of full liver function. However, this regression is only possible at early stages, since when advanced fibrosis or cirrhosis is established, all therapeutic approaches seem non-efficient. MFB can be eliminated by apoptosis, senescence or reversion to a quiescent (HSC) phenotype. Indeed, the first line treatment is, when possible, to counteract the underlying liver disease to stop fibrosis progression. For instance, patients with viral hepatitis should be treated with anti-viral therapies while corticosteroids are useful for autoimmune hepatitis. However, inactivated HSC remain primed for re-transdifferentiation, and may be even more responsive to recurrent fibrogenic stimuli than its original quiescent state. In advanced stages of fibrosis and cirrhosis the potential for reversibility declines [2], [5]. MFB and their products are primary targets for antifibrotic therapies, which in principle would address all types of fibrosis, including advanced fibrosis. Nevertheless, additional cellular elements that are either upstream of MFB or tightly linked to fibrogenic activation may provide a basis for complementary and more disease-specific antifibrotic approaches. A combined therapy may be a more effective approach, given the crosstalk between different cell types that generally underlies the fibrogenic activation [2], [3], [5], [54].
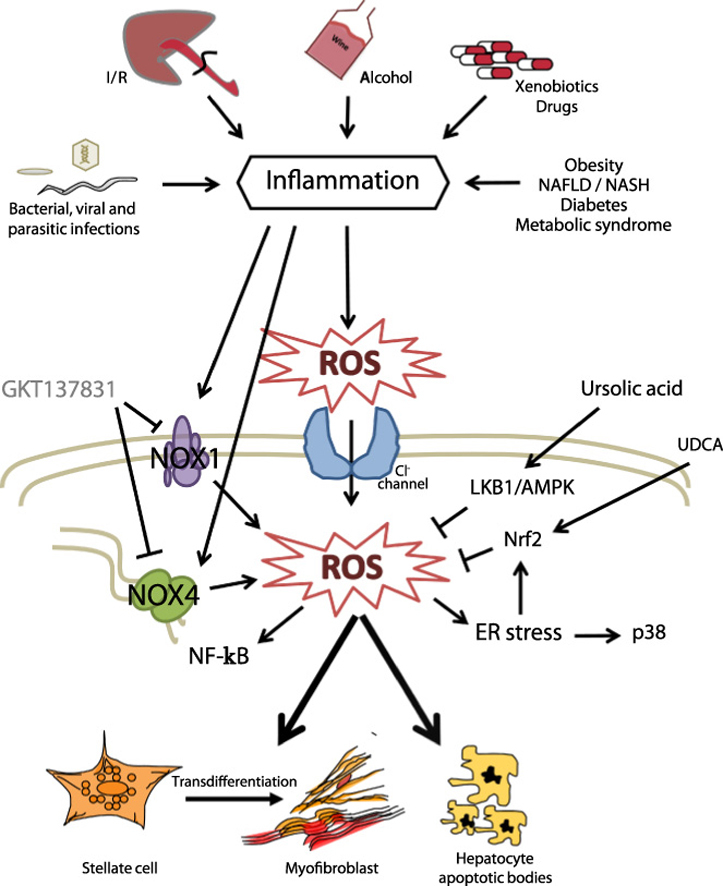
At present, there is no FDA approved drug for liver fibrosis treatment. Based on the knowledge of the pathogenic role of ROS, different strategies to prevent or reverse the oxidative damage are being developed in pre-clinical experiments to be used as therapeutic tools in liver fibrosis. Indeed, natural antioxidants, such as pyrroquinoline–quinone has been demonstrated to suppress oxidative stress and liver fibrogenesis in mice [55]. In the same line of evidence, natural compounds with antioxidant and anti-inflammatory properties have proved to be hepatoprotective in liver of rat with secondary biliary cirrhosis [56]. Mice lacking Methionine Adenosyltransferase 1A (MAT1A), which catalyses the production of S-Adenosylmethionine (AdoMet), a precursor of the glutathione (GSH) synthesis, spontaneously develop oxidative stress and non-alcoholic steatohepatitis [57], emphasizing the relevant role that maintaining GSH levels has in counteracting oxidative stress. In this same line of evidence, recent findings underscore a critical role for mitochondrial reduced glutathione (mGSH) in the therapeutic potential of superoxide scavengers and suggest that the combined approach of these agents with mGSH replenishment may be important in steatohepatitis and liver fibrosis [58]. A recent study has emphasized the role of chloride channels in the activation of HSC, allowing the entry of superoxide anion radicals [59]. Indeed, chloride channels may constitute a potential target for new anti-fibrotic drugs (Fig. 3).
Fig. 3.

Targeting liver fibrosis by hampering ROS/NOX and/or intracellular signals. [See text for further details.]
The experimental use of agents that prevent oxidative stress is contributing to a better understanding about the intracellular pathways that play essential roles in mediating or protecting against the consequences of an intracellular ROS increase in liver cells. Indeed, the use of ROS inhibitors allowed proving that HIV and HCV cooperatively promote hepatic fibrogenesis via induction of ROS and Nuclear Factor kappa-B (NFκB) [60]. Ursodeoxycholic acid (UDCA) plays a cytoprotective effect in primary biliary cirrhosis through activating Nuclear factor Erythroid-2-related transcription factor 2 (Nrf2), which plays a critical role in protecting against oxidative stress [61]. Indeed, UDCA improves circulating redox changes in primary biliary cirrhotic patients [62]. Ursolic acid, via LKB1-AMP-activated protein kinase signalling offers also protective effects on bile duct ligation-induced liver injury in mice, which is related to inhibition of oxidative stress [63]. Blockage of the IREα pathway, the branch of the unfolding protein response (UPR) mainly affected during liver fibrosis, in stellate cells significantly decreased both their activation and autophagic activity in a p38 MAPK-dependent manner, leading to a reduced fibrogenic response [64] (Fig. 3).
Considered the more precise role of NOX-derived ROS in hepatic fibrogenesis, the development of novel pharmacological NOX inhibitors is being assessed as the most promising potential anti-fibrotic therapeutics (Fig. 3). Historical NOX inhibitors (such as apocinin or dipheniliodinium-DPI) are unspecific and not isoform selective. Novel NOX inhibitors stemming from rational drug discovery approaches show improved specificity for NOX and even moderate isoform selectivity [65]. Interestingly, evidences for the role of dual NOX4/NOX1 pharmacological inhibitor GKT137831 in decreasing both the apparition of fibrogenic markers and hepatocyte apoptosis in vivo, upon bile duct ligation and CCl4 treatment, are reported [41], [66]; therefore, it is a promising therapeutic agent for future translational studies (Fig. 3). However, it is important to mention that NOX4 mediates suppressor effects of TGF-β in hepatocytes [50], [67] and recent results indicate that NOX4 would inhibit hepatocyte growth and liver tumorigenesis [68]. Indeed, stable knockdown of NOX4 expression in human liver tumour cells increased their in vitro cell proliferation and conferred them higher in vivo tumorigenic capacity in xenograft experiments in nude mice, resulting in earlier onset of tumour formation and increase in tumour size [68]. In vivo analysis in mice revealed that NOX4 expression was downregulated under physiological proliferative situations of the liver, such as regeneration after partial hepatectomy, as well as during pathological proliferative conditions, such as diethylnitrosamine-induced hepatocarcinogenesis [68]. Considering that liver fibrosis and cirrhosis predispose to the development of HCC, the collateral effects of inhibition of some physiological functions of NOX must be considered in future studies about the clinical safety of these compounds.
5. Conclusions
Current knowledge about the molecular mechanisms of liver fibrosis places inflammation and oxidative stress as one of the main causes for the initiation and progression of this disease. Different agents that cause chronic liver injuries provoke the production of ROS by different mechanisms, among them, NOXs may play an essential role. Different NOXs have been involved in fibrogenic responses, such as HSC activation to MFB or regulation of hepatocyte cell death. The experimental use, both in vitro and in vivo, of agents that prevent oxidative stress is contributing to a better understanding about the intracellular pathways that play essential roles in mediating or protecting against the consequences of an intracellular ROS increase in liver cells. Future expectations are focused on the use of specific NOX inhibitors that prevent HSC activation and protect hepatocyte injury, although further work is necessary to fully confirm the clinical safety of these compounds. However, it cannot be forgotten that liver fibrosis has multiple etiologies and, consequently, multiple mechanisms. Indeed, much further experimental work is necessary for a better understanding of the efficacy of ROS-chelating agents as therapeutic tools in this complex disease.
Acknowledgements
Research in our group is supported by grants from the Ministry of Economy and Competitiveness (MINECO), Spain (BFU2012-35538 and ISCIII-RTICC: RD12-0036-0029) and People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme, Spain (FP7/2007–2013) under REA Grant agreement no. PITN-GA-2012-316549-(IT-LIVER). We are also supported by the European Cooperation in Science and Technology, Belgium (COST Action BM1203/EU‐ROS). E.C.-M. was recipient of a predoctoral contract from the Ministry of Education, Culture and Sport, Spain (MEC) (AP2009-4739).
References
- 1.Xue Z.-F., Wu X.-M., Liu M. Hepatic regeneration and the epithelial to mesenchymal transition. World J. Gastroenterol. 2013;19:1380–1386. doi: 10.3748/wjg.v19.i9.1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mallat A., Lotersztajn S. Cellular mechanisms of tissue fibrosis. 5. Novel insights into liver fibrosis. Am. J. Physiol. Cell Physiol. 2013;305:C789–C799. doi: 10.1152/ajpcell.00230.2013. [DOI] [PubMed] [Google Scholar]
- 3.Schuppan D., Kim Y.O. Evolving therapies for liver fibrosis. J. Clin. Invest. 2013;123:1887–1901. doi: 10.1172/JCI66028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwaisako K., Jiang C., Zhang M., Cong M., Moore-Morris T.J., Park T.J. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E3297–E3305. doi: 10.1073/pnas.1400062111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Czaja A.J. Hepatic inflammation and progressive liver fibrosis in chronic liver disease. World J. Gastroenterol. 2014;20:2515–2532. doi: 10.3748/wjg.v20.i10.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnes J.L., Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int. 2011;79:944–956. doi: 10.1038/ki.2010.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fabregat I., Sancho P. The Transforming Growth Factor-Beta (TGF-β) in liver fibrosis. In: Moustakas A., Miyazawa K., editors. TGF-Β in Human Disease. Springer; 2013. pp. 255–277. [Google Scholar]
- 8.Brenner C., Galluzzi L., Kepp O., Kroemer G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013;59:583–594. doi: 10.1016/j.jhep.2013.03.033. [DOI] [PubMed] [Google Scholar]
- 9.Kitamura K., Nakamoto Y., Akiyama M., Fujii C., Kondo T., Kobayashi K. Pathogenic roles of tumor necrosis factor receptor p55-mediated signals in dimethylnitrosamine-induced murine liver fibrosis. Lab. Investig. J. Tech. Methods Pathol. 2002;82:571–583. doi: 10.1038/labinvest.3780452. [DOI] [PubMed] [Google Scholar]
- 10.Seki E., De Minicis S., Osterreicher C.H., Kluwe J., Osawa Y., Brenner D.A. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 11.Lambeth JD NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 12.Liu W., Baker S.S., Baker R.D., Zhu L. Antioxidant mechanisms in nonalcoholic fatty liver disease. Curr. Drug Targets. 2015 doi: 10.2174/1389450116666150427155342. [DOI] [PubMed] [Google Scholar]
- 13.Dunning S., Ur Rehman A., Tiebosch M.H., Hannivoort R.A., Haijer F.W., Woudenberg J. Glutathione and antioxidant enzymes serve complementary roles in protecting activated hepatic stellate cells against hydrogen peroxide-induced cell death. Biochim. Biophys. Acta. 2013;1832:2027–2034. doi: 10.1016/j.bbadis.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 14.Paik Y.-H., Kim J., Aoyama T., De Minicis S., Bataller R., Brenner D.A. Role of NADPH oxidases in liver fibrosis. Antioxid. Redox Signal. 2013;20:2854–2872. doi: 10.1089/ars.2013.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Novo E., Busletta C., Bonzo L.V., di, Povero D., Paternostro C., Mareschi K. Intracellular reactive oxygen species are required for directional migration of resident and bone marrow-derived hepatic pro-fibrogenic cells. J. Hepatol. 2011;54:964–974. doi: 10.1016/j.jhep.2010.09.022. [DOI] [PubMed] [Google Scholar]
- 16.Foo N.-P., Lin S.-H., Lee Y.-H., Wu M.-J., Wang Y.-J. α-Lipoic acid inhibits liver fibrosis through the attenuation of ROS-triggered signaling in hepatic stellate cells activated by PDGF and TGF-β. Toxicology. 2011;282:39–46. doi: 10.1016/j.tox.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 17.Abhilash P.A., Harikrishnan R., Indira M. Ascorbic acid supplementation down-regulates the alcohol induced oxidative stress, hepatic stellate cell activation, cytotoxicity and mRNA levels of selected fibrotic genes in guinea pigs. Free Radic. Res. 2012;46:204–213. doi: 10.3109/10715762.2011.647691. [DOI] [PubMed] [Google Scholar]
- 18.Yadav D., Hertan H.I., Schweitzer P., Norkus E.P., Pitchumoni C.S. Serum and liver micronutrient antioxidants and serum oxidative stress in patients with chronic hepatitis C. Am. J. Gastroenterol. 2002;97:2634–2639. doi: 10.1111/j.1572-0241.2002.06041.x. [DOI] [PubMed] [Google Scholar]
- 19.Pawlak K., Zolbach K., Borawski J., Mysliwiec M., Kovalchuk O., Chyczewski L. Chronic viral hepatitis C, oxidative stress and the coagulation/fibrinolysis system in haemodialysis patients. Thromb. Res. 2008;123:166–170. doi: 10.1016/j.thromres.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 20.MacDonald G.A., Bridle K.R., Ward P.J., Walker N.I., Houglum K., George D.K. Lipid peroxidation in hepatic steatosis in humans is associated with hepatic fibrosis and occurs predominately in acinar zone 3. J. Gastroenterol. Hepatol. 2001;16:599–606. doi: 10.1046/j.1440-1746.2001.02445.x. [DOI] [PubMed] [Google Scholar]
- 21.Seki S., Kitada T., Sakaguchi H. Clinicopathological significance of oxidative cellular damage in non-alcoholic fatty liver diseases. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2005;33:132–134. doi: 10.1016/j.hepres.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 22.Anavi S., Ni Z., Tirosh O., Fedorova M. Steatosis-induced proteins adducts with lipid peroxidation products and nuclear electrophilic stress in hepatocytes. Redox Biol. 2015;4:158–168. doi: 10.1016/j.redox.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weston C.J., Shepherd E.L., Claridge L.C., Rantakari P., Curbishley S.M., Tomlinson J.W. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J. Clin. Investig. 2015;125:501–520. doi: 10.1172/JCI73722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finkel T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samarakoon R., Dobberfuhl A.D., Cooley C., Overstreet J.M., Patel S., Goldschmeding R. Induction of renal fibrotic genes by TGF-β1 requires EGFR activation, p53 and reactive oxygen species. Cell Signal. 2013;25:2198–2209. doi: 10.1016/j.cellsig.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Holmström K.M., Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014;15:411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- 27.Guichard C., Moreau R., Pessayre D., Epperson T.K., Krause K.-H. NOX family NADPH oxidases in liver and in pancreatic islets: a role in the metabolic syndrome and diabetes? Biochem. Soc. Trans. 2008;36:920–929. doi: 10.1042/BST0360920. [DOI] [PubMed] [Google Scholar]
- 28.Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T.R., Horowitz J.C. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009;15:1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masamune A., Watanabe T., Kikuta K., Satoh K., Shimosegawa T. NADPH oxidase plays a crucial role in the activation of pancreatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;294:G99–108. doi: 10.1152/ajpgi.00272.2007. [DOI] [PubMed] [Google Scholar]
- 30.Sedeek M., Callera G., Montezano A., Gutsol A., Heitz F., Szyndralewiez C. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2010;299:F1348–F1358. doi: 10.1152/ajprenal.00028.2010. [DOI] [PubMed] [Google Scholar]
- 31.Bondi C.D., Manickam N., Lee D.Y., Block K., Gorin Y., Abboud H.E. NAD(P)H oxidase mediates TGF-β1-induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. 2010;21:93–102. doi: 10.1681/ASN.2009020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cucoranu I., Clempus R., Dikalova A., Phelan P.J., Ariyan S., Dikalov S. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 33.De Minicis S., Seki E., Paik Y.-H., Österreicher C.H., Kodama Y., Kluwe J. Role and cellular source of nicotinamide adenine dinucleotide phosphate oxidase in hepatic fibrosis. Hepatology. 2010;52:1420–1430. doi: 10.1002/hep.23804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cui W., Matsuno K., Iwata K., Ibi M., Matsumoto M., Zhang J. NOX1/nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase promotes proliferation of stellate cells and aggravates liver fibrosis induced by bile duct ligation. Hepatology. 2011;54:949–958. doi: 10.1002/hep.24465. [DOI] [PubMed] [Google Scholar]
- 35.Paik Y.-H., Iwaisako K., Seki E., Inokuchi S., Schnabl B., Osterreicher C.H. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology. 2011;53:1730–1741. doi: 10.1002/hep.24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhan S.-S., Jiang J.X., Wu J., Halsted C., Friedman S.L., Zern M.A. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. 2006;43:435–443. doi: 10.1002/hep.21093. [DOI] [PubMed] [Google Scholar]
- 37.Chan E.C., Peshavariya H.M., Liu G.-S., Jiang F., Lim S.-Y., Dusting G.J. Nox4 modulates collagen production stimulated by transforming growth factor β1 in vivo and in vitro. Biochem. Biophys. Res. Commun. 2013;430:918–925. doi: 10.1016/j.bbrc.2012.11.138. [DOI] [PubMed] [Google Scholar]
- 38.Sampson N., Koziel R., Zenzmaier C., Bubendorf L., Plas E., Jansen-Dürr P. ROS signaling by NOX4 drives fibroblast-to-myofibroblast differentiation in the diseased prostatic stroma. Mol. Endocrinol. 2011;25:503–515. doi: 10.1210/me.2010-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang F., Liu G.-S., Dusting G.J., Chan E.C. NADPH oxidase-dependent redox signaling in TGF-β-mediated fibrotic responses. Redox Biol. 2014;2:267–272. doi: 10.1016/j.redox.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Proell V., Carmona-Cuenca I., Murillo M.M., Huber H., Fabregat I., Mikulits W. TGF-beta dependent regulation of oxygen radicals during transdifferentiation of activated hepatic stellate cells to myofibroblastoid cells. Comp. Hepatol. 2007;6:1. doi: 10.1186/1476-5926-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang J.X., Chen X., Serizawa N., Szyndralewiez C., Page P., Schroder K. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831 a novel NOX4/NOX1 inhibitor in vivo. Free Radic. Biol. Med. 2012;53:289–296. doi: 10.1016/j.freeradbiomed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sancho P., Mainez J., Crosas-Molist E., Roncero C., Fernandez-Rodriguez C.M., Pinedo F. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0045285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bettaieb A., Jiang J.X., Sasaki Y., Chao T.-I., Kiss Z., Chen X. Hepatocyte NADPH oxidase 4 regulates stress signaling, fibrosis, and insulin sensitivity during development of steatohepatitis in mice. Gastroenterology. 2015 doi: 10.1053/j.gastro.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aram G., Potter J.J., Liu X., Wang L., Torbenson M.S., Mezey E. Deficiency of nicotinamide adenine dinucleotide phosphate, reduced form oxidase enhances hepatocellular injury but attenuates fibrosis after chronic carbon tetrachloride administration. Hepatology. 2009;49:911–919. doi: 10.1002/hep.22708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang J.X., Venugopal S., Serizawa N., Chen X., Scott F., Li Y. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology. 2010;139:1375–1384. doi: 10.1053/j.gastro.2010.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sancho P., Martín-Sanz P., Fabregat I. Reciprocal regulation of NADPH oxidases and the cyclooxygenase-2 pathway. Free Radic. Biol. Med. 2011;51:1789–1798. doi: 10.1016/j.freeradbiomed.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 47.Jiang J.X., Venugopal S., Serizawa N., Chen X., Scott F., Li Y. NOX2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology. 2010;139 doi: 10.1053/j.gastro.2010.05.074. 1375–84.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herrera B., Murillo M.M., Alvarez-Barrientos A., Beltrán J., Fernández M., Fabregat I. Source of early reactive oxygen species in the apoptosis induced by transforming growth factor-beta in fetal rat hepatocytes. Free Radic. Biol. Med. 2004;36:16–26. doi: 10.1016/j.freeradbiomed.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 49.Carnesecchi S., Deffert C., Donati Y., Basset O., Hinz B., Preynat-Seauve O. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid. Redox Signal. 2011;15:607–619. doi: 10.1089/ars.2010.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carmona-Cuenca I., Roncero C., Sancho P., Caja L., Fausto N., Fernández M. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J. Hepatol. 2008;49:965–976. doi: 10.1016/j.jhep.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 51.Caja L., Sancho P., Bertran E., Iglesias-Serret D., Gil J., Fabregat I. Overactivation of the MEK/ERK pathway in liver tumor cells confers resistance to TGF-β-induced cell death through impairing up-regulation of the NADPH oxidase NOX4. Cancer Res. 2009;69:7595–7602. doi: 10.1158/0008-5472.CAN-09-1482. [DOI] [PubMed] [Google Scholar]
- 52.Sancho P., Bertran E., Caja L., Carmona-Cuenca I., Murillo M.M., Fabregat I. The inhibition of the epidermal growth factor (EGF) pathway enhances TGF-beta-induced apoptosis in rat hepatoma cells through inducing oxidative stress coincident with a change in the expression pattern of the NADPH oxidases (NOX) isoforms. Biochim. Biophys. Acta. 2009;1793:253–263. doi: 10.1016/j.bbamcr.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 53.Sancho P., Fabregat I. The NADPH oxidase inhibitor VAS2870 impairs cell growth and enhances TGF-β-induced apoptosis of liver tumor cells. Biochem. Pharmacol. 2011;81:917–924. doi: 10.1016/j.bcp.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 54.Tsochatzis E.A., Bosch J., Burroughs A.K. Liver cirrhosis. Lancet. 2014;383:1749–1761. doi: 10.1016/S0140-6736(14)60121-5. [DOI] [PubMed] [Google Scholar]
- 55.Jia D., Duan F., Peng P., Sun L., Ruan Y., Gu J. Pyrroloquinoline-quinone suppresses liver fibrogenesis in mice. PloS One. 2015;10:e0121939. doi: 10.1371/journal.pone.0121939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Serviddio G., Bellanti F., Stanca E., Lunetti P., Blonda M., Tamborra R. Silybin exerts antioxidant effects and induces mitochondrial biogenesis in liver of rat with secondary biliary cirrhosis. Free Radic. Biol. Med. 2014;73:117–126. doi: 10.1016/j.freeradbiomed.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 57.Martínez-Chantar M.L., Corrales F.J., Martínez-Cruz L.A., García-Trevijano E.R., Huang Z.-Z., Chen L. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002;16:1292–1294. doi: 10.1096/fj.02-0078fje. [DOI] [PubMed] [Google Scholar]
- 58.Von Montfort C., Matias N., Fernandez A., Fucho R., Conde de la Rosa L., Martinez-Chantar M.L. Mitochondrial GSH determines the toxic or therapeutic potential of superoxide scavenging in steatohepatitis. J. Hepatol. 2012;57:852–859. doi: 10.1016/j.jhep.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Den Hartog G.J.M., Qi S., van Tilburg J.H.O., Koek G.H., Bast A. Superoxide anion radicals activate hepatic stellate cells after entry through chloride channels: a new target in liver fibrosis. Eur. J. Pharmacol. 2014;724:140–144. doi: 10.1016/j.ejphar.2013.12.033. [DOI] [PubMed] [Google Scholar]
- 60.Lin W., Wu G., Li S., Weinberg E.M., Kumthip K., Peng L.F. HIV and HCV cooperatively promote hepatic fibrogenesis via induction of reactive oxygen species and NFkappaB. J. Biol. Chem. 2011;286:2665–2674. doi: 10.1074/jbc.M110.168286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kawata K., Kobayashi Y., Souda K., Kawamura K., Sumiyoshi S., Takahashi Y. Enhanced hepatic Nrf2 activation after ursodeoxycholic acid treatment in patients with primary biliary cirrhosis. Antioxid. Redox Signal. 2010;13:259–268. doi: 10.1089/ars.2009.2903. [DOI] [PubMed] [Google Scholar]
- 62.Grattagliano I., Palmieri V.O., Portincasa P., Minerva F., Palasciano G. Long-term ursodeoxycholate improves circulating redox changes in primary biliary cirrhotic patients. Clin. Biochem. 2011;44:1400–1404. doi: 10.1016/j.clinbiochem.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 63.Yang Y., Zhao Z., Liu Y., Kang X., Zhang H., Meng M. Suppression of oxidative stress and improvement of liver functions in mice by ursolic acid via LKB1-AMP-activated protein kinase signaling. J. Gastroenterol. Hepatol. 2015;30:609–618. doi: 10.1111/jgh.12723. [DOI] [PubMed] [Google Scholar]
- 64.Hernández-Gea V., Hilscher M., Rozenfeld R., Lim M.P., Nieto N., Werner S. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013;59:98–104. doi: 10.1016/j.jhep.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Altenhöfer S., Radermacher K.A., Kleikers P.W.M., Wingler K., Schmidt H.H.H.W. Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid. Redox Signal. 2014 doi: 10.1089/ars.2013.5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aoyama T., Paik Y.-H., Watanabe S., Laleu B., Gaggini F., Fioraso-Cartier L. Nicotinamide adenine dinucleotide phosphate oxidase (NOX) in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatolology. 2012;56:2316–2327. doi: 10.1002/hep.25938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Senturk S., Mumcuoglu M., Gursoy-Yuzugullu O., Cingoz B., Akcali K.C., Ozturk M. Transforming growth factor-beta induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology. 2010;52:966–974. doi: 10.1002/hep.23769. [DOI] [PubMed] [Google Scholar]
- 68.Crosas-Molist E., Bertran E., Sancho P., López-Luque J., Fernando J., Sánchez A. The NADPH oxidase NOX4 inhibits hepatocyte proliferation and liver cancer progression. Free Radic. Biol. Med. 2014;69:338–347. doi: 10.1016/j.freeradbiomed.2014.01.040. [DOI] [PubMed] [Google Scholar]