

Summary
After combining the summary results of SNPs in 106 centrosomal genes from eight published lung cancer GWASs, we found that two potential functional SNPs (rs151606 and rs12212247) in FGFR1OP were significantly associated with lung cancer risk.
Abstract
Centrosome abnormalities are often observed in premalignant lesions and in situ tumors and have been associated with aneuploidy and tumor development. We investigated the associations of 9354 single-nucleotide polymorphisms (SNPs) in 106 centrosomal genes with lung cancer risk by first using the summary data from six published genome-wide association studies (GWASs) of the Transdisciplinary Research in Cancer of the Lung (TRICL) (12 160 cases and 16 838 controls) and then conducted in silico replication in two additional independent lung cancer GWASs of Harvard University (984 cases and 970 controls) and deCODE (1319 cases and 26 380 controls). A total of 44 significant SNPs with false discovery rate (FDR) ≤ 0.05 were mapped to one novel gene FGFR1OP and two previously reported genes (TUBB and BRCA2). After combined the results from TRICL with those from Harvard and deCODE, the most significant association (P combined = 8.032×10−6) was with rs151606 within FGFR1OP. The rs151606 T>G was associated with an increased risk of lung cancer [odds ratio (OR) = 1.10, 95% confidence interval (95% CI) = 1.05–1.14]. Another significant tagSNP rs12212247 T>C (P combined = 9.589×10−6) was associated with a decreased risk of lung cancer (OR = 0.93, 95% CI = 0.90–0.96). Further in silico functional analyzes revealed that rs151606 might affect transcriptional regulation and result in decreased FGFR1OP expression (P trend = 0.022). The findings shed some new light on the role of centrosome abnormalities in the susceptibility to lung carcinogenesis.
Introduction
Lung cancer represents the number one cancer-related mortality worldwide, with more than 1.6 million cases diagnosed and 1.3 million deaths per year (1). In the United States, it is the primary cause of deaths from cancers in both men and women, leading to more deaths than from breast, colorectal and prostate cancers all combined. Although lung cancer is commonly considered a disease caused by environmental exposures, genetic factors also play a role in the etiology (2). Genomic regions at chromosomes 15q25.1 (3,4), 5p15.33 (4–6), 6p21.33 (4), 3q28 (4,7,8), 13q13.1 (8) and 22q12.1 (8) have been identified to be associated with lung cancer susceptibility by genome-wide association studies (GWASs) in populations of European descent.
Centrosomes are the pivotal regulating element of cell division and act as a reaction center where the cell cycle key regulating elements assemble and play an important role in cell polarization and ciliogenesis (9,10). Recent studies have revealed that deregulation of centrosome-related proteins induce centrosome abnormalities (11), which in turn result in tumor development through the induction of chromosome instability and aneuploidy (12,13). A growing body of evidence has linked centrosome abnormalities to the developments of solid tumors and hematopoietic malignancies (10), and the underlying mechanisms are now an active area of research (14–16).
Centrosome abnormalities and chromosome instability are also frequently observed in human lung cancer (17). Genes involved in centrosome dysregulation have emerged as the candidate targets to study the mechanism of initiation and progression of lung cancer. In this study, we hypothesized that genetic variants of centrosomal genes are associated with lung cancer risk. To test this hypothesis, we performed a meta-analysis of the association results of SNPs in centrosomal genes from eight lung cancer GWASs from Transdisciplinary Research in Cancer of the Lung (TRICL) and ILCCO, including 14 463 cases and 44 188 controls.
Materials and methods
Study populations
The study populations have been detailed previously (8,18). Briefly, this study included six previously reported lung cancer GWASs of from the TRICL, which was built upon the collaborative network of the International Lung Cancer Consortium (ILCCO) (8,18) including 12 160 lung cancer cases and 16 838 controls of European descent: the MD Anderson Cancer Center (MDACC) GWAS, the Institute of Cancer Research (ICR) GWAS, the National Cancer Institute (NCI) GWAS, the International Agency for Research on Cancer (IARC) GWAS, Toronto study from Lunenfeld-Tanenbaum Research Institute study (Toronto) GWAS and the German Lung Cancer Study (GLC) GWAS, Germany. The additional datasets of another two independent GWASs of Caucasian populations were from Harvard Lung Cancer Study (19) (984 cases and 970 controls) and Icelandic Lung Cancer Study (deCODE) (1319 cases and 26 380 controls) (20) from the ILCCO. A written informed consent was obtained from each participant, and this study was approved by the institutional review boards for each of the participating institutions.
Genotyping platforms and quality controls
For these GWASs, genotyping was performed using Illumina HumanHap 317, 317+240S, 370Duo, 550, 610 or 1M arrays. Imputation was performed by using data from the 1000 Genomes Project (phase I integrated release 3, March 2012) (21) as reference and IMPUTE2 v2.1.1 (22), MaCH v1.0 (23) or minimac (version 2012.10.3) (24) software. Poorly imputed SNPs defined as an information score < 0.40 with IMPUTE2 or an r 2 < 0.30 with MaCH were excluded from the final analyses. Standard quality control on samples was performed on all scans, excluding individuals with low call rate (< 90%) and extremely high or low heterozygosity (P < 1.0×10−4), as well as all individuals evaluated to be of non-European ancestry (using the HapMap phase II CEU, JPT/CHB and YRI populations as a reference).
Genes and SNPs selection
The centrosomal genes were collected from the Molecular Signatures Database (http://software.broadinstitute.org/gsea/msigdb), which is a collection of annotated gene sets for use with gene set enrichment analysis. Overall, 106 centrosomal genes located on autosomal chromosomes were selected (Supplementary Table 1, available at Carcinogenesis Online). The genotyped or imputed common SNPs located within 2 kB up- and downstream of centrosomal genes were extracted from the GWAS datasets and the final meta-analysis included 9354 SNPs with genotyping call rate ≥90%, minor allele frequency ≥5%, and Hardy Weinberg Equilibrium exact P ≥ 10–5. As previously reported (8,25), SNPs from the 1000 Genomes project release Phase 1 integrated release version 3 and filtered with only SNPs showing imputation accuracy > 0.3 were retained. The detailed workflow is shown in Supplementary Figure 1, available at Carcinogenesis Online.
In silico functional validation
To predict the potential functions of risk-associated SNPs and their high-linkage disequilibrium (LD) (r 2 ≥ 0.60) variants, we used three in silico tools: SNPinfo (26), RegulomeDB (27) and F-SNP (28). Expression quantitative trait loci (eQTL) analysis was carried out using lymphoblastoid cell data from 1000 Genomes Project (21) European subpopulation (EUR, 373 individuals) (phase I integrated release 3, March 2012).
Statistical methods
For each GWAS, ORs and associated 95% CIs were calculated by unconditional logistic regression using R (v2.6), Stata (v10, State College, Texas, USA) and PLINK (v1.06) (29) software. With the inverse variance method, meta-analysis under fixed and random-effects models was conducted on the results of log-additive model of 9354 SNPs. Cochran’s Q statistic to test for heterogeneity and the I 2 statistic to quantify the proportion of the total variation due to heterogeneity were calculated (30). Fixed effects model was applied when there was no heterogeneity among GWASs (Q-test P > 0.100 and I 2 < 50%); otherwise, random effects model was applied. The false discovery rate (FDR) procedure was employed to control for multiple testing (31) with FDR ≤ 0.050. The correlation between SNPs and corresponding mRNA expression levels was estimated by using a linear regression model.
LocusZoom (32) was applied to generate regional association plots, using the 1000 Genomes European (EUR) reference data (phase I integrated release 3, March 2012) to compute LD. Haploview v4.2 (33) was employed to construct the LD plots. SNPs whose r 2 in linkage with any of the causal SNPs is less than 0.60 are also considered independent. All analyses were conducted with SAS (version 9.1.3; SAS Institute, Cary, NC, USA) unless specified otherwise.
Results
Meta-analysis of the main effects of SNPs in 106 centrosomal genes
The samples sizes of the used GWASs in this study are summarized in Table 1. In this study, we firstly carried out the analysis within the six TRICL GWASs, which included 12 160 lung cancer case subjects and 16 838 unaffected controls. After stringent quality control, we analyzed 9354 SNPs from 106 centrosomal genes (Figure 1). There are 843 SNPs and 96 SNPs with nominal P < 0.050 and < 0.001 under an additive (per allele) model, respectively; 44 SNPs remained significant at the FDR threshold ≤ 0.050. These top SNPs are mapped to TUBB, FGFR1OP and BRCA2, respectively (Table 2). We removed SNPs that had a high pairwise LD with those in previously reported GWASs (4,8,34) (TUBB at 6p21.33 and BRCA2 at 13q13.1), and only SNPs mapped to FGFR1OP were included for the additional analyses. The regional association plots demonstrated that SNP rs12212247 showed high LD (r 2 ≥ 0.60) with other 31 SNPs in FGFR1OP, except for rs151606 (Figure 2; Supplementary Table 2, available at Carcinogenesis Online). Finally, two tagSNPs (rs151606 and rs12212247) in FGFR1OP were selected for the further analysis. As shown in Table 3, both SNPs had a high imputation quality in each dataset.
Table 1.
Characteristics of the study populations included in the participating genome-wide association studies
| Variable | ICRa | MDACCb | IARCc | NCId | Torontoe | GLCf | Harvardg | deCODEh | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case | Control | Case | Control | Case | Control | Case | Control | Case | Control | Case | Control | Case | Control | Case | Control | |
| Overall | 1952 | 5200 | 1150 | 1134 | 2533 | 3791 | 5713 | 5736 | 331 | 499 | 481 | 478 | 984 | 970 | 1319 | 26380 |
| AD | 465 | 5200 | 619 | 1134 | 517 | 2824 | 1841 | 5736 | 90 | 499 | 186 | 478 | 597 | 970 | 547 | 26380 |
| SQ | 611 | 5200 | 306 | 1134 | 911 | 2968 | 1447 | 5736 | 50 | 499 | 97 | 478 | 216 | 970 | 259 | 26380 |
AD, adenocarcinoma; SQ, squamous cell carcinoma.
aICR: the Institute of Cancer Research Genome-wide Association Study, UK.
bMDACC: the MD Anderson Cancer Center Genome-wide Association Study, USA.
cIARC: the International Agency for Research on Cancer Genome-wide Association Study, France.
dNCI: the National Cancer Institute Genome-wide Association Study, USA.
eToronto: the Lunenfeld-Tanenbaum Research Institute Genome-wide Association Study, Toronto, Canada.
fGLC: German Lung Cancer Study, Germany.
gHarvard: Harvard Lung Cancer Study, USA.
hdeCODE: Icelandic Lung Cancer Study, Iceland.
Figure 1.

Manhattan plot of genome-wide association results. SNPs are plotted on the x-axis according to their positions on each chromosome. On the y-axis, the association P values with lung cancer risk are shown (as –log10 P values). Horizontal red line represents FDR threshold 0.05. Horizontal blue line represents nominal P values of 0.05.
Table 2.
Associations between SNPs in centrosomal genes and lung cancer risk with FDR P ≤ 0.050
| SNP | Gene | Chr | Position (hg19) | Allele a | EAF | Q b | I 2 | Effects c | OR (95%CI) | P | FDR |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs115897636 | TUBB | 6 | 30689001 | T/A | 0.12 | 0.869 | 0 | ++++++ | 1.17 (1.11–1.24) | 4.23E−09 | <.0001 |
| rs114884831 | TUBB | 6 | 30687614 | G/C | 0.11 | 0.725 | 0 | ++++++ | 1.18 (1.12–1.24) | 4.98E−09 | <.0001 |
| rs115605279 | TUBB | 6 | 30688575 | G/C | 0.11 | 0.724 | 0 | ++++++ | 1.18 (1.12–1.24) | 5.05E−09 | <.0001 |
| rs116812176 | TUBB | 6 | 30693816 | A/G | 0.20 | 0.456 | 0 | ++++++ | 1.12 (1.07–1.17) | 3.60E−07 | 0.001 |
| rs114380302 | TUBB | 6 | 30692965 | G/A | 0.20 | 0.448 | 0 | ++++++ | 1.12 (1.07–1.17) | 3.78E−07 | 0.001 |
| rs116338781 | TUBB | 6 | 30688427 | G/T | 0.20 | 0.453 | 0 | ++++++ | 1.12 (1.07–1.17) | 4.88E−07 | 0.001 |
| rs114460911 | TUBB | 6 | 30693633 | A/G | 0.20 | 0.522 | 0 | ++++++ | 1.11 (1.07–1.16) | 6.09E−07 | 0.001 |
| rs151606 | FGFR1OP | 6 | 167430482 | A/T | 0.34 | 0.552 | 0 | ++++++ | 1.12 (1.07–1.17) | 1.18E−06 | 0.001 |
| rs114075700 | TUBB | 6 | 30694374 | T/C | 0.18 | 0.485 | 0 | ++++++ | 1.11 (1.07–1.17) | 2.01E−06 | 0.002 |
| rs115969492 | TUBB | 6 | 30693121 | A/G | 0.33 | 0.780 | 0 | ++++++ | 1.10 (1.05–1.15) | 9.78E−06 | 0.009 |
| rs239935 | FGFR1OP | 6 | 167411788 | A/G | 0.48 | 0.309 | 16 | ++++++ | 1.07 (1.04–1.11) | 5.03E−05 | 0.025 |
| rs400837 | FGFR1OP | 6 | 167411008 | C/T | 0.47 | 0.338 | 12 | ++++++ | 1.07 (1.04–1.11) | 6.28E−05 | 0.025 |
| rs424185 | FGFR1OP | 6 | 167410907 | T/C | 0.47 | 0.340 | 12 | ++++++ | 1.07 (1.04–1.11) | 6.78E−05 | 0.025 |
| rs376097 | FGFR1OP | 6 | 167410878 | G/A | 0.47 | 0.340 | 12 | ++++++ | 1.07 (1.04–1.11) | 6.78E−05 | 0.025 |
| rs239934 | FGFR1OP | 6 | 167412048 | A/G | 0.49 | 0.379 | 6 | ++++++ | 1.07 (1.04–1.11) | 7.24E−05 | 0.025 |
| rs1322077 | FGFR1OP | 6 | 167424293 | T/C | 0.46 | 0.334 | 13 | ------ | 0.93 (0.90–0.97) | 7.35E−05 | 0.025 |
| rs9457251 | FGFR1OP | 6 | 167426707 | T/C | 0.46 | 0.320 | 15 | ------ | 0.93 (0.90–0.97) | 7.54E−05 | 0.025 |
| rs9457249 | FGFR1OP | 6 | 167426438 | A/G | 0.46 | 0.320 | 15 | ------ | 0.93 (0.90–0.97) | 7.58E−05 | 0.025 |
| rs9459836 | FGFR1OP | 6 | 167419105 | G/A | 0.46 | 0.321 | 15 | ------ | 0.93 (0.90–0.97) | 8.56E−05 | 0.025 |
| rs13195812 | FGFR1OP | 6 | 167443902 | C/T | 0.45 | 0.317 | 15 | ------ | 0.93 (0.90–0.97) | 8.59E−05 | 0.025 |
| rs9459849 | FGFR1OP | 6 | 167444160 | G/T | 0.45 | 0.313 | 16 | ------ | 0.93 (0.90–0.97) | 8.69E−05 | 0.025 |
| rs9459841 | FGFR1OP | 6 | 167431949 | G/A | 0.45 | 0.315 | 15 | ------ | 0.93 (0.90–0.97) | 8.77E−05 | 0.025 |
| rs9459840 | FGFR1OP | 6 | 167431910 | G/A | 0.45 | 0.315 | 15 | ------ | 0.93 (0.90–0.97) | 8.77E−05 | 0.025 |
| rs4710171 | FGFR1OP | 6 | 167430186 | A/G | 0.45 | 0.315 | 15 | ------ | 0.93 (0.90–0.97) | 8.79E−05 | 0.025 |
| rs1060404 | FGFR1OP | 6 | 167429467 | A/G | 0.45 | 0.315 | 15 | ------ | 0.93 (0.90–0.97) | 8.80E−05 | 0.025 |
| rs12212247 | FGFR1OP | 6 | 167413539 | T/C | 0.46 | 0.329 | 13 | ------ | 0.93 (0.90–0.97) | 8.93E−05 | 0.025 |
| rs2237272 | FGFR1OP | 6 | 167443443 | C/T | 0.45 | 0.313 | 16 | ------ | 0.93 (0.90–0.97) | 9.17E−05 | 0.025 |
| rs6904946 | FGFR1OP | 6 | 167433948 | T/C | 0.45 | 0.310 | 16 | ------ | 0.93 (0.90–0.97) | 9.19E−05 | 0.025 |
| rs10484531 | FGFR1OP | 6 | 167454434 | G/A | 0.45 | 0.311 | 16 | ------ | 0.93 (0.90–0.97) | 9.21E−05 | 0.025 |
| rs9457256 | FGFR1OP | 6 | 167436461 | T/C | 0.45 | 0.307 | 17 | ------ | 0.93 (0.90–0.97) | 9.26E−05 | 0.025 |
| rs3752520 | FGFR1OP | 6 | 167436159 | T/C | 0.45 | 0.307 | 17 | ------ | 0.93 (0.90–0.97) | 9.26E−05 | 0.025 |
| rs9457252 | FGFR1OP | 6 | 167433925 | G/A | 0.45 | 0.307 | 17 | ------ | 0.93 (0.90–0.97) | 9.27E−05 | 0.025 |
| rs6929466 | FGFR1OP | 6 | 167422922 | C/T | 0.45 | 0.302 | 17 | ------ | 0.93 (0.90–0.97) | 9.53E−05 | 0.025 |
| rs9295385 | FGFR1OP | 6 | 167448181 | A/G | 0.45 | 0.310 | 16 | ------ | 0.93 (0.90–0.97) | 9.63E−05 | 0.025 |
| rs10455982 | FGFR1OP | 6 | 167448728 | C/T | 0.45 | 0.307 | 17 | ------ | 0.93 (0.90–0.97) | 9.74E−05 | 0.025 |
| rs10946204 | FGFR1OP | 6 | 167451129 | T/C | 0.45 | 0.308 | 16 | ------ | 0.93 (0.90–0.97) | 9.75E−05 | 0.025 |
| rs10946203 | FGFR1OP | 6 | 167446735 | A/C | 0.45 | 0.297 | 18 | ------ | 0.93 (0.90–0.97) | 9.88E−05 | 0.025 |
| rs9534269 | BRCA2 | 13 | 32939286 | T/G | 0.26 | 0.252 | 24 | ----+- | 0.93 (0.89–0.96) | 1.03E−04 | 0.025 |
| rs6900701 | FGFR1OP | 6 | 167434112 | A/G | 0.45 | 0.331 | 13 | ------ | 0.93 (0.90–0.97) | 1.09E−04 | 0.026 |
| rs2237276 | FGFR1OP | 6 | 167442115 | C/T | 0.45 | 0.299 | 18 | ------ | 0.93 (0.90–0.97) | 1.14E−04 | 0.026 |
| rs9457254 | FGFR1OP | 6 | 167434135 | G/A | 0.45 | 0.330 | 13 | ------ | 0.93 (0.90–0.97) | 1.14E−04 | 0.026 |
| rs9457259 | FGFR1OP | 6 | 167444281 | C/T | 0.45 | 0.323 | 14 | ------ | 0.93 (0.90–0.97) | 1.38E−04 | 0.031 |
| rs9943876 | BRCA2 | 13 | 32927894 | C/T | 0.30 | 0.438 | 0 | ----+- | 0.93 (0.90–0.97) | 1.48E−04 | 0.032 |
| rs2237275 | FGFR1OP | 6 | 167442994 | C/A | 0.44 | 0.308 | 16 | ------ | 0.93 (0.90–0.97) | 1.70E−04 | 0.036 |
Chr, chromosome; EAF, effect allele frequency; FDR, false discovery rate; OR, odds ratio; SNP, single nucleotide polymorphism.
aReference allele/effect allele.
bFixed effect models were used when no heterogeneity was found between studies (Q test P > 0.10 and I 2 < 50.0%); otherwise, random effect models were used.
c‘+’ means positive association and ‘-’ means negative association.
Figure 2.
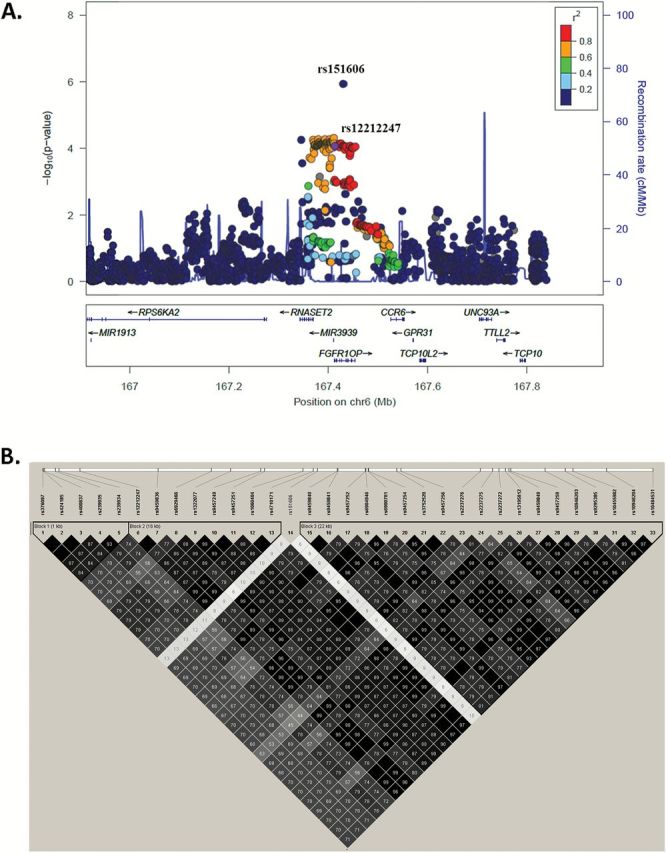
Regional association plots. Data points are colored according to their level of LD of the each SNP with the tagging SNPs. (A) SNPs in the region 500kb up- or downstream of the marker SNP and (B) SNPs with FDR ≤ 0.050. In A, the left-hand y-axis shows the association P value of individual SNPs, which is plotted as −log10 (P) against chromosomal base pair position; the right-hand y-axis shows the recombination rate estimated from the hg19/1000 Genomes European population.
Table 3.
Summary of the association results of two SNPs in the eight lung cancer GWASs
| Study population | Sample size | Imputation quality | rs151606 A > T | Imputation quality | rs12212247 T > C | |||
|---|---|---|---|---|---|---|---|---|
| Cases | Controls | OR (95% CI) | P | OR (95% CI) | P | |||
| TRICL combineda | 12 160 | 16 838 | 1.12 (1.07–1.17) | 1.18E−06 | 0.93 (0.90–0.97) | 8.93E−05 | ||
| ICRb | 1952 | 5200 | 0.74 | 1.19 (1.09–1.30) | 1.24E−04 | 0.99 | 0.93 (0.86–1.00) | 4.44E−02 |
| MDACCc | 1150 | 1134 | 0.45 | 1.10 (0.92–1.32) | 3.10E−01 | 0.96 | 0.95 (0.84–1.07) | 3.94E−01 |
| IARCd | 2533 | 3791 | 0.44 | 1.04 (0.93–1.17) | 5.23E−01 | 0.98 | 0.97 (0.90–1.04) | 3.52E−01 |
| NCIe | 5713 | 5736 | 0.72 | 1.10 (1.03–1.18) | 3.28E−03 | 0.99 | 0.94 (0.89–0.99) | 1.88E−02 |
| Torontof | 331 | 499 | 0.70 | 1.19 (0.91–1.57) | 2.06E−01 | 0.99 | 0.82 (0.66–1.02) | 7.60E−02 |
| GLCg | 481 | 478 | 0.52 | 1.16 (0.89–1.50) | 2.78E−01 | 0.98 | 0.78 (0.65–0.94) | 9.02E−03 |
| Replication combineda | 2303 | 27 350 | 1.02 (0.93–1.11) | 7.19E−01 | 0.93 (0.87–1.00) | 3.90E−02 | ||
| Harvardh | 984 | 970 | 0.75 | 1.15 (0.95–1.39) | 1.50E−01 | 1.00 | 0.88 (0.76–1.01) | 5.91E−02 |
| DeCODEi | 1319 | 26 380 | 0.75 | 0.98 (0.89–1.08) | 7.50E−01 | 1.00 | 0.95 (0.88–1.03) | 1.96E−01 |
| All combineda | 14 463 | 44 188 | 1.10 (1.05–1.14) | 8.03E−06 | 0.93 (0.90–0.96) | 9.59E−06 | ||
GWAS, genome-wide association study; SNP, single-nucleotide polymorphism.
aThe combined OR and P value were estimated using a fixed effects model.
bICR: the Institute of Cancer Research Genome-wide Association Study, UK.
cMDACC: the MD Anderson Cancer Center Genome-wide Association Study, USA.
dIARC: the International Agency for Research on Cancer Genome-wide Association Study, France;
eNCI: the National Cancer Institute Genome-wide Association Study, USA.
fToronto: the Lunenfeld-Tanenbaum Research Institute Genome-wide Association Study, Toronto, Canada.
gGLC: German Lung Cancer Study, Germany.
hHarvard: Harvard Lung Cancer Study, USA.
ideCODE: Icelandic Lung Cancer Study, Iceland.
Of these two SNPs, rs151606 A>T was shown to be associated with an increased risk of lung cancer (OR = 1.12, 95% CI = 1.07–1.17, P = 1.177×10–6), while rs12212247 T>C was associated with a decreased risk (OR = 0.93, 95% CI = 0.90–0.97, P = 8.926×10–5). There was no heterogeneity observed for these risk estimates in these six GWASs (Figure 3).
Figure 3.

Forest plots of effect size and direction for tagSNPs with all cases from TRICL consortium. FGFR1OP rs151606 in all cases, P combined = 8.032×10−6 (A); FGFR1OP rs151606 in adenocarcinoma cases, P combined = 0.058 (B); FGFR1OP rs151606 in squamous cell carcinoma cases, P combined = 0.011 (C); FGFR1OP rs12212247 in all cases, P combined = 9.589×10−6 (D); FGFR1OP rs12212247 in adenocarcinoma cases, P combined = 0.007 (E); FGFR1OP rs12212247 in squamous cell carcinoma cases, P combined = 0.043 (F). Each box and horizontal line represent the OR point estimate and 95% CI derived from the additive model. The area of each box is proportional to the statistical weight of the study. Diamonds represent the summary ORs obtained from the combined analysis with 95% confidence intervals indicated by their widths. The meta-analysis includes eight GWASs [the Institute of Cancer Research (ICR) GWAS, the MD Anderson Cancer Center (MDACC) GWAS, the International Agency for Research on Cancer (IARC) GWAS, the National Cancer Institute (NCI) GWAS, the Lunenfeld-Tanenbaum Research Institute (Toronto) GWAS, German Lung Cancer Study (GLC), Harvard lung cancer study (Harvard) and Icelandic Lung Cancer Study (deCODE)].
We sought to expand these findings in another two independent lung cancer GWASs from Harvard University (984 cases and 970 controls) and deCODE (1319 cases and 26 380 controls). When the two studies were combined, rs12212247 and its 31 high LD SNPs within FGFR1OP were found with the same effect direction with a nominal P = 0.039. In addition, the protective effect of SNP rs151606 was insignificant (Table 3; Supplementary Table 3, available at Carcinogenesis Online). However, when we combined the summary results of all the eight studies, we observed a significant evidence for the association of rs151606 and lung cancer risk (OR = 1.10, 95% CI = 1.05–1.14, P combined = 8.032×10−6) and rs12212247 (OR = 0.93, 95% CI = 0.90–0.96, P combined = 9.589×10−6) (Table 3; Figure 3), given only 9354 SNPs were tested for their associations with lung cancer risk in the present study.
Stratified analyses
Like many other cancers, lung cancer histologic subtypes can have dramatically different clinical behaviors. In the further analyses stratified by histology, only adenocarcinoma (AD) and squamous cell carcinoma (SQ) were included. SNP rs151606 was associated only with the risk of SQ (OR = 1.09, 95% CI = 1.02–1.16, P combined = 0.011), but not with AD (OR = 1.06, 95% CI = 1.00–1.13, P combined = 0.058); the strongest effect of rs12212247 was detected in AD (OR = 0.94, 95%CI = 0.89–0.98, P combined = 0.007), and then in SQ (OR = 0.95, 95%CI = 0.90–1.00, P combined = 0.043) (Figure 3).
In silico functional validation
In silico eQTL analysis was performed by using the lymphoblastoid cell lines from 373 individuals of European descendent. The rs151606 genotypes demonstrated significant association with decreased mRNA expression of FGFR1OP in both additive (P trend = 0.022) and recessive models (P trend = 0.031) (Figure 4). According to the prediction results of the online tool F-SNP, SNP rs151606 might be associated with mRNA expression by influencing the binding activity of transcriptional factors (i.e. HNF-3b, XFD-2). Although rs12212247 might have an effect on the activity of transcription factor binding site (TFBS) as predicted by SNPinfo and RegulomeDB score of 2b (Supplementary Table 4, available at Carcinogenesis Online), which indicated that transcription factor TAF1 binding might be influenced according to the Encyclopedia of DNA Elements (ENCODE) ChIP-Seq data from human A549 cells (35), we did not find any significant association of this SNP with FGFR1OP mRNA expression levels in either model (Figure 4).
Figure 4.
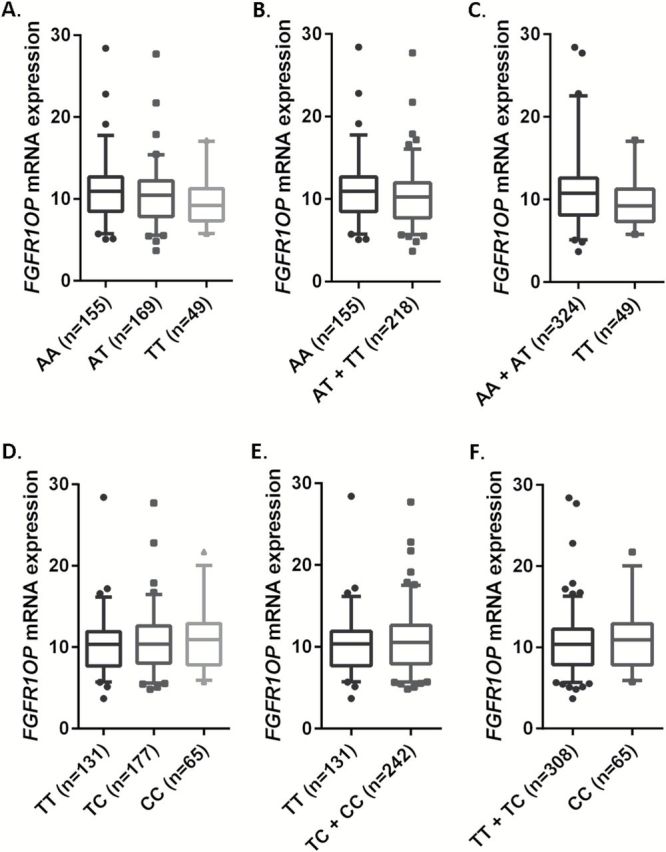
The correlations between identified SNPs and FGFR1OP mRNA expression. rs151606 (A, additive model, P = 0.022; B, dominant model, P = 0.088; C, recessive model, P = 0.031) and rs12212247 (D, additive model, P = 0.344; E, dominant model, P = 0.465; F, recessive model, P = 0.405).
Discussion
The centrosome is the crucial microtubule-organizing center in animal cells and comprises of a pair of centrioles surrounded by the pericentriolar material (36). Centrosome deficiencies, including the occurrence of extra centrioles, and increased ability to nucleate microtubules, are common in a high percentage of many tumors. While centrosome deficiencies in cancer may arise as a result of tumorigenesis, early centrosome aberrations may also lead to increased risk of malignancy (37). Our study firstly attempted to systematically examine the effects of genetic variants in the centrosomal genes on lung cancer susceptibility. Notably, we found two potentially functional SNPs in FGFR1OP to be associated with lung cancer risk in the final meta-analysis of 14 463 lung cancer patients and 44 188 control subjects. Our current study is the first to provide an important insight into the association of variants in 6q27 and carcinogenesis, suggesting that FGFR1OP (rs151606 and rs12212247) could represent a novel lung cancer locus in Caucasian populations.
FGFR1OP (fibroblast growth factor receptor 1 oncogene partner) is a centriolar satellite cargo protein, by which centriolar satellites play the role of key mediators of centrosome functions (38). It has been reported that FGFR1OP is involved in G1/S transition and thus necessary for cell-cycle progression and survival (39). Two chromosomal translocations, fusing this gene with the FGFR1 and RET genes, have been found in hematopoietic cancers (40,41). Published GWASs have identified several SNPs in the nearby region (6q27) of FGFR1OP to be associated with the risk of multiple autoimmune diseases, including Crohn’s disease (rs2301436 in FGFR1OP) (42), Graves’ disease (rs9355610 near RNASET2) (43), rheumatoid arthritis (rs3093024 and rs3093023 near CCR6) (44,45) and vitiligo (rs2236313 in RNASET2 and rs6902119 near CCR6) (46,47). Recent studies have also suggested a link between autoimmune diseases and some cancers (48–50). However, little is known about the association between the genetic variants in FGFR1OP and cancer risk. In this study, we observed that FGFR1OP rs151606 T allele was associated with decreased levels of mRNA expression and increased risk of lung cancer. These results indicate that the association between SNP rs151606 and lung cancer risk might due to its effect on the expression of FGFR1OP, as suggested the eQTL evidence for a role of FGFR1OP in the assembling of mitotic spindle (10,13,51). However, the exact molecular mechanism underlying the associations among lower expression of FGFR1OP on spindle multipolarity, centrosome amplification and lung cancer risk warrants additional investigation.
Except for dysregulation of gene expressions, exposure to environmental risk factors has also been shown to induce centrosome abnormality (52). Several studies have reported that exposure to chrysotile asbestos fibers, chromate particles, arsenite, and benzo[a]pyrene diol epoxide, which is a carcinogen present in tobacco smoke as well as in environmental pollution, might cause centrosome abnormalities and multipolar spindles in human lung cells (53–57). Early occurrence of centrosome alterations have been found in the uninvolved lung tissues adjacent to the tumor of NSCLCs, representing an early event in pulmonary carcinogenesis by increasing chromosome instability (58,59). Based on these previous reports and our current findings, analysis of an interaction between SNPs and main environmental risk factors (i.e. smoking status) in future studies might provide novel evidence for the effects of genetic variants in centrosomal genes on lung cancer risk.
Previous studies have identified several genetic variants in centrosomal genes (NIN, TUBG1 and APC) that contributed to breast (60) or pancreatic (61) cancer susceptibility among the Caucasian populations. However, in the present study, we did not find any significance for those SNPs but identified four other SNPs within NIN (rs67977855, rs45558232, rs59096640 and rs4534750) showing moderate associations with lung cancer risk (P ≤ 0.05) based on the six TRICL GWASs results (data not shown). Such inconsistence might due to the heterogeneity among cancers or populations.
There are some potential limitations of the present study. Firstly, the gene-set selection mainly depended on the quality and integrity of curated biological pathways. As the biological understanding of the function of diverse genes in pathways has been steadily improved, the emerging new data on functions of these genes will inevitably expand the currently available lists of ‘canonical’ pathways. Some important centrosomal genes might be omitted in this study. Considering this, we comprehensively considered multiple centrosomal gene sets and managed to make an all-inclusive listing as possible as we could. Secondly, defining whether a particular SNP is of biological function was limited by the applied in silico tools. To overcome this limitation, we combined the prediction results of multiple bioinformatics tools to explain our data. Thirdly, our eQTL analyses were limited to lymphoblastoid cell lines with publically available data, and gene expression analysis in tissues will give biologically plausible and more convincing results of the effect of those two identified SNPs.
In summary, the present study found significant associations between SNPs in FGFR1OP and lung cancer risk. The results suggested that the SNPs within the centrosomal genes may serve as potential markers to predict lung cancer risk in European populations. Our discoveries shed some new light on the role of centrosome abnormalities in human carcinogenesis. Further validation and functional evaluation of these genetic variants are needed to verify our findings.
URLs
Transdisciplinary Research In Cancer of the Lung (TRICL), http://u19tricl.org/; Genetic Associations and MEchanisms in ONcology (GAME-ON) consortium, http://epi.grants.cancer.gov/gameon/; International Lung Cancer Consortium (ILCCO), http://ilcco.iarc.fr/; 1000 Genomes Project, http://www.1000genomes.org/; IMPUTE2, http://mathgen.stats.ox.ac.uk/impute/impute_v2.html; MaCH, http://www.sph.umich.edu/csg/abecasis/MACH/; Minimac, http://genome.sph.umich.edu/wiki/Minimac/; MsigDB, http://www.broadinstitute.org/gsea/msigdb/index.jsp; LocusZoom, http://csg.sph.umich.edu/locuszoom/; RegulomeDB, http://regulome.stanford.edu/; SNPinfo, http://snpinfo.niehs.nih.gov/snpinfo/snpfunc.htm; F-SNP, http://compbio.cs.queensu.ca/F-SNP/.
Supplementary material
Supplementary Tables 1–4 and Figure 1 can be found at http://carcin.oxfordjournals.org/
Funding
As Duke Cancer Institute members, Q.W., M.W.O., and K.O. acknowledge support from the Duke Cancer Institute as part of the P30 Cancer Center Support Grant (NIH CA014236). Q.W. was also supported by a start-up fund from Duke Cancer Institute, Duke University Medical Center.
TRICL
Transdisciplinary Research in Cancer of the Lung (TRICL) Study, U19-CA148127 on behalf of the Genetic Associations and Mechanisms in Oncology (GAME-ON) Network. The Toronto study was supported by Canadian Cancer Society Research Institute(020214), Ontario Institute of Cancer and Cancer Care Ontario Chair Award to RH The ICR study was supported by Cancer Research UK (C1298/A8780 andC1298/A8362—Bobby Moore Fund for Cancer Research UK) and NCRN,HEAL and Sanofi-Aventis. Additional funding was obtained from NIH grants (5R01CA055769, 5R01CA127219, 5R01CA133996 and 5R01CA121197). The Liverpool Lung Project (LLP) was supported by The Roy Castle Lung Cancer Foundation, UK. The ICR and LLP studies made use of genotyping data from the Wellcome Trust Case Control Consortium 2 (WTCCC2); a full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. Sample collection for the Heidelberg lung cancer study was in part supported by a grant (70–2919) from the Deutsche Krebshilfe. The work was additionally supported by a Helmholtz-DAAD fellowship (A/07/97379 to MNT) and by the NIH (U19CA148127). The KORA Surveys were financed by the GSF, which is funded by the German Federal Ministry of Education, Science, Research and Technology and the State of Bavaria. The Lung Cancer in the Young study (LUCY) was funded in part by the National Genome Research Network (NGFN), the DFG (BI576/2-1; BI 576/2-2), the Helmholtzgemeinschaft (HGF) and the Federal office for Radiation Protection (BfS: STSch4454). Genotyping was performed in the Genome Analysis Center (GAC) of the Helmholtz Zentrum Muenchen. Support for the Central Europe, HUNT2/Tromsø and CARET genome-wide studies was provided by Institut National du Cancer, France. Support for the HUNT2/Tromsø genome-wide study was also provided by the European Community (Integrated Project DNA repair, LSHG-CT- 2005–512113), the Norwegian Cancer Association and the Functional Genomics Programme of Research Council of Norway. Support for the Central Europe study, Czech Republic, was also provided by the European Regional Development Fund and the State Budget of the Czech Republic (RECAMO, CZ.1.05/2.1.00/03.0101). Support for the CARET genome-wide study was also provided by grants from the US National Cancer Institute, NIH (R01 CA111703 and UO1 CA63673), and by funds from the Fred Hutchinson Cancer Research Center. Additional funding for study coordination, genotyping of replication studies and statistical analysis was provided by the US National Cancer Institute (R01 CA092039). The lung cancer GWAS from Estonia was partly supported by a FP7 grant (REGPOT245536), by the Estonian Government (SF0180142s08), by EU RDF in the frame of Centre of Excellence in Genomics and Estoinian Research Infrastructure’s Roadmap and by University of Tartu (SP1GVARENG). The work reported in this article was partly undertaken during the tenure of a Postdoctoral Fellowship from the IARC (for MNT). The Environment and Genetics in Lung Cancer Etiology (EAGLE), the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study (ATBC), and the Prostate, Lung, Colon, Ovary Screening Trial (PLCO)studies and the genotyping of ATBC, the Cancer Prevention Study II Nutrition Cohort (CPS-II) and part of PLCO were supported by the Intramural Research Program of NIH, NCI, Division of Cancer Epidemiology and Genetics. ATBC was also supported by US Public Health Service contracts (N01-CN-45165, N01-RC-45035 and N01-RC-37004) from the NCI. PLCO was also supported by individual contracts from the NCI to the University of Colorado Denver (NO1-CN-25514), Georgetown University (NO1-CN-25522), Pacific Health Research Institute (NO1-CN-25515), Henry Ford Health System (NO1-CN-25512), University of Minnesota (NO1-CN-25513), Washington University (NO1-CN-25516), University of Pittsburgh (NO1-CN-25511), University of Utah (NO1-CN-25524), Marshfield Clinic Research Foundation (NO1-CN-25518), University of Alabama at Birmingham (NO1-CN-75022, Westat, Inc. NO1-CN-25476), University of California, Los Angeles (NO1-CN-25404). The Cancer Prevention Study II Nutrition Cohort was supported by the American Cancer Society. The NIH Genes, Environment and Health Initiative (GEI) partly funded DNA extraction and statistical analyses (HG-06-033-NCI-01 and RO1HL091172-01), genotyping at the Johns Hopkins University Center for Inherited Disease Research (U01HG004438 and NIH HHSN268200782096C) and study coordination at the GENEVA Coordination Center (U01 HG004446) for EAGLE and part of PLCO studies. Funding for the MD Anderson Cancer Study was provided by NIH grants (P50 CA70907, R01CA121197, R01CA127219, U19 CA148127, R01 CA55769 and K07CA160753) and CPRIT grant (RP100443). Genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is funded through a federal contract from the NIH to The Johns Hopkins University (HHSN268200782096C). The Harvard Lung Cancer Study was supported by the NIH (National Cancer Institute) grants CA092824, CA090578 and CA074386.
deCODE
The deCODE project was funded in part by the National Institutes of Health (DA017932). Approval for the deCODE study was granted by the Icelandic National Bioethics Committee (ref. 12-122-V7) and the Icelandic Data Protection Authority (refs. 2001/25 and 2006/518).
Conflict of Interest Statement: None declared.
Supplementary Material
Glossary
Abbreviations
- eQTL
expression quantitative trait loci
- FDR
false discovery rate
- GWASs
genome-wide association studies
- LD
linkage disequilibrium
- SNPs
single-nucleotide polymorphisms
References
- 1. Jemal A., et al. (2011) Global cancer statistics. CA. Cancer J. Clin., 61, 69–90. [DOI] [PubMed] [Google Scholar]
- 2. Sun S., et al. (2007) Lung cancer in never smokers–a different disease. Nat. Rev. Cancer, 7, 778–790. [DOI] [PubMed] [Google Scholar]
- 3. Hung R.J., et al. (2008) A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature, 452, 633–637. [DOI] [PubMed] [Google Scholar]
- 4. Amos C.I., et al. (2008) Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat. Genet., 40, 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McKay J.D., et al. (2008) Lung cancer susceptibility locus at 5p15.33. Nat. Genet., 40, 1404–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pande M., et al. (2011) Novel genetic variants in the chromosome 5p15.33 region associate with lung cancer risk. Carcinogenesis, 32, 1493–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Y., et al. (2011) Variation in TP63 is associated with lung adenocarcinoma in the UK population. Cancer Epidemiol. Biomarkers Prev., 20, 1453–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Y., et al. (2014) Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat. Genet., 46, 736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gergely F., et al. (2008) Multiple centrosomes: together they stand, divided they fall. Genes Dev., 22, 2291–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zyss D., et al. (2009) Centrosome function in cancer: guilty or innocent? Trends Cell Biol., 19, 334–346. [DOI] [PubMed] [Google Scholar]
- 11. Fukasawa K. (2007) Oncogenes and tumour suppressors take on centrosomes. Nat. Rev. Cancer, 7, 911–924. [DOI] [PubMed] [Google Scholar]
- 12. Ganem N.J., et al. (2009) A mechanism linking extra centrosomes to chromosomal instability. Nature, 460, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nigg E.A., et al. (2009) Centrioles, centrosomes, and cilia in health and disease. Cell, 139, 663–678. [DOI] [PubMed] [Google Scholar]
- 14. Anderhub S.J., et al. (2012) Centrosome amplification in tumorigenesis. Cancer Lett., 322, 8–17. [DOI] [PubMed] [Google Scholar]
- 15. Pihan G.A. (2013) Centrosome dysfunction contributes to chromosome instability, chromoanagenesis, and genome reprograming in cancer. Front. Oncol., 3, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Godinho S.A., et al. (2014) Oncogene-like induction of cellular invasion from centrosome amplification. Nature, 510, 167–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shinmura K., et al. (2012) Centrosome abnormality and human lung cancer. Lung Dis., 171–188. [Google Scholar]
- 18. Timofeeva M.N., et al. (2012) Influence of common genetic variation on lung cancer risk: meta-analysis of 14 900 cases and 29 485 controls. Hum. Mol. Genet., 21, 4980–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Su L., et al. (2006) Genotypes and haplotypes of matrix metalloproteinase 1, 3 and 12 genes and the risk of lung cancer. Carcinogenesis, 27, 1024–1029. [DOI] [PubMed] [Google Scholar]
- 20. Thorgeirsson T.E., et al. (2008) A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature, 452, 638–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Consortium G.P. (2012) An integrated map of genetic variation from 1,092 human genomes. Nature, 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Howie B.N., et al. (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet., 5, e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Y., et al. (2010) MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol., 34, 816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Howie B., et al. (2012) Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet., 44, 955–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Y., et al. (2015) Deciphering associations for lung cancer risk through imputation and analysis of 12 316 cases and 16 831 controls. Eur. J. Hum. Genet., 23, 1723–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu Z., et al. (2009) SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res., 37, W600–W605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boyle A.P., et al. (2012) Annotation of functional variation in personal genomes using RegulomeDB. Genome Res., 22, 1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee P.H., et al. (2008) F-SNP: computationally predicted functional SNPs for disease association studies. Nucleic Acids Res., 36, D820–D824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Purcell S., et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Higgins J.P., et al. (2003) Measuring inconsistency in meta-analyses. BMJ, 327, 557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Benjamini Y., et al. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Statis. Soc. B (Methodological), 57, 289–300. [Google Scholar]
- 32. Pruim R.J., et al. (2010) LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics, 26, 2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barrett J.C., et al. (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics, 21, 263–265. [DOI] [PubMed] [Google Scholar]
- 34. Broderick P., et al. (2009) Deciphering the impact of common genetic variation on lung cancer risk: a genome-wide association study. Cancer Res., 69, 6633–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Consortium E.P. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bettencourt-Dias M., et al. (2007) Centrosome biogenesis and function: centrosomics brings new understanding. Nat. Rev. Mol. Cell Biol., 8, 451–463. [DOI] [PubMed] [Google Scholar]
- 37. Marx J. (2001) Cell biology. Do centrosome abnormalities lead to cancer? Science, 292, 426–429. [DOI] [PubMed] [Google Scholar]
- 38. Tollenaere M.A., et al. (2015) Centriolar satellites: key mediators of centrosome functions. Cell. Mol. Life Sci., 72, 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Acquaviva C., et al. (2009) The centrosomal FOP protein is required for cell cycle progression and survival. Cell Cycle, 8, 1217–1227. [DOI] [PubMed] [Google Scholar]
- 40. Popovici C., et al. (1999) The t(6;8)(q27;p11) translocation in a stem cell myeloproliferative disorder fuses a novel gene, FOP, to fibroblast growth factor receptor 1. Blood, 93, 1381–1389. [PubMed] [Google Scholar]
- 41. Mulligan L.M. (2014) RET revisited: expanding the oncogenic portfolio. Nat. Rev. Cancer, 14, 173–186. [DOI] [PubMed] [Google Scholar]
- 42. Barrett J.C., et al. (2008) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet., 40, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chu X., et al. (2011) A genome-wide association study identifies two new risk loci for Graves’ disease. Nat. Genet., 43, 897–901. [DOI] [PubMed] [Google Scholar]
- 44. Kochi Y., et al. (2010) A regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibility. Nat. Genet., 42, 515–519. [DOI] [PubMed] [Google Scholar]
- 45. Stahl E.A., et al. (2010) Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet., 42, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jin Y., et al. (2010) Common variants in FOXP1 are associated with generalized vitiligo. Nat. Genet., 42, 576–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Quan C., et al. (2010) Genome-wide association study for vitiligo identifies susceptibility loci at 6q27 and the MHC. Nat. Genet., 42, 614–618. [DOI] [PubMed] [Google Scholar]
- 48. Teulings H.E., et al. (2013) Decreased risk of melanoma and nonmelanoma skin cancer in patients with vitiligo: a survey among 1307 patients and their partners. Br. J. Dermatol., 168, 162–171. [DOI] [PubMed] [Google Scholar]
- 49. Joseph C.G., et al. (2014) Association of the autoimmune disease scleroderma with an immunologic response to cancer. Science, 343, 152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Franks A.L., et al. (2012) Multiple associations between a broad spectrum of autoimmune diseases, chronic inflammatory diseases and cancer. Anticancer Res., 32, 1119–1136. [PMC free article] [PubMed] [Google Scholar]
- 51. Bettencourt-Dias M., et al. (2011) Centrosomes and cilia in human disease. Trends Genet., 27, 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chan J.Y. (2011) A clinical overview of centrosome amplification in human cancers. Int. J. Biol. Sci., 7, 1122–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cortez B.A., et al. (2008) Chrysotile effects on human lung cell carcinoma in culture: 3-D reconstruction and DNA quantification by image analysis. BMC Cancer, 8, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xie H., et al. (2007) Neoplastic transformation of human bronchial cells by lead chromate particles. Am. J. Respir. Cell Mol. Biol., 37, 544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Holmes A.L., et al. (2010) Chronic exposure to zinc chromate induces centrosome amplification and spindle assembly checkpoint bypass in human lung fibroblasts. Chem. Res. Toxicol., 23, 386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liao W.T., et al. (2007) Arsenic promotes centrosome abnormalities and cell colony formation in p53 compromised human lung cells. Toxicol. Appl. Pharmacol., 225, 162–170. [DOI] [PubMed] [Google Scholar]
- 57. Shinmura K., et al. (2008) Induction of centrosome amplification and chromosome instability in p53-deficient lung cancer cells exposed to benzo[a]pyrene diol epoxide (B[a]PDE). J. Pathol., 216, 365–374. [DOI] [PubMed] [Google Scholar]
- 58. Jung C.K., et al. (2007) Centrosome abnormalities in non-small cell lung cancer: correlations with DNA aneuploidy and expression of cell cycle regulatory proteins. Pathol. Res. Pract., 203, 839–847. [DOI] [PubMed] [Google Scholar]
- 59. Matsuura S., et al. (2013) SGOL1 variant B induces abnormal mitosis and resistance to taxane in non-small cell lung cancers. Sci. Rep., 3, 3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Olson J.E., et al. (2011) Centrosome-related genes, genetic variation, and risk of breast cancer. Breast Cancer Res. Treat., 125, 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Couch F.J., et al. (2010) Association of mitotic regulation pathway polymorphisms with pancreatic cancer risk and outcome. Cancer Epidemiol. Biomarkers Prev., 19, 251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.