

Summary
Colonic Lgr5+ stem cells are highly susceptible to AOM-induced DNA damage and actively induce damage repair enzyme expression and untargeted apoptosis.
Abstract
Perturbations in DNA damage, DNA repair, apoptosis and cell proliferation in the base of the crypt where stem cells reside are associated with colorectal cancer (CRC) initiation and progression. Although the transformation of leucine-rich repeat-containing G protein-coupled receptor 5 (Lgr5)+ cells is an extremely efficient route towards initiating small intestinal adenomas, the role of Lgr5+ cells in CRC pathogenesis has not been well investigated. Therefore, we further characterized the properties of colonic Lgr5+ cells compared to differentiated cells in Lgr5-EGFP-IRES-creERT2 knock-in mice at the initiation stage of carcinogen azoxymethane (AOM)-induced tumorigenesis using a quantitative immunofluorescence microscopy approach. At 12 and 24h post-AOM treatment, colonic Lgr5+ stem cells (GFPhigh) were preferentially damaged by carcinogen, exhibiting a 4.7-fold induction of apoptosis compared to differentiated (GFPneg) cells. Furthermore, with respect to DNA repair, O6-methylguanine DNA methyltransferase (MGMT) expression was preferentially induced (by 18.5-fold) in GFPhigh cells at 24h post-AOM treatment compared to GFPneg differentiated cells. This corresponded with a 4.3-fold increase in cell proliferation in GFPhigh cells. These data suggest that Lgr5+ stem cells uniquely respond to alkylation-induced DNA damage by upregulating DNA damage repair, apoptosis and cell proliferation compared to differentiated cells in order to maintain genomic integrity. These findings highlight the mechanisms by which colonic Lgr5+ stem cells respond to cancer-causing environmental factors.
Introduction
The transformation of leucine-rich repeat-containing G protein-coupled Receptor 5 (Lgr5+) stem cells drives intestinal neoplasia in the Apc flox/flox mouse model, indicating that Lgr5+ crypt stem cells are the cell-of-origin of cancer (1). Colon cancer has been suggested to follow a cancer stem cell (CSC) hierarchical model (2,3). Although cycling Lgr5+ stem cells fuel the rapid turnover of the adult intestinal epithelium (4,5), and its properties are not shared by other crypt cells (6,7), the role of Lgr5+ cells in colorectal cancer pathogenesis has not been well investigated. Therefore, it is important to identify the properties of Lgr5+ cells at the initiation stage of colon tumorigenesis because transformation of Lgr5+ stem cells is an extremely efficient route towards initiating intestinal cancer (1). Recently, DNA damage responses in Lgr5+ cells have been reported (2,8,9), however, to date, colonic Lgr5+ cytokinetics have not been examined in the context of an alkylating agent induced-DNA damage model of colorectal cancer.
Critical to the prevention of colon cancer is the removal of O6-methylguanine (O6-meG) DNA adducts, either through repair, e.g. via O6-methylguanine-DNA methyltransferase (MGMT) or targeted apoptosis (10). MGMT is an inducible repair enzyme (11) that acts by transferring the methyl group from guanine in DNA to a cysteine residue on the repair protein (12), resulting in rapid removal of promutagenic O6-meG DNA adducts and the irreversible inactivation of the repair enzyme (11). Cells with unrepaired DNA adducts may be eliminated through the activation of the apoptotic cascade, resulting in their selective removal (13). In general, during the initiation of tumorigenesis following carcinogen exposure, there is an immediate apoptotic response to DNA damage in the colonic epithelium (14). This is consistent with the fact that O6-meG DNA lesions trigger apoptosis (10).
The life-time risk of cancer is strongly correlated with the total number of stem cell divisions (15). Although the effect of DNA alkylating agent administration on O6-meG DNA adduct repair, apoptosis and proliferation in colonic crypts has been reported in preclinical models (8,9,16,17), there is no information on the interrelationship among these phenotypes in DNA damaged adult colonic Lgr5+ stem cells. We therefore determined how colonic Lgr5+ stem cells respond to the administration of AOM, a well-characterized experimental colon carcinogen and DNA alkylating agent (18), in terms of O6-meG DNA adduct removal by MGMT, apoptosis and cell cycle arrest. In this study, we provide evidence that Lgr5+ stem cells exhibit elevated AOM induced-DNA damage compared to differentiated cells. We also show that Lgr5+ stem cells profoundly induce MGMT to delete O6-meG and promote nontargeted apoptosis upon AOM exposure. These data document for the first time how the rapidly cycling Lgr5+ stem cell population responds to environmental factors at the initiation stage of colon tumorigenesis.
Materials and methods
Animals, diet and study design
The animal use protocol was approved by the University Animal Care Committee of Texas A&M University and conformed to NIH guidelines. Lgr5-EGFP-IRES-creERT2 (1) knock-in mice 6–7-week-old were acclimated for 1 week and then provided with a semi-purified diet (Supplementary Table 1, available at Carcinogenesis Online) for 3 weeks prior to injection with AOM (Sigma Chemical, [St. Louis, MO]; 10mg/kg body weight). Mice were injected with EdU (Life Technologies) 2h prior to killing. Twelve (n = 8) and 24h (n = 8) following a single intraperitoneal injection of AOM, animals were killed by CO2 asphyxiation. Control mice (n = 3) received a single saline injection. Immediately after termination, the colon was rapidly removed, flushed with ice-cold saline and immediately fixed in 4% paraformaldehyde for immunofluorescence analyses. Supplementary Figure 1, available at Carcinogenesis Online, shows the timeline of the treatments and the experimental design.
In vivo DNA damage and repair measurement
Formalin-fixed paraffin-embedded 4 μm colon sections were deparaffinized, rehydrated through graded ethanol and stained with antibodies using standard procedures. DNA double strand breaks (DSBs) were measured by immunofluorescence using a rabbit monoclonal phospho-gamma H2AX (γH2AX) Ser139 antibody (9718, Cell Signaling; dilution 1:200), Lgr5+ stem cells were labeled using goat polyclonal GFP antibody (ab6673, Abcam; dilution 1:400) and O6-meG DNA adduct removal was estimated by the induction of MGMT expression using a mouse monoclonal MGMT antibody (ab54306, Abcam; prediluted). Secondary antibodies were antirabbit Alexa 647 (711-605-152, Jackson ImmunoResearch: dilution 1:400) for γH2AX, antigoat 488 (705-545-147, Jackson ImmunoResearch) for GFP and antimouse Alexa 546 (A10036, Life Technologies) for MGMT. The DNA damage (or repair) index was determined by dividing the number of γH2AX (or MGMT) positive cells by the total number of cells in each crypt column and multiplying by 100.
In vivo apoptosis measurement
To investigate whether alkylating agent-induced DNA damage triggered apoptotic cell death in colonic Lgr5+ stem cells, apoptotic bodies were visualized using the TACS 2 TdT-Fluor in situ apoptosis detection kit (Trevigen) as per the manufacturer’s instructions. Negative control slides were incubated without TdT enzyme. The apoptotic index was determined by dividing the number of apoptotic cells by the total number of cells in the crypt column and multiplying by 100. Serial sections were also stained with hematoxylin and eosin (H&E) and analyzed using a light microscope. Apoptotic cells were identified by characteristic morphology, i.e. cell shrinkage, nuclear condensation and blebbing, and formation of apoptotic bodies (19).
In vivo apoptosis-BE measurement
To document the ability of AOM to induce bystander effect (BE) in stem cells, apoptotic cells were classified as BE-dependent or BE-independent. BE-dependent apoptosis was defined as apoptotic cells without DNA damage adjacent to damaged or apoptotic/damaged cells. In comparison, BE-independent apoptosis was defined as apoptotic cells with no adjacent damaged cells. Thus, BE-dependent apoptotic cells were classified by proximity, i.e. P1, P2 and P3 represent the proximity of the apoptotic cell (1, 2 or 3 cells away) from the damaged cell.
In vivo measurement of cell proliferation
To investigate the effects of alkylating agent-induced DNA damage on cell cycle in colonic epithelial cells, proliferative activity was measured using the Click-iT EdU Alexa Fluor 555 Imaging kit (Life Technologies) as per the manufacturer’s instructions. Negative control slides were incubated without Alexa Fluor.
Slide scoring
Images of colonic crypts were captured on an inverted TE 300 Nikon Eclipse fluorescence microscope equipped with 40×/1.30 Nikon Plan Fluor oil immersion objective and a Photometrics Cool snap EZ digital CCD camera. The external light source was powered by a mercury lamp. Images were processed using NIS Image software, version 3.2 (Nikon). A total of 426 GFPhigh crypts from eight mice were counted at 12 and 24h post-AOM exposure and 150 GFPhigh crypts from three saline injected mice (control) were examined.
Statistics
GraphPad Prism6 was used to analyze DNA adduct removal, apoptosis and proliferation and to produce graphs. Two-way analysis of variance (ANOVA) was used to determine the effect of carcinogen in Lgr5+ stem cells compared to differentiated cells over time. Comparisons between different time points were analyzed using one-way ANOVA. A P-value < 0.05 was considered statistically significant.
Results
Lgr5+ stem cells are preferentially damaged by carcinogen
To elucidate the unique properties of colonic Lgr5+ stem cells in terms of their response to DNA damage, we assessed Lgr5+ stem cell homeostasis in the context of AOM-induced tumorigenesis. Lgr5+ stem cells and differentiated cells were tracked using the Lgr5-EGFP-IRES-creERT2 knock-in mouse. In this model, GFP expression is driven by the Lgr5 locus, leading to high GFP expression in Lgr5+ stem cells (GFPhigh), which is distinguishable from Lgr5− cells (GFPneg) within crypts (20). Fluorescence microscopy was used to visualize GFPhigh (Lgr5+) cells within the context of the colonic crypt. In addition, sequencing results confirmed that GFPhigh cells highly expressed Lgr5 and other stem cell markers, whereas GFPneg cells expressed differentiated cell markers, e.g. Atoh1 (Supplementary Table 2, available at Carcinogenesis Online).
O6-meG is the dominant mutagenic adduct introduced by AOM (21). O6-meG:T mispairs can stall DNA replication when not repaired before replication, resulting in DSBs (22). DSBs are always followed by phosphorylation of γH2AX (23). Hence, γH2AX foci in the nucleus are a useful marker of the DSBs induced by S-phase dependent genotoxins during replication (23,24). We hypothesized that AOM would induce more DSBs in GFPhigh cells compared to GFPneg, due to the fact that Lgr5+ cells are actively proliferating and AOM induced-DSBs are formed during replication (24). We therefore assessed the cytotoxic effects of AOM by quantifying γH2AX (phospho S139) labeling. As shown in Figure 1A, γH2AX levels in the proximal colon were nearly undetectable similar to saline control indicating that the distal colon is the primary target of AOM. Following AOM exposure, colonic cells in the distal colon were intensely stained for γH2AX at 12h, which persisted through the 24h time point. DNA damage levels were 5.7-fold higher (P < 0.01) in GFPhigh stem cells as compared to GFPneg cells (Figure 1B).
Figure 1.
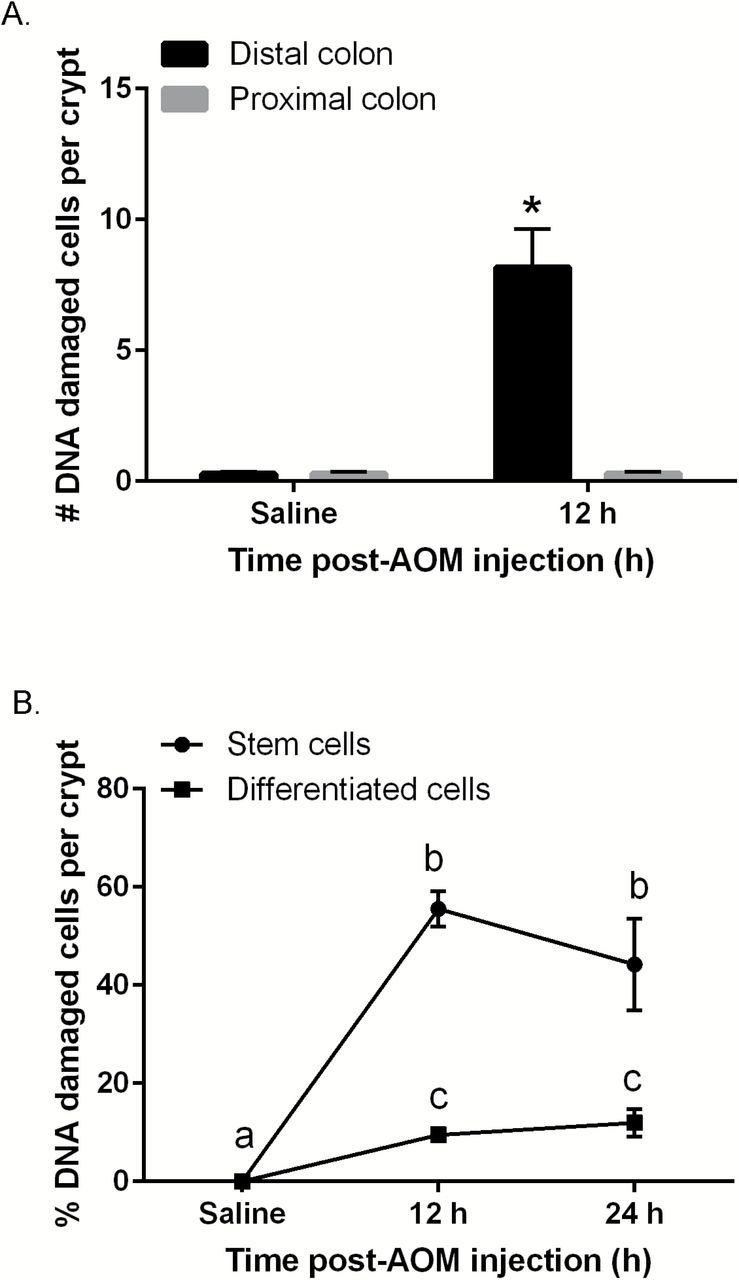
Carcinogen (AOM)-induced DNA DSBs in mouse colonic crypts at 12 and 24h post-AOM injection (% of saline control). (A) Quantitative comparison of the number of γH2AX+ (DNA damaged) cells per crypt in the distal and proximal colon in saline versus AOM injected mice. (B) Quantitative comparison of the % of γH2AX+ stem cells (GFPhigh) and differentiated cells (GFPneg) per crypt. A total of 426 Lgr5 GFP+ crypts from eight mice were counted at 12h, 150 Lgr5 GFP+ crypts from three mice were counted at 24h and 150 crypts from three saline control mice were examined. DAPI stained nuclei along the mouse colonic crypt were counted and stem cells were determined by counting cells double stained for GFP and DAPI within Lgr5 GFP+ crypts. Statistically significant differences between time points were determined using two-way ANOVA followed by Fisher’s LSD multiple comparison testing. Different letters or * indicate significant differences between treatment groups (P < 0.05).
Lgr5+ stem cells preferentially promote damage-induced apoptosis in response to carcinogen
It has been reported that O6-meG can generate stalled replication and DSBs, resulting in the induction of apoptosis (22). In addition, stem cells exhibit a lower apoptotic threshold due to mitochondrial priming (25,26). This is noteworthy, because an excess number of stem cells can cause cancer (27) and ‘apoptosis resistance’ has been reported in normal and CSCs (28). Therefore, we quantified the levels of AOM-induced apoptotic GFPhigh and GFPneg cells per crypt in the distal colon. For this purpose, we used the TUNEL assay to colocalize apoptosis with γH2AX and GFP (Lgr5+ cells). To corroborate the frequency of epithelial cells undergoing apoptosis, paraffin-embedded sections were also assessed by H&E staining (Supplementary Figure 3A and C, available at Carcinogenesis Online). Consistent with previous findings (19), H&E and TUNEL data were highly correlated (Supplementary Figure 3B, available at Carcinogenesis Online). GFPhigh cells preferentially exhibited AOM-induced apoptosis, e.g. 4.7-fold higher levels (P < 0.0001), compared to GFPneg cells at 12h (Figure 2A and B). In contrast, apoptosis in GFPneg cells after exposure to carcinogen was more modest. Next, using linear regression analysis, we determined whether AOM-induced apoptosis was associated with the levels of DNA damage induced by AOM. As shown in Figure 2C, only GFPhigh cells exhibited proportionally increased apoptosis (slope = 0.42, P = 0.03) in response to DNA damage, whereas GFPneg cells were non-responsive. We subsequently quantified the number of damaged Lgr5+ stem cells that were targeted for apoptotic deletion, i.e. GFPhigh, TUNEL+, γH2AX+ triple positive cells, since the selective deletion of damaged Lgr5+ stem cells by promoting apoptosis could mitigate the clonal expansion of DNA-damaged Lgr5+ stem cells. As shown in Figure 2D, no statistically significant differences between GFPhigh cells and GFPneg cells (P = 0.54) were observed in terms of targeted apoptosis (TUNEL+, γH2AX+ cells) both at 12 and 24h. However, γH2AX−, GFPhigh cells exhibited a 7.7-fold higher induction of apoptosis (nontargeted apoptosis—TUNEL+, γH2AX− cells) as compared to γH2AX−, GFPneg cells at 12h (P = 0.001) (Figure 2D right). These data indicate that the majority (63%) of induced apoptosis in GFPhigh cells was nontargeted. Specifically, 28% of GFPhigh cells exhibited nontargeted apoptosis (12h, Figure 2D right) and 13% exhibited targeted apoptosis (12h, Figure 2D left).
Figure 2.
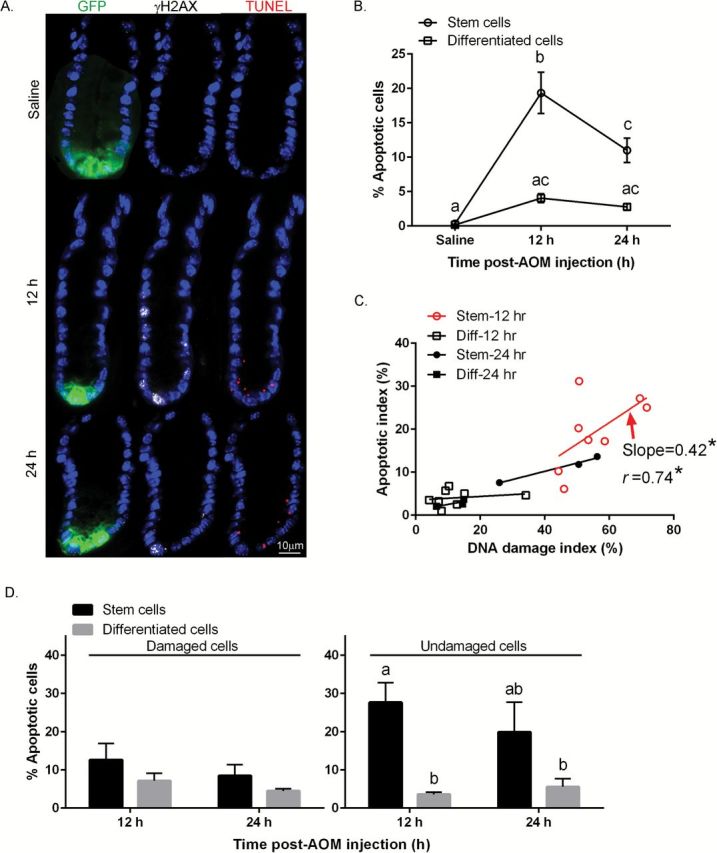
Comparison of AOM-induced apoptosis in stem cells and differentiated cells. (A) Representative images (objective, 40×) of GFP+ (GFPhigh stem cells, green), γH2AX+ (DSBs, white) and TUNEL+ (apoptotic body, red) cells are shown. For comparative purposes, immunofluorescence staining of colonic crypts in the distal colon in saline (control) and AOM injected mice is shown. (B) % TUNEL+ (apoptotic) stem cells and differentiated cells per crypt. (C) Association between the % of AOM-induced apoptotic cells and % of γH2AX+ cells per crypt in stem and differentiated (Diff) cells. Each point represents an individual animal. The P value was calculated using an F test. Correlation computes the value of the Pearson correlation coefficient, r, ranges from −1 to +1. The linear regression was fitted using GraphPad Prism 6.0. Apoptotic index = # of TUNEL+ stem or differentiated cells/total # of stem or differentiated cells per crypt X 100 at 12 and 24h post-AOM injection; Damage index = # of γH2AX+ stem or differentiated cells/total # of stem or differentiated cells per crypt X 100 at 12 and 24h post-AOM injection. Slope where the difference from zero is statistically significant is marked in red (Stem, 12h). (D) (left panel) % targeted apoptosis (double positive TUNEL+ and γH2AX+ stem or differentiated cells/γH2AX+ stem or differentiated cells) at 12 and 24h post-AOM injection; (right panel) % nontargeted apoptosis (TUNEL+ and γH2AX− stem or differentiated cells/γH2AX− stem or differentiated cells) at 12 and 24h post-AOM injection. Different letters or * indicate significant differences between treatment groups (P < 0.05). Refer to Figure 1 legend for animal numbers.
AOM exposure promotes an apoptosis-bystander effect in Lgr5+ stem cells
In order to further shed mechanistic insight into the cause of the nontargeted apoptosis stem cell phenotype (TUNEL+, γH2AX−, Lgr5+, Figure 2D), we defined BE-dependent apoptosis as apoptotic cells without DNA damage adjacent to damaged or apoptotic/damaged cells. In comparison, BE-independent apoptosis was defined as apoptotic cells with no adjacent damaged cells (Figure 3A, left). Our findings indicate that TUNEL+, γH2AX−, Lgr5+ stem cells were predominantly (70%) localized to regions adjacent to γH2AX+ cells (Figure 3B, left). The proximity of γH2AX+ cells to TUNEL+, γH2AX− Lgr5+ stem cells suggest the involvement of an AOM-induced BE (Figures 3A and B, right).
Figure 3.

(A) Representative image of direct BE (apoptotic cell located one or two cells away from damaged cell) and indirect BE (apoptotic cells with no damaged cells in the field of interest) at 12h post-AOM injection. An apoptotic cell immediately adjacent to a damaged cell is defined as P1; an apoptotic cell located one cell away from a damaged cell is defined as P2 (right). Representative images (objective, 40×) of TUNEL+ (apoptotic body, red) cells next to γH2AX+ (DSBs, white) cells are shown counter-stained with DAPI (blue). (B) Percentage of BE-dependent and BE-independent apoptotic Lgr5+ stem versus differentiated cells (left) and percentage of BE-dependent apoptotic cells in relation to its proximity to a damaged cell.
Lgr5+ stem cells upregulate DNA repair enzyme expression in response to carcinogen
The major pathway for the repair of O6-meG is via the MGMT mechanism (29). Because this alkyltransferase is induced by alkylating agents, quantitative immunohistochemical analysis can be used to estimate MGMT activity upon AOM injection (30). As shown in Figure 4A and B, MGMT expression in GFPhigh cells in the distal colon was increased by 18.5-fold 24h after AOM injection, whereas in GFPneg cells, no induction was detected. Using linear regression analysis, both GFPhigh cells (slope = 1.08, P = 0.03) and GFPneg cells (slope = 0.87, P = 0.006) at 24h exhibited an induction of MGMT expression that was associated with the amount of DNA damage induced by AOM (Figure 4C), whereas at 12h, no statistical differences were detected either in Lgr5+ stem and differentiated cells. Next, we quantified the number of damaged cells expressing MGMT, i.e. exhibiting targeted repair (GFPhigh, γH2AX+ and MGMT+ triple positive), since the removal of mutagenic and cytotoxic adducts via induced-MGMT expression is negatively associated with G to A mutations in K-ras in colorectal tumorigenesis (31). With respect to the γH2AX+ cell compartment, only γH2AX+ cells exhibited a statistically induced MGMT expression in GFPhigh cells at 24h, whereas no significant changes were detected in GFPneg cells (Figure 4D left). In comparison, MGMT expression in γH2AX− cells was negligible both in GFPhigh and GFPneg cells (Figure 4D right).
Figure 4.
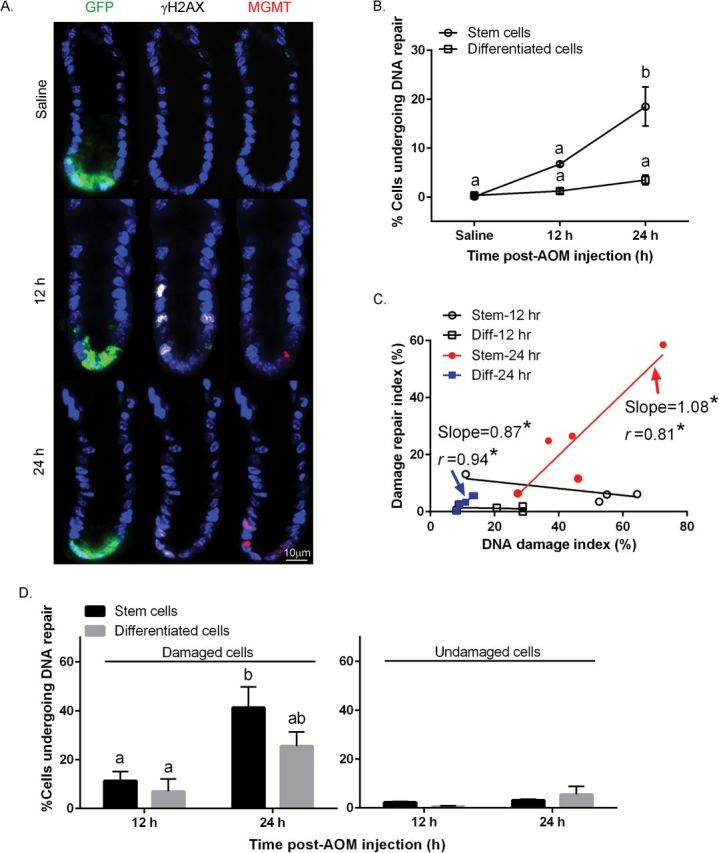
Comparison of AOM-induced MGMT expression in stem cells and differentiated cells. (A) Representative images (objective, 40×) of GFP+ (Lgr5+ stem cells), γH2AX+ (DSBs) and MGMT+ (repair enzyme) cells are shown. For comparative purposes, immunofluorescence staining of colonic crypt in the distal colon in saline (control) and AOM injected mice is shown. (B) % MGMT+ stem cells and differentiated cells per crypt. (C) Association between the % of AOM-induced MGMT+ (DNA repair) cells and % of γH2AX+ cells per crypt in stem and differentiated (Diff) cells. Repair index = # of MGMT+ stem or differentiated cells/total # of stem or differentiated cells per crypt X 100 at 12 and 24h post-AOM injection; DNA damage index = # of γH2AX+ stem or differentiated cells/total # of stem or differentiated cells per crypt X 100 at 12 and 24h post-AOM injection. Slope where the difference from zero is statistically significant is marked in red (Stem, 24h) or blue (Diff, 24h). (D) (left panel) % of γH2AX+ cells expressing MGMT (both γH2AX+ and MGMT+ stem or differentiated cells/γH2AX+ stem or differentiated cells) at 12 and 24h (right) post-AOM injection; (right panel) % of γH2AX− cells expressing MGMT (both γH2AX− and MGMT+ stem or differentiated cells/γH2AX− stem or differentiated cells) at 12 and 24h post-AOM injection. Different letters or * indicate significant differences between treatment groups (P < 0.05). Refer to Figure 1 legend for animal numbers.
DSBs can be repaired by at least four independent pathways including homologous repair (HR), non-homologous end joining (NHEJ), alternative-NHEJ (alt-NHEJ) and single-strand annealing (32). Therefore, we used BRCA1 and Rad51 as markers of HR to define AOM-induced postreplicative repair process of Lgr5+ stem cells. BRCA1 and Rad51 are well known markers for potsreplication repair, especially HR in the presence of radiation (8). However, we were not able to detect an induction of HR marked by BRCA1 and Rad51 (Supplementary Figure 2, available at Carcinogenesis Online).
Lgr5+ stem cells exhibit increased proliferation following AOM exposure
Elevated crypt epithelial cell proliferation is considered a risk factor for colon cancer (33). For example, assessment of colonic crypt cell proliferation, including detection of an increased proliferative state and expansion of the proliferative zone, are considered putative intermediate markers of colon cancer risk (34). Furthermore, it has been shown that alkylating agent exposure results in increased colonic crypt cellularity, colonic crypt cell proliferation and the crypt proliferative zone (35). Therefore, we determined whether AOM injection results in increased GFPhigh cell proliferation in the presence of AOM in the distal colon. As shown in Figures 5A and B, no reduction in cell cycle was observed immediately following AOM injection (12h) both in GFPhigh and GFPneg cells. However, a significant increase in cell proliferation (EdU labeling) was detected in GFPhigh cells at 24h (Figure 5B) and the increase in proliferation was associated with the induction of DNA damage (slope = 0.98, P = 0.01) as assessed by linear regression analysis (Figure 5C). In contrast, this association was not detected in GFPneg cells. Figure 5D (left) also shows that the percentage of GFPhigh, γH2AX+, EdU+ triple positive Lgr5+ stem cells increased overtime. This is consistent with the fact that Lgr5+ cells are actively cycling and have a greater chance of accumulating DNA DSBs as compared to differentiated cells (24). The percentage of γH2AX−, EdU+ cells did not exhibit a cell type or temporal response (Figure 5D right).
Figure 5.
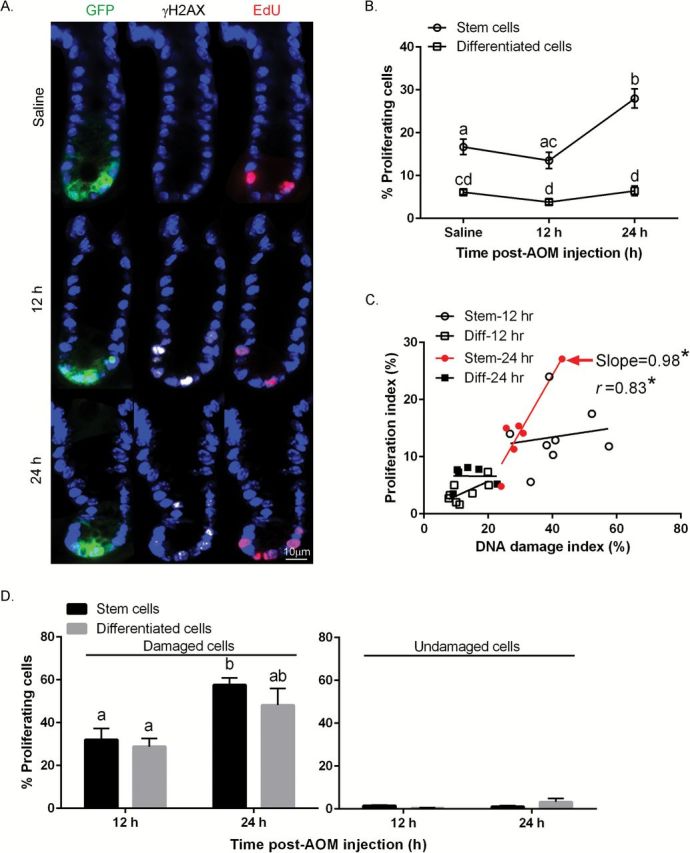
Comparison of AOM-induced proliferation in stem cells and differentiated cells. (A) Representative images (objective, 40×) of GFP+ (Lgr5+ stem cells), γH2AX+ (DSBs) and EdU+ (proliferation) cells are shown. For comparative purposes, immunofluorescence staining of colonic crypts in the distal colon in saline (control) and AOM injected mice is shown. (B) % EdU+ stem cells and differentiated cells per crypt. (C) Association between the % of AOM-induced proliferating cells and % of γH2AX+ cells per crypt in both stem and differentiated (Diff) cells. Proliferating index = # of EdU+ stem or differentiated cells/total # of stem or differentiated cells per crypt X 100 at 12 and 24h post-AOM injection; DNA damage index = # of γH2AX+ stem or differentiated cells/total # of stem or differentiated cells per crypt X 100 at 12 and 24h post-AOM injection. Slope where the difference from zero is statistically significant is marked in red (stem, 24h). (D) (left panel) % of γH2AX+ cells also positive for EdU (both γH2AX+ and EdU+ stem or differentiated cells/γH2AX+ stem or differentiated cells) at 12 and 24h post-AOM injection; (right panel) % of γH2AX− cells positive for EdU (both γH2AX− and EdU+ stem or differentiated cells/γH2AX− stem or differentiated cells) at 12 and 24h post-AOM injection. Different letters or * indicate significant differences between treatment groups (P < 0.05). Refer to Figure 1 legend for animal numbers.
Discussion
Historically, alkylating agents are known to produce gastrointestinal tumors in rodents (18,36), and to cause cancer in humans as a result of lifetime environmental exposures (37). Of the spectrum of adducts produced by AOM, O6-meG accounts for ~8% of the total DNA methyl adducts, is stable and persists in the absence of the DNA repair enzyme MGMT (21). If left unrepaired, this adduct remains in the genome, triggering a futile repair loop which can eventually result in highly toxic DSBs, which are intermediates in apoptotic and DSB repair pathways (22). DSBs are the most dominant cytotoxic effect generated by AOM (21), and are typically detected at 12h following exposure (17). This form of alkylation-induced DNA damage is clinically relevant because AOM hyperactivates β-catenin and K-ras signaling, which are dysregulated in sporadic colorectal cancer in humans (38,39). In addition, this preclinical model recapitulates the pathogenesis of human sporadic colon cancer (40).
Colon cancer has been suggested to follow a CSC hierarchical model (1,41). Several studies have monitored DNA damage in stem cells, including MNNG-induced (42) and radiation-induced (3,43) mouse stem cells, and small intestinal Lgr5+ stem cells at the initiation stage of tumorigenesis (9). Moreover, the transformation of actively cycling, long-lived intestinal Lgr5+ stem cells drives intestinal cancer (20). Overexpression of Lgr5 has been shown to occur in human colorectal adenomas and cancers (44), as well as in other solid tumors (45). However, to date, no investigation has determined the in vivo effect of AOM exposure with respect to colonic Lgr5+ stem cells. In the present study, we report for the first time the biological properties of AOM-damaged colonic Lgr5+ stem cells at the initiation stage of tumorigenesis. For this purpose, the relationship between DNA damage, apoptosis, repair and cell cycle arrest was examined in Lgr5+ stem cells versus differentiated cells within the colonic crypt at 12 and 24h following the administration of carcinogen.
We have previously demonstrated that (a) AOM induces DNA damage to a greater degree in actively proliferating cells at the base of the crypt (16); (b) AOM-induced apoptosis is primarily targeted to the base of the crypt (16); and (c) AOM-induced MGMT protein expression increases over time in the bottom one-third of the crypt as compared to the differentiated cell compartment (17). In the present study, we extend these findings by demonstrating that: DNA damage occurs to a greater degree in Lgr5+ stem cells (located at the base of the crypt) as compared to the differentiated cell compartment (Figure 1). It is assumed that AOM-induced γH2AX phosphorylation occurs during DNA replication in rapidly cycling Lgr5+ stem cells before the damage has been repaired, i.e. restored to normal (46). This finding is consistent with the fact that alkylating agent-induced γH2AX foci are selectively formed in proliferating cells (24). In addition, we also demonstrate that colonic Lgr5+ stem cells uniquely respond to AOM by inducing preferential apoptosis (Figure 2B), which is a response to increased levels of γH2AX (Figure 2C), whereas differentiated cells do not exhibit this response. These findings are consistent with the fact that stem cells exhibit a rapid apoptotic response due to mitochondrial priming in a p53-dependent manner (26). It is noteworthy that AOM-induced apoptosis is p53-dependent whereas spontaneous apoptosis is p53-independent (47). However, it remains to be determined whether the induced apoptosis in Lgr5+ stem cells is promoted due to p53-dependent mitochondrial priming.
Interestingly, ‘nontargeted’ apoptosis (TUNEL+, γH2AX−) in Lgr5+ stem cells (Figure 2D right) was 2.2-fold higher than ‘targeted’ apoptosis (TUNEL+, γH2AX+) (Figure 2D left), implying an inefficient removal of damaged Lgr5+ stem cells via programmed cell death. Apoptotic cell death might be considered protective, because it selectively eliminates damaged cells that can contribute to carcinogenesis. However, nontargeted cell death in the presence of residual DNA damage could promote cell proliferation and generate defective progenitor cells, which is potentially a tumor-promoting factor (8). It has also been demonstrated that nontargeted apoptosis can mediate DNA damage and induce chromosomal instability in neighboring cells (48,49). Therefore, primary prevention strategies resulting in a favorable enhancement of apoptosis in damaged Lgr5+ stem cells would be expected to reduce colon cancer risk.
Data in Figure 2D indicate that the majority of apoptosis occurs in undamaged GFPhigh Lgr5+ stem cells following AOM exposure. Based on the proximity of γH2AX+ cells to TUNEL+, γH2AX− Lgr5+ stem cells, we have proposed the involvement of an AOM-induced BE (Figure 3B). This unexpected outcome suggests that undamaged ‘bystander’ Lgr5+ stem cells are being indirectly influenced by the intestinal niche. For example, numerous studies have shown that irradiated/damaged colonic cells can induce secondary apoptosis (50) and γH2AX foci in non-irradiated cells via BEs (51). This process, in part, may be mediated by lipid rafts (52) via the absorption of exosomes by naïve bystander cells (53) or intestinal commensal bacteria by triggering macrophages (49). It is noteworthy that bystander cells exhibit a significant pro-apoptotic gene expression profile compared to cells directly impacted by radiation (50). From a mechanistic perspective, soluble mediators induced by AOM such as PGE2 may partly mediate this process (54,55). Additional studies are required to further elucidate the mechanism of the alkylation-induced BEs.
With respect to cell proliferation, we also determined that Lgr5+ stem cells exhibiting DNA damage were actively cycling (Figure 5D left), and that proliferation was positively associated with DNA damage (Figure 5C), implying that Lgr5+ stem cells have a high potential to accumulate mutations which enhance the risk of tumorigenesis. These findings provide a model for future therapeutic studies designed to ameliorate the effects of DNA damage in Lgr5+ stem cells. Importantly, the level of γH2AX+, GFPhigh cells (56%) per crypt was 4.1-fold higher (Figure 1) than EdU+, GFPhigh cells (14%) per crypt (Figure 5B) at 12h. This implies that DSBs may also be formed independently of cell division. γH2AX induced by BEs might explain the relatively high number of γH2AX+, GFPhigh cells compared to γH2AX+, GFPhigh. This may in part be mediated by apurinic sites generated from release of N-methylpurines which can be converted into DSBs by the activity of the apurinic endonuclease (48).
A goal of the present study was to assess the localization of the MGMT repair enzyme in both the Lgr5+ stem cell and differentiated cell compartments, which is not possible when using scraped mucosa for enzyme assays. MGMT methylation status has been shown to influence the risk of colon cancer development (56) and it is known that MGMT expression is regulated by p53 in human astrocytic cells (57) and human brain tumors (58). This is noteworthy because p53 in Lgr5+ stem cells is a critical regulator of AOM/DSS-induced tumorigenesis (unpublished data) and MGMT has been shown to function similarly in humans and rats (59). Therefore, it was interesting to note that Lgr5+ stem cells preferentially induced MGMT in response to DNA damage as compared to differentiated cells. It is possible that this increased expression may represent both an increase in protein expression and also an accumulation of inactive protein targeted for degradation, a distinction that would have to be further evaluated by measuring enzyme activity. Our findings also indicate that Lgr5+ stem cells can modulate cancer risk by promoting DNA repair (Figure 4B and C) and by promoting targeted apoptosis (Figure 2D left). Interestingly, we did not detect evidence of an AOM-induced postreplicative repair process in Lgr5+ stem cells (Supplementary Figure 2, available at Carcinogenesis Online). Collectively these results help clarify the homeostatic responses of Lgr5+ stem cells at the initiation stage of tumorigenesis.
In summary, we demonstrate for the first time that colonic Lgr5+ stem cells actively induce repair enzyme following AOM-induced DNA damage. This phenotype is consistent with the enhanced xenobiotic resistance reported in normal stem cells, attributed in part to a more efficient DNA repair response (60). Importantly, the stem-like state is deeply linked to resilience and stress response, a relationship that appears to hold for their neoplastic counterpart, the CSC (61–64). Thus, we propose that the monitoring of Lgr5+ stem cell targeting responses provides a powerful tool to interrogate primary prevention strategies, e.g. diet and exercise, to specifically eradicate damaged Lgr5+ stem cells. Ultimately, this strategy will provide a better understanding of the origin of colon cancer and the development of diagnostic tests that can detect cancer development at its earliest stages, which will improve overall survival.
Supplementary material
Supplementary Tables 1 and 2 and Figures 1–3 can be found at http://carcin.oxfordjournals.org/
Funding
National Institutes of Health (CA164623, CA129444, CA168312 and P30ES023512); American Institute for Cancer Research.
Conflict of Interest Statement: None declared.
Supplementary Material
Glossary
Abbreviations
- AOM
azoxymethane
- BE
bystander effect
- CSC
cancer stem cell
- DSB
double strand break
- HR
homologous repair
- Lgr5
leucine-rich repeat-containing G protein-coupled Receptor 5
- MGMT
O6-methylguanine-DNA methyltransferase
References
- 1. Barker N., et al. (2009) Crypt stem cells as the cells-of-origin of intestinal cancer. Nature, 457, 608–611. [DOI] [PubMed] [Google Scholar]
- 2. Li Q., et al. (2011) The response of intestinal stem cells and epithelium after alemtuzumab administration. Cell. Mol. Immunol., 8, 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Asfaha S., et al. (2015) Krt19(+)/Lgr5(-) cells are radioresistant cancer-initiating stem cells in the colon and intestine. Cell Stem Cell, 16, 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sato T., et al. (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature, 459, 262–265. [DOI] [PubMed] [Google Scholar]
- 5. Schepers A.G., et al. (2011) Lgr5 intestinal stem cells have high telomerase activity and randomly segregate their chromosomes. EMBO J., 30, 1104–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barker N., et al. (2010) Leucine-rich repeat-containing G-protein-coupled receptors as markers of adult stem cells. Gastroenterology, 138, 1681–1696. [DOI] [PubMed] [Google Scholar]
- 7. Li L., et al. (2010) Coexistence of quiescent and active adult stem cells in mammals. Science, 327, 542–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hua G., et al. (2012) Crypt base columnar stem cells in small intestines of mice are radioresistant. Gastroenterology, 143, 1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Metcalfe C., et al. (2014) Lgr5+ stem cells are indispensable for radiation-induced intestinal regeneration. Cell Stem Cell, 14, 149–159. [DOI] [PubMed] [Google Scholar]
- 10. Meikrantz W., et al. (1998) O6-alkylguanine DNA lesions trigger apoptosis. Carcinogenesis, 19, 369–372. [DOI] [PubMed] [Google Scholar]
- 11. Boldogh I., et al. (1998) Regulation of expression of the DNA repair gene O6-methylguanine-DNA methyltransferase via protein kinase C-mediated signaling. Cancer Res., 58, 3950–3956. [PubMed] [Google Scholar]
- 12. Montesano R., et al. (1990) Alkylation repair in human tissues. Basic Life Sci., 53, 437–452. [DOI] [PubMed] [Google Scholar]
- 13. Thompson C.B. (1995) Apoptosis in the pathogenesis and treatment of disease. Science, 267, 1456–1462. [DOI] [PubMed] [Google Scholar]
- 14. Hirose Y., et al. (1996) Early alterations of apoptosis and cell proliferation in azoxymethane-initiated rat colonic epithelium. Jpn. J. Cancer Res., 87, 575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tomasetti C., et al. (2015) Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science, 347, 78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hong M.Y., et al. (1999) Relationship between DNA adduct levels, repair enzyme, and apoptosis as a function of DNA methylation by azoxymethane. Cell Growth Differ., 10, 749–758. [PubMed] [Google Scholar]
- 17. Hong M.Y., et al. (2000) Dietary fish oil reduces O6-methylguanine DNA adduct levels in rat colon in part by increasing apoptosis during tumor initiation. Cancer Epidemiol. Biomarkers Prev., 9, 819–826. [PubMed] [Google Scholar]
- 18. Hawks A., et al. (1974) The alkylation of nucleic acids of rat and mouse in vivo by the carcinogen 1,2-dimethylhydrazine. Br. J. Cancer, 30, 440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu Y., et al. (2002) The colonic response to genotoxic carcinogens in the rat: regulation by dietary fibre. Carcinogenesis, 23, 1131–1137. [DOI] [PubMed] [Google Scholar]
- 20. Barker N., et al. (2007) Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature, 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- 21. Kondo N., et al. (2010) DNA damage induced by alkylating agents and repair pathways. J. Nucleic Acids, 2010, 543531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karran P., et al. (1992) Self-destruction and tolerance in resistance of mammalian cells to alkylation damage. Nucleic Acids Res., 20, 2933–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuo L.J., et al. (2008) Gamma-H2AX - a novel biomarker for DNA double-strand breaks. In Vivo, 22, 305–309. [PubMed] [Google Scholar]
- 24. Staszewski O., et al. (2008) Kinetics of gamma-H2AX focus formation upon treatment of cells with UV light and alkylating agents. Environ. Mol. Mutagen., 49, 734–740. [DOI] [PubMed] [Google Scholar]
- 25. Dumitru R., et al. (2012) Human embryonic stem cells have constitutively active Bax at the Golgi and are primed to undergo rapid apoptosis. Mol. Cell, 46, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu J.C., et al. (2013) High mitochondrial priming sensitizes hESCs to DNA-damage-induced apoptosis. Cell Stem Cell, 13, 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Verzi M.P., et al. (2010) Stem cells: the intestinal-crypt casino. Nature, 467, 1055–1056. [DOI] [PubMed] [Google Scholar]
- 28. Kruyt F.A., et al. (2010) Apoptosis and cancer stem cells: Implications for apoptosis targeted therapy. Biochem. Pharmacol., 80, 423–430. [DOI] [PubMed] [Google Scholar]
- 29. Pegg A.E., et al. (1992) Repair of DNA containing O6-alkylguanine. FASEB J., 6, 2302–2310. [DOI] [PubMed] [Google Scholar]
- 30. Zaidi N.H., et al. (1996) Quantitative immunohistochemical estimates of O6-alkylguanine-DNA alkyltransferase expression in normal and malignant human colon. Clin. Cancer Res., 2, 577–584. [PubMed] [Google Scholar]
- 31. Esteller M., et al. (2000) Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res., 60, 2368–2371. [PubMed] [Google Scholar]
- 32. Ciccia A., et al. (2010) The DNA damage response: making it safe to play with knives. Mol. Cell, 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chapkin R.S., et al. (1999) Colonic cell proliferation and apoptosis in rodent species. Modulation by diet. Adv. Exp. Med. Biol., 470, 105–118. [DOI] [PubMed] [Google Scholar]
- 34. Colussi C., et al. (2001) 1,2-Dimethylhydrazine-induced colon carcinoma and lymphoma in msh2(-/-) mice. J. Natl. Cancer Inst., 93, 1534–1540. [DOI] [PubMed] [Google Scholar]
- 35. Richards TC. (1977) Early changes in the dynamics of crypt cell populations in mouse colon following administration of 1,2-dimethylhydrazine. Cancer Res., 37, 1680–1685. [PubMed] [Google Scholar]
- 36. James J.T., et al. (1983) Methylated DNA adducts in the large intestine of ICR/Ha and C57BL/Ha mice given 1,2-dimethylhydrazine. J. Natl. Cancer Inst., 70, 541–546. [PubMed] [Google Scholar]
- 37. Hall C.N., et al. (1991) The detection of alkylation damage in the DNA of human gastrointestinal tissues. Br. J. Cancer, 64, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hata K., et al. (2004) Tumor formation is correlated with expression of beta-catenin-accumulated crypts in azoxymethane-induced colon carcinogenesis in mice. Cancer Sci., 95, 316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Takahashi M., et al. (2004) Gene mutations and altered gene expression in azoxymethane-induced colon carcinogenesis in rodents. Cancer Sci., 95, 475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Deschner E.E., et al. (1977) Colonic neoplasms in mice produced with six injections of 1,2-dimethylhydrazine. Oncology, 34, 255–257. [DOI] [PubMed] [Google Scholar]
- 41. Chandler J.M., et al. (2010) Cancerous stem cells: deviant stem cells with cancer-causing misbehavior. Stem Cell Res. Ther., 1, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tominaga Y., et al. (1997) Alkylation-induced apoptosis of embryonic stem cells in which the gene for DNA-repair, methyltransferase, had been disrupted by gene targeting. Carcinogenesis, 18, 889–896. [DOI] [PubMed] [Google Scholar]
- 43. Bhanja P., et al. (2009) Protective role of R-spondin1, an intestinal stem cell growth factor, against radiation-induced gastrointestinal syndrome in mice. PLoS One, 4, e8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fan X.S., et al. (2010) Expression of Lgr5 in human colorectal carcinogenesis and its potential correlation with beta-catenin. Int. J. Colorectal Dis., 25, 583–590. [DOI] [PubMed] [Google Scholar]
- 45. McClanahan T., et al. (2006) Identification of overexpression of orphan G protein-coupled receptor GPR49 in human colon and ovarian primary tumors. Cancer Biol. Ther., 5, 419–426. [DOI] [PubMed] [Google Scholar]
- 46. O’Neill J.P. (2000) DNA damage, DNA repair, cell proliferation, and DNA replication: how do gene mutations result? Proc. Natl. Acad. Sci. USA, 97, 11137–11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hu Y., et al. (2005) Absence of acute apoptotic response to genotoxic carcinogens in p53-deficient mice is associated with increased susceptibility to azoxymethane-induced colon tumours. Int. J. Cancer, 115, 561–567. [DOI] [PubMed] [Google Scholar]
- 48. Coquerelle T., et al. (1995) Overexpression of N-methylpurine-DNA glycosylase in Chinese hamster ovary cells renders them more sensitive to the production of chromosomal aberrations by methylating agents–a case of imbalanced DNA repair. Mutat. Res., 336, 9–17. [DOI] [PubMed] [Google Scholar]
- 49. Yang Y., et al. (2013) Colon macrophages polarized by commensal bacteria cause colitis and cancer through the bystander effect. Transl. Oncol., 6, 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Furlong H., et al. (2013) Apoptosis is signalled early by low doses of ionising radiation in a radiation-induced bystander effect. Mutat. Res., 741-742, 35–43. [DOI] [PubMed] [Google Scholar]
- 51. Dickey J.S., et al. (2011) H2AX phosphorylation in response to DNA double-strand break formation during bystander signalling: effect of microRNA knockdown. Radiat. Prot. Dosimetry, 143, 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Burdak-Rothkamm S., et al. (2007) ATR-dependent radiation-induced gamma H2AX foci in bystander primary human astrocytes and glioma cells. Oncogene, 26, 993–1002. [DOI] [PubMed] [Google Scholar]
- 53. Al-Mayah A.H., et al. (2012) Possible role of exosomes containing RNA in mediating nontargeted effect of ionizing radiation. Radiat. Res., 177, 539–545. [DOI] [PubMed] [Google Scholar]
- 54. Riehl T.E., et al. (2006) Azoxymethane protects intestinal stem cells and reduces crypt epithelial mitosis through a COX-1-dependent mechanism. Am. J. Physiol. Gastrointest. Liver Physiol., 291, G1062–G1070. [DOI] [PubMed] [Google Scholar]
- 55. Zhou H., et al. (2005) Mechanism of radiation-induced bystander effect: role of the cyclooxygenase-2 signaling pathway. Proc. Natl. Acad. Sci. USA, 102, 14641–14646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kycler W., et al. (2012) Analysis of O6-methylguanine-DNA methyltransferase methylation status in sporadic colon polyps. Rep. Pract. Oncol. Radiother., 17, 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Blough M.D., et al. (2007) O6-methylguanine-DNA methyltransferase regulation by p53 in astrocytic cells. Cancer Res., 67, 580–584. [DOI] [PubMed] [Google Scholar]
- 58. Russell S.J., et al. (1995) p53 mutations, O6-alkylguanine DNA alkyltransferase activity, and sensitivity to procarbazine in human brain tumors. Cancer, 75, 1339–1342. [DOI] [PubMed] [Google Scholar]
- 59. Gerson S.L., et al. (1995) Determinants of O6-alkylguanine-DNA alkyltransferase activity in human colon cancer. Clin. Cancer Res., 1, 519–525. [PubMed] [Google Scholar]
- 60. Kenyon J., et al. (2007) The role of DNA damage repair in aging of adult stem cells. Nucleic Acids Res., 35, 7557–7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dean M., et al. (2005) Tumour stem cells and drug resistance. Nat. Rev. Cancer, 5, 275–284. [DOI] [PubMed] [Google Scholar]
- 62. Donnenberg V.S., et al. (2005) Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J. Clin. Pharmacol., 45, 872–877. [DOI] [PubMed] [Google Scholar]
- 63. Medema J.P. (2013) Cancer stem cells: the challenges ahead. Nat. Cell Biol., 15, 338–344. [DOI] [PubMed] [Google Scholar]
- 64. Pisco A.O., et al. (2015) Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: ‘What does not kill me strengthens me’. Br. J. Cancer, 112, 1725–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.