

Abstract
Motor neuron disease (MND), also known as amyotrophic lateral sclerosis, is a relentlessly progressive neurodegenerative condition that is invariably fatal, usually within 3 to 5 years of diagnosis. The aetio-pathogenesis of MND remains unresolved and no effective treatments exist. The only Food and Drug Administration approved disease modifying therapy is riluzole, a glutamate antagonist, which prolongs survival by up to 3 mo. Current management is largely symptomatic/supportive. There is therefore a desperate and unmet clinical need for discovery of disease mechanisms to guide novel therapeutic strategy. In this review, we start by introducing the organizational anatomy of the motor system, before providing a clinical overview of its dysfunction specifically in MND. We then summarize insights gained from pathological, genetic and animal models and conclude by speculating on optimal strategies to drive the step change in discovery, which is so desperately needed in this arena.
Keywords: Motor neuron disease, Amyotrophic lateral sclerosis, Neurodegeneration, Disease models
Core tip: Motor neuron disease (MND) is a fatal neurodegenerative disorder with no known cure. Here we discuss the organization of the motor system and the clinical presentation of MND. We detail the diagnostic criteria for MND including electrophysiological studies and potential future diagnostic markers of disease. We discuss the staging of disease progression in MND. We then provide an overview of disease management and end with insights into molecular pathogenesis of the disease and the use of disease models.
ORGANIZATIONAL ANATOMY OF THE MOTOR SYSTEM
The staggering complexity of the vertebrate nervous system is directed largely at the generation and regulation of movement by the careful choreography of muscles responsible for walking, talking and breathing. The motor system can be categorized most simply into upper and lower divisions. Betz cells within both frontal lobe motor cortices are classically large pyramidal upper motor neurons (MNs). Their smaller cortical counterparts densely populate the motor and premotor cortices. Upper MNs control lower MNs in the spinal cord either directly (monosynaptic input) or indirectly (through spinal interneurons). Descending MNs in the spinal cord travel in laterally partitioned corticospinal tracts, most of which cross the midline at the level of the lower brainstem medullary pyramids to synapse contralaterally within the spinal cord. There are also anterior corticospinal tracts, which do not cross at the medullary pyramids but remain ipsilateral. Notably, a minority of spinal cord regions are innervated by these anterior corticospinal projections, which branch and innervate on both sides of the spinal cord, crossing at the appropriate spinal segment. Direct synaptic connection between upper and lower MNs is likely a recent development in evolution, given that it is exclusive to higher primates.
Lower MNs are anatomically positioned in the ventral horns of the spinal cord and motor nuclei within the brainstem; these in turn synapse at neuromuscular junctions and muscle spindles forming a final common pathway for voluntary movement. Spinal MNs are large, polarized cells with long axons, and are the conduit through which the motor cortex in the brain activates contraction of skeletal muscles. These multipolar cells can project axons over a meter long and each innervate up to 1000 muscle fibres. Remarkably, their extensive dentritic arborisation can accommodate up to 10000 synaptic terminals, receiving input from descending upper MNs and spinal interneurons. Despite certain generic properties, distinct molecular phenotypes of MNs exist. Even seemingly simple motor actions require collaboration and coordination of multiple MN subtypes, which are anatomically organized into motor columns and further grouped into motor pools in a muscle-specific manner. The generation of MN subtype diversity is an absolute pre-requisite to survival. In total, the human body has more than 100000 spinal MNs, which innervate 600 peripheral muscle targets organized into bilateral pairs. MNs can be classified according to the type of motor unit they generate into alpha, beta, and gamma. Alpha MNs abound in the motor system and innervate extrafusal skeletal muscle to generate contractile force and movement. Alpha MNs can be further codified by the contractile properties of muscle fibers they innervate into fast-twitch fatigable, fast twitch fatigue resistant, and slow twitch fatigue resistant[1]. Beta MNs innervate both intra- and extrafusal fibres, although these are the least well-understood MN class. Gamma MNs innervate intrafusal muscle fibers of the spindle, modulating their sensitivity to stretch[2,3]. Compared to alpha MNs, gamma MNs possess smaller cell somae, slower axonal conduction velocities, less complex dendritic arrangements and they lack monosynaptic input from proprioceptive sensory neurons[4-8]. This degree of structural and functional diversity commands distinct developmental lineage restriction programs for each different class of MN.
MN subclasses are spatially allocated into groups that reflect both their developmental origins and also their adult function. This coupling of developmental origin to adult function is depicted in Figure 1. MNs are developmentally partitioned into discrete motor columns, which extend along the rostro-caudal (R-C) neural tube. Within a column, the group of MNs responsible for innervating a single skeletal muscle is termed a motor pool, each of which is also arranged by an anatomical logic related to the muscle target(s) of its projections. The medial motor column (MMC) contains MNs that innervate dorsal epaxial muscles, which mainly subserve postural functions. Hypaxial motor column (HMC) MNs project to the ventral hypaxial muscles, which are mainly involved in respiration. The lateral motor columns (LMC) are responsible for innervating limb muscles. The preganglionic motor column (PGC) is present at thoracic levels and MNs originating from here innervate sympathetic ganglia. The MMCs run throughout the R-C extent of the spinal cord, while the LMCs, HMCs and PGCs occur only at brachio-lumbar (LMCs) and thoracic (HMCs and PGCs) foci (Figure 1). Against this background, the simple term “MN” thus fails to capture myriad subtype differences including rostrocaudal position, motor column and axonal trajectory. This striking complexity is an absolute pre-requisite to normal motor function.
Figure 1.
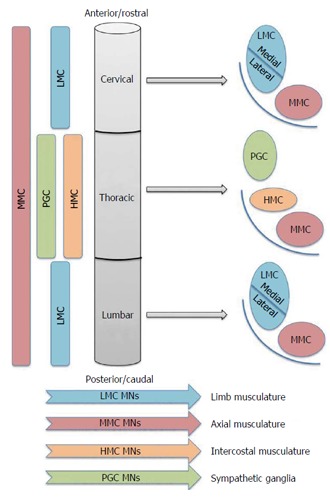
The motor columns of the spinal cord. The LMCs innervate the muscles of the upper and lower limbs, the MMC innervates axial musculature and the HMC and PGC are in the thoracic spinal cord and innervate the intercostal musculature and sympathetic ganglia respectively. LMC: Lateral motor column; MMC: Medial motor column; HMC: Hypaxial motor column; PGC: Preganglionic motor column; MNs: Motor neurons.
MN DISEASE - A CLINICAL PERSPECTIVE
MN disease (MND) causes progressive MN degeneration in the anterior horn of the spinal cord, brain stem and motor cortex[9-12], invariably leading to fatal paralysis usually through respiratory failure[13,14]. The lifetime risk of MND is 1:400 in those of European ancestry[15]. Most cases (90%) are sporadic and affect men more than women. It can present at any age, but with a peak incidence in the sixth to seventh decades of life. Familial MND is caused by mutations in a variety of genes, about 60% of which are now identified[16-18]. Clinically, the patient history and examination typically suggest evidence of upper and lower MN dysfunction in the absence of sensory or autonomic symptoms or signs. A striking clinical feature of this condition is the near universal sparing of the oculomotor nerves and the MNs in the sacral spinal cord that are responsible for pelvic sphincter control, called Onufrowicz nucleus.
Although initial presentation is quite variable, limb muscle weakness often begins focally (over 60% of cases, approximately equally distributed over upper and lower limb) and spreads in an orderly/stereotyped fashion, although overall patterns of motor weakness do vary quite widely between patients. While not pathognomonic, the so called “split-hand phenomenon” is certainly a well-recognized feature of MND, clinically presenting as lateral hand muscle atrophy (i.e., thenar eminence and first dorsal interosseous) with comparative normality of the medial hand muscles. Approximately 30% of patients present with bulbar symptoms, which include dysarthria, dysphagia and sialorrhoea. Sialorrhoea is caused by inability to swallow secretions due to a combination of tongue spasticity, weakness of the facial, mouth and pharyngeal muscles, and loss of oropharyngeal co-ordination and function[19]. Pseudobulbar palsy is also a recognized feature of MND, which can manifest clinically with spasticity of the tongue or of speech, a brisk jaw jerk, a positive gag reflex and mood incongruent emotionality. Muscle cramps and hypersalivation are common symptoms, and head drop, bilateral tongue wasting and widespread fasciculations important physical signs. Fasciculations can be a prominent and early sign in the disease[20]. Although only a minority of patients with MND initially present with acute respiratory failure, the majority do progress to this; indeed it is often the cause of their ultimate demise. The El Escorial criteria can facilitate diagnosis of MND. Combined upper MN and lower MN dysfunction can be difficult to detect in early disease, sometimes explaining diagnostic uncertainty both between conceivable differential diagnoses (Table 1) and different MND subtypes (Table 2). Although a period of observation can be valuable for diagnostic clarification in this context (as concurrent upper MN and lower MN involvement will typically become more evident as the disease progresses), one must take into careful consideration the importance of making a timely diagnosis. Most MND patients who present with predominantly upper MN pathology will develop lower MN signs within 3 or 4 years. The clinical diagnosis of MND is usually fairly self-evident, however it is critical not to miss any possible differential diagnoses listed in Table 2, as suggested by the history, examination and paraclinical tests.
Table 1.
Possible differential diagnoses and diagnostic clues to discriminate from motor neuron disease[23]
| Alternative diagnosis | Diagnostic clue |
| Cervical (myelo) neuropathy | Cervicalgia, osteopaenia/osteoporosis, abnormal cervical MRI |
| Benign fasciculations | Absence of weakness, limited distribution, young age |
| Nutritional (B12 or Cu deficiency) | Usually have sensory impairment |
| Motor predominant CIDP | Relapsing-remitting course, evidence of demyelination on NCS, IVIG-responsive |
| Multifocal motor neuropathy with conduction block | Weakness with little wasting, distal and slowly progressive, absent bulbar involvement, conduction block on NCS |
| Autoimmune and paraneoplastic | e.g., stiff person’s syndrome: GAD, amphiphysin, gephyrin antibodies, EMG differences |
| HIV, HTLV1 | HIV: History, sensory neuropathy, opportunistic infections |
| Parsonage-Turner syndrome (or brachial neuritis) | Preceded by pain, preceding vaccination/viral illness, process arrests and followed by recovery, usually upper limb |
| Inclusion body myositis | Distribution - forearm and quadriceps, raised CK, muscle biopsy |
| Hirayama’s disease | Upper limb, young males from Asia, unilateral, may arrest after a few years |
| Radiation-induced motor neuropathies | History and distribution of radiotherapy |
| Kennedy’s disease | Family history (X-linked), gynecomastia |
| Spinal muscular atrophy | Only affects lower MNs |
| Primary progressive multiple sclerosis | MRI and/or cerebrospinal fluid (oligoclonal bands) |
| Adrenoleucodystrophy | Family history (X-linked), adult onset, slowly progressive, usually have sensory ataxia and sphincteric involvement |
| Hexosaminidase A deficiency | Family history, dystonia, ataxia, psychosis |
| Poliomyelitis or post-polio syndrome | Clinical history and NCS/EMG |
| Hereditary spastic paraparesis | Family history and genetic testing |
Cu: Copper; CIDP: Chronic inflammatory demyelinating polyneuropathy; NCS: Nerve conduction studies; IVIG: Intravenous immunoglobulin; GAD: Glutamic acid decarboxylase; EMG: Electromyography; HIV: Human immunodeficiency virus; HTLV: Human T-cell lymphotropic virus; MRI: Magnetic resonance imaging; CK: Creatinine kinase.
Table 2.
Motor neuron disease subtypes, discriminating features and possible differential diagnoses
| MND subtype | Clinical features | Possible differential diagnoses |
| ALS | Affect both upper MNs and lower MNs | Cervical myeloneuropathy |
| Onset 50 or 60 s | HIV | |
| Median survival 3 to 5 yr | ||
| PLS | Only affect upper MNs 3 yr from onset | Cervical myelopathy |
| Onset 50 s | Nutritional (B12 or Cu deficiency) | |
| Profound spasticity | Primary progressive multiple sclerosis | |
| Progressive quadriparesis | Hereditary spastic paraparesis | |
| Late cranial nerve involvement | Stiff person syndrome | |
| Rarely bulbar onset | Tropical spastic paraparesis (HTLV1) | |
| Slow progression | Adrenomyeloneuropathy | |
| Median survival 5 to 10 yr | Hexosaminidase A deficiency | |
| Corticobasal degeneration | ||
| PMA | Only affect upper MNs 3 yr from onset | Benign fasciculations |
| Focal asymmetric distal weakness, followed by proximal involvement | Post-polio syndrome | |
| Late bullar/respiratory involvement | Adult onset spinal muscular atrophy | |
| Earlier onset than ALS | Inclusion body myositis | |
| Raised CK (< 10 × normal) | ||
| Median survival 3 to 5 yr |
HIV: Human immunodeficiency virus; Cu: Copper; CK: Creatinine kinase; ALS: Amyotrophic lateral sclerosis; PLS: Primary lateral sclerosis; HTLV: Human T-cell lymphotropic virus; PMA: Progressive muscular atrophy.
The Revised El Escorial diagnostic criteria and the Awaji electrodiagnostic criteria are well established for the clinical diagnosis of MND and evaluate evidence for progressive degeneration of upper MNs and lower MNs in the absence of other disease processes that could explain the clinical findings[21-23]. There are three diagnostic categories: Clinically definite, probable or possible MND. Importantly, the Awaji criteria established equivalent importance of both clinical and electrophysiological findings when detecting chronic neurogenic changes[24]. A study prior to the introduction of the Awaji criteria found that 29% of MND patients died without a diagnosis of definite MND[25]. The Awaji diagnostic criteria have been shown to increase the sensitivity of MND diagnosis[24,26]. As the diagnosis is made on the basis of upper MN and lower MN involvement in bulbar and spinal regions, the addition of electrophysiology for more sensitive detection of lower MN involvement facilitates the diagnosis. Evidence for neurogenic changes on the electromyography (EMG) should be sought[23]. Chronic neurogenic change may be demonstrated by motor unit potentials (MUPs) of increased amplitude and duration usually with increased number of phases and decreased motor unit recruitment or using a narrow pass filter to detect unstable or complex MUPs. Fibrillation potentials with positive sharp waves may be observed and fasciculation potentials with complex morphology, in the presence of chronic neurogenic change on needle EMG, may also be seen. The Revised El Escorial and Awaji criteria have proved very useful for diagnosis, especially for determining patient inclusion for clinical trials, however for use in clinical practice it is proposed that these criteria should be updated, to reflect the phenotypic heterogeneity of MND, the stage of disease and the presence of familial disease[27].
Similarly the use of investigations to support upper MN involvement would add further diagnostic certainty. Transcranial magnetic stimulation (TMS) is a technique used to measure corticomotoneuronal function with the parameters of motor threshold, motor evoked potential amplitude, central motor conduction time, cortical silent period, intracortical inhibition and facilitation[28]. Early cortical hyperexcitability, which may reflect glutamate excitotoxicity, precedes lower MN involvement in MND, and through the course of the disease this hyperexcitability decreases[28-32]. Threshold tracking TMS has the potential for use as a diagnostic marker and distinguishes MND from non-MND disorders with a sensitivity of 73.21% and specificity of 80.88% at an early disease stage[33]. Three hypotheses for MN death have been proposed: (1) a “dying-forward” phenomenon, where diseases initiates in upper MNs, leading to excitotoxic death of lower MNs; (2) a “dying-back” phenomenon, where disease begins at the lower MN level and progresses back to the upper MNs; or (3) an independent-degeneration phenomenon. The finding that cortical hyperexcitability starts below lower MN involvement supports the “dying-forward” hypothesis. Furthermore neuroimaging techniques, such as diffusion tensor magnetic resonance imaging, are showing promise for determining motor cortex and corticospinal tract involvement in disease, and could be used as biomarkers of disease and predictors of prognosis[34].
Various staging systems have been devised to measure disease progression in MND[35-40]. Individuals can progress through the disease at very variable rates[41,42], and as each clinical stage is reached at a consistent proportion through the disease process, staging can be used to make more useful comparisons between patients[35,36]. Furthermore, incremental stages correspond to decreasing function and health utility, and can be used in cost-benefit analyses of new treatments[38]. An important application of staging is as an endpoint in clinical trial design. The goal is to develop therapies which would prolong time in the earlier stages of disease, when function and quality of life are better, as compared to the later stages.
Cognitive impairment is recognized in up to half of patients with MND, usually detectable on neuropsychological testing rather than from routine clinical evaluation. However, frank dementia of the frontotemporal lobar degeneration (FTLD) type is increasingly diagnosed against the background of pathological and genetic discoveries that have mechanistically linked these two conditions together over the last decade[43]. Conversely, some patients presenting with FTLD will have clinical and para-clinical evidence of MND and the mode of presentation here is likely determined by the same pathomechanistic process starting/predominating at different neuraxial sites. Approximately 15% of MND patients have a clinical diagnosis of FTLD and 15% of FTLD patients have a diagnosis of MND[43,44].
Both European Federation of Neurological Societies and American Academy of Neurology guidelines for the management of MND patients have guided management to some degree in the United Kingdom[45-48]. Following a review decision in November 2014, the national institute for health and care excellence (NICE) is currently developing a guideline for the management of MND. This will ultimately replace the current NICE guideline on non-invasive ventilation in MND. The MND Association website offers a comprehensive list of available regional and national/international guidelines in specific MND-related areas, with direct links to documents. Indeed the support of the MND Association in all respects is frequently fed back as being highly valued by patients and carers. Most patients will experience hypoventilation/orthopnea as the disease progresses, justifying proactive interval monitoring of respiratory performance (including nocturnal oximetry, dynamic forced vital capacity, and maximal inspiratory pressure). Noninvasive positive pressure ventilation should be accessible when needed. Importantly, the management of MND should be in a multidisciplinary clinical setting, including experts in neurology, respiratory medicine, nutrition, psychology/psychiatry, speech therapy, physical and occupational therapy, social work, and case management. Other supportive measures include reactive and proactive interval examination of swallowing function as MND increases risk of aspiration. It is noteworthy that parotid/submandibular botulinum toxin injections can be helpful for sialorrhoea[19,49]. Consideration of a percutaneous gastrostomy tube can help to maintain body weight and hydration in MND. Pseudobulbar affect is often treated off-licence with selective serotonin reuptake inhibitors or tricyclic antidepressants. In October 2010, Food and Drug Administration approved a dextromethorphan-quinidine combination for symptomatic relief of pseudobulbar affect.
LESSONS FROM PATHOLOGICAL, GENETIC, ANIMAL AND CELLULAR MODELS
Various experimental strategies including in-vivo studies, cell based in-vitro approaches and human post-mortem neuropathological specimens from MND patients have been employed in order to improve understanding of this disease. Human stem cell strategies are becoming an increasingly important component of the armoury of investigative tools used to study disease mechanisms and identify potential therapeutic targets[50,51].
Historically, the most intensively studied cause of familial MND has been mutations in the copper/zinc superoxide dismutase (SOD1) gene, which account for approximately 15% of cases of familial MND and less than 5% of sporadic MND cases. The mutant SOD1 protein characteristically maintains its dismutase function, but appears to cause MN degeneration through alternative mechanisms, including a possible toxic gain of function[52]. Well over 100 individual point mutations located throughout the primary structure of SOD1 are sufficient to cause disease, suggesting protein-folding abnormalities as a possible initiating event. Transgenic mice globally expressing mutant forms of human SOD1 exhibit selective MN degeneration, which broadly mirrors the pathology of human sporadic and familial MND. Unfortunately, despite countless pre-clinical and clinical trials based on SOD1 models, not one of these has led to a significant therapeutic advance in MND. A landmark study in 2006 then discovered that the pathological hallmark of > 95% MND cases (sporadic and familial) is cytoplasmic misaccumulation of ubiquitinated and hyperphosphorylated transactive response DNA-binding protein (TDP-43)[53], a highly conserved, ubiquitously expressed and multifunctional nuclear protein with both DNA and RNA binding capacities[54-56]. A striking observation made in this work was that TDP-43 appeared mislocalised from the nucleus to the cytoplasm in MND and FTLD, although the pathophysiological significance of this remains incompletely understood. Interestingly, TDP-43 immunoreactive inclusions are found in both neurons and glia in MND and FTLD, hence their proposed taxonomic reclassification as TDP-43 “proteinopathies”. SOD1 mutations do not produce this common hallmark of MND and may not therefore be pathomechanistically representative of the majority of MND. Different subtypes of FTLD are based upon the protein found in pathological inclusions: In 45% of cases this is TDP-43, in another 45% of cases this is tau, and in 10% of cases this is fused in sarcoma (FUS)[43,57].
Other recent discoveries identified MND-causing gene mutations in TDP-43 and FUS[58,59]; findings that both complement and extend previous pathological studies. Furthermore, two recent contemporaneous studies have identified another MND-causing intronic mutation that introduces long hexanucleotide repeats into C9orf72 pre-mRNA[60,61], which is the most frequent genetic cause of MND and a common cause of FTLD. TDP-43 and FUS are both RNA-binding proteins. Collectively, these discoveries implicate a dysregulation of RNA metabolism as playing a crucial role in MND pathogenesis. In addition to these genes, several further mutations have been discovered including in the following genes: PGRN, UBQLN2, SQSTM1, PFN1, ANG, VCP, MATR3, TUB4A. Taken together, gene mutations and pathological studies implicate both protein misfolding/aggregation and perturbed RNA regulation as key underlying pathways in the molecular pathogenesis of MND[43,58,59,62-66].
A widely held view regarding the pathogenesis of neurodegenerative disease posits that selective injury to a disease-specific subclass of neurons is mechanistically cell autonomous. This “neuron-centric” view has been increasingly challenged by pivotal mice-chimera studies using lineage-specific expression of mutant SOD1 and subsequent related investigation, which confirmed a major non cell-autonomous role for astrocytes and microglia in SOD1-related MND pathogenesis[67-69]. Non cell-autonomous injury has also recently been implicated in sporadic MND, raising the possibility of common pathogenic mechanisms[70,71].
The discovery of induced pluripotent stem cells (iPSC) enables patient-specific fibroblasts to be virally transduced with up to 4 transcription factors and “reprogrammed” into embryonic-like stem cells[72]. Using insights from developmental neurobiology, these cells can subsequently be treated with a programme of extrinsic cues to direct their differentiation into a range of regionally defined neurons and glia for further study[73-76]. Importantly, a variety of studies have confirmed the capacity of these terminally differentiated cells to recapitulate key pathological hallmarks of a range of different neurodegenerative diseases[71,77-79]. In particular, several important studies have already demonstrated that iPSC-derived neurons and glia from patients with monogenic and sporadic MND show pathological phenotypes when compared to their control counterparts. Furthermore, this reductionist and human in vitro model system allows assays that directly elucidate non cell autonomous mechanisms of disease[80]. Several studies have also confirmed the utility in this model system as a pre-clinical test-bed for drug discovery[81-83], including the practical feasibility of high throughput automated approaches[84].
FUTURE STRATEGIES
We conclude that the integration of human experimental approaches is required to drive the desperately needed discovery of disease mechanisms and therapeutic strategy in MND. Unfortunately animal models have failed to deliver a significant therapeutic advance in MND, despite numerous efforts and important discoveries. Human iPSC models can better approximate clinical MND not only by virtue of species, but also because they express mutations at accurate pathophysiological levels and thus bypass the need for artificial overexpression, knock down or knock out experiments. A multitude of studies have now validated the human iPSC technology for disease modeling of both developmental and adult-onset conditions and drug discovery. However, this remains an in vitro system and thus lacks the dynamic cellular and signaling environments of an in vivo model. The integration of transgenic animal models that recapitulate MND pathogenesis together with patient-specific iPSCs represents an unprecedented opportunity to capture the complexity of pathogenic events underlying this devastating condition. By combining these approaches at the pre-clinical phase, we firmly believe that the translational yield of clinical trials will increase in MND.
Footnotes
Supported by A Wellcome Trust Research Training Fellowship (107196/Z/15/Z); Wellcome Trust Clinician Scientist and an Anne Rowling Fellow in Regenerative Neurology.
Conflict-of-interest statement: No potential conflicts of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: November 28, 2015
First decision: December 3, 2015
Article in press: January 11, 2016
P- Reviewer: de Carvalho M, Pan HC S- Editor: Qiu S L- Editor: A E- Editor: Liu SQ
References
- 1.Burke RE, Levine DN, Tsairis P, Zajac FE. Physiological types and histochemical profiles in motor units of the cat gastrocnemius. J Physiol. 1973;234:723–748. doi: 10.1113/jphysiol.1973.sp010369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuffler SW, Hunt CC, Quilliam JP. Function of medullated small-nerve fibers in mammalian ventral roots; efferent muscle spindle innervation. J Neurophysiol. 1951;14:29–54. doi: 10.1152/jn.1951.14.1.29. [DOI] [PubMed] [Google Scholar]
- 3.Hunt CC, Kuffler SW. Stretch receptor discharges during muscle contraction. J Physiol. 1951;113:298–315. doi: 10.1113/jphysiol.1951.sp004573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Westbury DR. A comparison of the structures of alpha and gamma-spinal motoneurones of the cat. J Physiol. 1982;325:79–91. doi: 10.1113/jphysiol.1982.sp014137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burke RE, Strick PL, Kanda K, Kim CC, Walmsley B. Anatomy of medial gastrocnemius and soleus motor nuclei in cat spinal cord. J Neurophysiol. 1977;40:667–680. doi: 10.1152/jn.1977.40.3.667. [DOI] [PubMed] [Google Scholar]
- 6.Shneider NA, Brown MN, Smith CA, Pickel J, Alvarez FJ. Gamma motor neurons express distinct genetic markers at birth and require muscle spindle-derived GDNF for postnatal survival. Neural Dev. 2009;4:42. doi: 10.1186/1749-8104-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eccles JC, Eccles RM, Iggo A, Lundberg A. Electrophysiological studies on gamma motoneurones. Acta Physiol Scand. 1960;50:32–40. doi: 10.1111/j.1748-1716.1960.tb02070.x. [DOI] [PubMed] [Google Scholar]
- 8.Friese A, Kaltschmidt JA, Ladle DR, Sigrist M, Jessell TM, Arber S. Gamma and alpha motor neurons distinguished by expression of transcription factor Err3. Proc Natl Acad Sci USA. 2009;106:13588–13593. doi: 10.1073/pnas.0906809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saito M, Tomonaga M, Narabayashi H. Histochemical study of the muscle spindles in parkinsonism, motor neuron disease and myasthenia. An examination of the pathological fusimotor endings by the acetylcholinesterase technic. J Neurol. 1978;219:261–271. doi: 10.1007/BF00312979. [DOI] [PubMed] [Google Scholar]
- 10.Swash M, Fox KP. The pathology of the human muscle spindle: effect of denervation. J Neurol Sci. 1974;22:1–24. doi: 10.1016/0022-510x(74)90050-1. [DOI] [PubMed] [Google Scholar]
- 11.Swash M, Leader M, Brown A, Swettenham KW. Focal loss of anterior horn cells in the cervical cord in motor neuron disease. Brain. 1986;109(Pt 5):939–952. doi: 10.1093/brain/109.5.939. [DOI] [PubMed] [Google Scholar]
- 12.Vucic S, Rothstein JD, Kiernan MC. Advances in treating amyotrophic lateral sclerosis: insights from pathophysiological studies. Trends Neurosci. 2014;37:433–442. doi: 10.1016/j.tins.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Radunović A, Mitsumoto H, Leigh PN. Clinical care of patients with amyotrophic lateral sclerosis. Lancet Neurol. 2007;6:913–925. doi: 10.1016/S1474-4422(07)70244-2. [DOI] [PubMed] [Google Scholar]
- 14.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 15.Alonso A, Logroscino G, Jick SS, Hernán MA. Incidence and lifetime risk of motor neuron disease in the United Kingdom: a population-based study. Eur J Neurol. 2009;16:745–751. doi: 10.1111/j.1468-1331.2009.02586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marangi G, Traynor BJ. Genetic causes of amyotrophic lateral sclerosis: new genetic analysis methodologies entailing new opportunities and challenges. Brain Res. 2015;1607:75–93. doi: 10.1016/j.brainres.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, van den Berg LH. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012;124:339–352. doi: 10.1007/s00401-012-1022-4. [DOI] [PubMed] [Google Scholar]
- 19.Young CA, Ellis C, Johnson J, Sathasivam S, Pih N. Treatment for sialorrhea (excessive saliva) in people with motor neuron disease/amyotrophic lateral sclerosis. Cochrane Database Syst Rev. 2011;(5):CD006981. doi: 10.1002/14651858.CD006981.pub2. [DOI] [PubMed] [Google Scholar]
- 20.de Carvalho M. Pathophysiological significance of fasciculations in the early diagnosis of ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1 Suppl 1:S43–S46. doi: 10.1080/14660820050515539. [DOI] [PubMed] [Google Scholar]
- 21.Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci. 1994;124 Suppl:96–107. doi: 10.1016/0022-510x(94)90191-0. [DOI] [PubMed] [Google Scholar]
- 22.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 23.de Carvalho M, Dengler R, Eisen A, England JD, Kaji R, Kimura J, Mills K, Mitsumoto H, Nodera H, Shefner J, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008;119:497–503. doi: 10.1016/j.clinph.2007.09.143. [DOI] [PubMed] [Google Scholar]
- 24.Carvalho MD, Swash M. Awaji diagnostic algorithm increases sensitivity of El Escorial criteria for ALS diagnosis. Amyotroph Lateral Scler. 2009;10:53–57. doi: 10.1080/17482960802521126. [DOI] [PubMed] [Google Scholar]
- 25.Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman OM. Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie House diagnostic criteria: A population-based study. Arch Neurol. 2000;57:1171–1176. doi: 10.1001/archneur.57.8.1171. [DOI] [PubMed] [Google Scholar]
- 26.Costa J, Swash M, de Carvalho M. Awaji criteria for the diagnosis of amyotrophic lateral sclerosis: a systematic review. Arch Neurol. 2012;69:1410–1416. doi: 10.1001/archneurol.2012.254. [DOI] [PubMed] [Google Scholar]
- 27.Agosta F, Al-Chalabi A, Filippi M, Hardiman O, Kaji R, Meininger V, Nakano I, Shaw P, Shefner J, van den Berg LH, et al. The El Escorial criteria: strengths and weaknesses. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:1–7. doi: 10.3109/21678421.2014.964258. [DOI] [PubMed] [Google Scholar]
- 28.Vucic S, Ziemann U, Eisen A, Hallett M, Kiernan MC. Transcranial magnetic stimulation and amyotrophic lateral sclerosis: pathophysiological insights. J Neurol Neurosurg Psychiatry. 2013;84:1161–1170. doi: 10.1136/jnnp-2012-304019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eisen A, Weber M. The motor cortex and amyotrophic lateral sclerosis. Muscle Nerve. 2001;24:564–573. doi: 10.1002/mus.1042. [DOI] [PubMed] [Google Scholar]
- 30.Vucic S, Kiernan MC. Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain. 2006;129:2436–2446. doi: 10.1093/brain/awl172. [DOI] [PubMed] [Google Scholar]
- 31.Mills KR, Nithi KA. Corticomotor threshold is reduced in early sporadic amyotrophic lateral sclerosis. Muscle Nerve. 1997;20:1137–1141. doi: 10.1002/(sici)1097-4598(199709)20:9<1137::aid-mus7>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 32.Menon P, Kiernan MC, Vucic S. Cortical hyperexcitability precedes lower motor neuron dysfunction in ALS. Clin Neurophysiol. 2015;126:803–809. doi: 10.1016/j.clinph.2014.04.023. [DOI] [PubMed] [Google Scholar]
- 33.Menon P, Geevasinga N, Yiannikas C, Howells J, Kiernan MC, Vucic S. Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: a prospective study. Lancet Neurol. 2015;14:478–484. doi: 10.1016/S1474-4422(15)00014-9. [DOI] [PubMed] [Google Scholar]
- 34.Turner MR, Agosta F, Bede P, Govind V, Lulé D, Verstraete E. Neuroimaging in amyotrophic lateral sclerosis. Biomark Med. 2012;6:319–337. doi: 10.2217/bmm.12.26. [DOI] [PubMed] [Google Scholar]
- 35.Roche JC, Rojas-Garcia R, Scott KM, Scotton W, Ellis CE, Burman R, Wijesekera L, Turner MR, Leigh PN, Shaw CE, et al. A proposed staging system for amyotrophic lateral sclerosis. Brain. 2012;135:847–852. doi: 10.1093/brain/awr351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balendra R, Jones A, Jivraj N, Steen IN, Young CA, Shaw PJ, Turner MR, Leigh PN, Al-Chalabi A. Use of clinical staging in amyotrophic lateral sclerosis for phase 3 clinical trials. J Neurol Neurosurg Psychiatry. 2015;86:45–49. doi: 10.1136/jnnp-2013-306865. [DOI] [PubMed] [Google Scholar]
- 37.Balendra R, Jones A, Jivraj N, Knights C, Ellis CM, Burman R, Turner MR, Leigh PN, Shaw CE, Al-Chalabi A. Estimating clinical stage of amyotrophic lateral sclerosis from the ALS Functional Rating Scale. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:279–284. doi: 10.3109/21678421.2014.897357. [DOI] [PubMed] [Google Scholar]
- 38.Jones AR, Jivraj N, Balendra R, Murphy C, Kelly J, Thornhill M, Young C, Shaw PJ, Leigh PN, Turner MR, et al. Health utility decreases with increasing clinical stage in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:285–291. doi: 10.3109/21678421.2013.872149. [DOI] [PubMed] [Google Scholar]
- 39.Chiò A, Hammond ER, Mora G, Bonito V, Filippini G. Development and evaluation of a clinical staging system for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2015;86:38–44. doi: 10.1136/jnnp-2013-306589. [DOI] [PubMed] [Google Scholar]
- 40.Tramacere I, Dalla Bella E, Chiò A, Mora G, Filippini G, Lauria G. The MITOS system predicts long-term survival in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2015;86:1180–1185. doi: 10.1136/jnnp-2014-310176. [DOI] [PubMed] [Google Scholar]
- 41.Talbot K. Motor neuron disease: the bare essentials. Pract Neurol. 2009;9:303–309. doi: 10.1136/jnnp.2009.188151. [DOI] [PubMed] [Google Scholar]
- 42.Turner MR, Parton MJ, Shaw CE, Leigh PN, Al-Chalabi A. Prolonged survival in motor neuron disease: a descriptive study of the King’s database 1990-2002. J Neurol Neurosurg Psychiatry. 2003;74:995–997. doi: 10.1136/jnnp.74.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65:586–590. doi: 10.1212/01.wnl.0000172911.39167.b6. [DOI] [PubMed] [Google Scholar]
- 45.Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, Kalra S, Katz JS, Mitsumoto H, Rosenfeld J, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1227–1233. doi: 10.1212/WNL.0b013e3181bc01a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, Kalra S, Katz JS, Mitsumoto H, Rosenfeld J, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1218–1226. doi: 10.1212/WNL.0b013e3181bc0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller RG, Anderson F, Brooks BR, Mitsumoto H, Bradley WG, Ringel SP. Outcomes research in amyotrophic lateral sclerosis: lessons learned from the amyotrophic lateral sclerosis clinical assessment, research, and education database. Ann Neurol. 2009;65 Suppl 1:S24–S28. doi: 10.1002/ana.21556. [DOI] [PubMed] [Google Scholar]
- 48.Andersen PM, Abrahams S, Borasio GD, de Carvalho M, Chio A, Van Damme P, Hardiman O, Kollewe K, Morrison KE, Petri S, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force. Eur J Neurol. 2012;19:360–375. doi: 10.1111/j.1468-1331.2011.03501.x. [DOI] [PubMed] [Google Scholar]
- 49.Jenkins TM, Hollinger H, McDermott CJ. The evidence for symptomatic treatments in amyotrophic lateral sclerosis. Curr Opin Neurol. 2014;27:524–531. doi: 10.1097/WCO.0000000000000135. [DOI] [PubMed] [Google Scholar]
- 50.Wichterle H, Przedborski S. What can pluripotent stem cells teach us about neurodegenerative diseases? Nat Neurosci. 2010;13:800–804. doi: 10.1038/nn.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han SS, Williams LA, Eggan KC. Constructing and deconstructing stem cell models of neurological disease. Neuron. 2011;70:626–644. doi: 10.1016/j.neuron.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 52.Chen H, Qian K, Du Z, Cao J, Petersen A, Liu H, Blackbourn LW, Huang CL, Errigo A, Yin Y, et al. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell. 2014;14:796–809. doi: 10.1016/j.stem.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 54.Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem. 2001;276:36337–36343. doi: 10.1074/jbc.M104236200. [DOI] [PubMed] [Google Scholar]
- 55.Buratti E, Brindisi A, Pagani F, Baralle FE. Nuclear factor TDP-43 binds to the polymorphic TG repeats in CFTR intron 8 and causes skipping of exon 9: a functional link with disease penetrance. Am J Hum Genet. 2004;74:1322–1325. doi: 10.1086/420978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mercado PA, Ayala YM, Romano M, Buratti E, Baralle FE. Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res. 2005;33:6000–6010. doi: 10.1093/nar/gki897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 63.Valdmanis PN, Kabashi E, Dyck A, Hince P, Lee J, Dion P, D’Amour M, Souchon F, Bouchard JP, Salachas F, et al. Association of paraoxonase gene cluster polymorphisms with ALS in France, Quebec, and Sweden. Neurology. 2008;71:514–520. doi: 10.1212/01.wnl.0000324997.21272.0c. [DOI] [PubMed] [Google Scholar]
- 64.Valdmanis PN, Kabashi E, Dion PA, Rouleau GA. ALS predisposition modifiers: knock NOX, who’s there? SOD1 mice still are. Eur J Hum Genet. 2008;16:140–142. doi: 10.1038/sj.ejhg.5201961. [DOI] [PubMed] [Google Scholar]
- 65.Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IO, Pietrzyk J, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345:1192–1194. doi: 10.1126/science.1256800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y, McKnight SL. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345:1139–1145. doi: 10.1126/science.1254917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamanaka K, Boillee S, Roberts EA, Garcia ML, McAlonis-Downes M, Mikse OR, Cleveland DW, Goldstein LS. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc Natl Acad Sci USA. 2008;105:7594–7599. doi: 10.1073/pnas.0802556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 70.Meyer K, Ferraiuolo L, Miranda CJ, Likhite S, McElroy S, Renusch S, Ditsworth D, Lagier-Tourenne C, Smith RA, Ravits J, et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc Natl Acad Sci USA. 2014;111:829–832. doi: 10.1073/pnas.1314085111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29:824–828. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 73.Patani R, Lewis PA, Trabzuni D, Puddifoot CA, Wyllie DJ, Walker R, Smith C, Hardingham GE, Weale M, Hardy J, et al. Investigating the utility of human embryonic stem cell-derived neurons to model ageing and neurodegenerative disease using whole-genome gene expression and splicing analysis. J Neurochem. 2012;122:738–751. doi: 10.1111/j.1471-4159.2012.07825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Patani R, Hollins AJ, Wishart TM, Puddifoot CA, Alvarez S, de Lera AR, Wyllie DJ, Compston DA, Pedersen RA, Gillingwater TH, et al. Retinoid-independent motor neurogenesis from human embryonic stem cells reveals a medial columnar ground state. Nat Commun. 2011;2:214. doi: 10.1038/ncomms1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patani R, Compston A, Puddifoot CA, Wyllie DJ, Hardingham GE, Allen ND, Chandran S. Activin/Nodal inhibition alone accelerates highly efficient neural conversion from human embryonic stem cells and imposes a caudal positional identity. PLoS One. 2009;4:e7327. doi: 10.1371/journal.pone.0007327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gupta K, Patani R, Baxter P, Serio A, Story D, Tsujita T, Hayes JD, Pedersen RA, Hardingham GE, Chandran S. Human embryonic stem cell derived astrocytes mediate non-cell-autonomous neuroprotection through endogenous and drug-induced mechanisms. Cell Death Differ. 2012;19:779–787. doi: 10.1038/cdd.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, Mandal PK, Vera E, Shim JW, Kriks S, et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 2013;13:691–705. doi: 10.1016/j.stem.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee G, Ramirez CN, Kim H, Zeltner N, Liu B, Radu C, Bhinder B, Kim YJ, Choi IY, Mukherjee-Clavin B, et al. Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nat Biotechnol. 2012;30:1244–1248. doi: 10.1038/nbt.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ebert AD, Yu J, Rose FF, Mattis VB, Lorson CL, Thomson JA, Svendsen CN. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277–280. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. 2008;3:637–648. doi: 10.1016/j.stem.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 81.Yang YM, Gupta SK, Kim KJ, Powers BE, Cerqueira A, Wainger BJ, Ngo HD, Rosowski KA, Schein PA, Ackeifi CA, et al. A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell. 2013;12:713–726. doi: 10.1016/j.stem.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sareen D, O’Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5:208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maury Y, Côme J, Piskorowski RA, Salah-Mohellibi N, Chevaleyre V, Peschanski M, Martinat C, Nedelec S. Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat Biotechnol. 2015;33:89–96. doi: 10.1038/nbt.3049. [DOI] [PubMed] [Google Scholar]