

Abstract
The brain is a complex network system that has the capacity to support emotion, thought, action, learning and memory, and is characterized by constant activity, constant structural remodeling, and constant attempt to compensate for this remodeling. The basic insight that emerges from complex network organization is that substantively different networks can share common key organizational principles. Moreover, the interdependence of network organization and behavior has been successfully demonstrated for several specific tasks. From this viewpoint, increasing experimental/clinical observations suggest that mental disorders are neural network disorders. On one hand, single psychiatric disorders arise from multiple, multifactorial molecular and cellular structural/functional alterations spreading throughout local/global circuits leading to multifaceted and heterogeneous clinical symptoms. On the other hand, various mental diseases may share functional deficits across the same neural circuit as reflected in the overlap of symptoms throughout clinical diagnoses. An integrated framework including experimental measures and clinical observations will be necessary to formulate a coherent and comprehensive understanding of how neural connectivity mediates and constraints the phenotypic expression of psychiatric disorders.
Keywords: Neuron, Network, Synapse, Schizophrenia, Bipolar, Depression, Stress, Pain, Collapsin response mediator proteins
Core tip: Increasing evidences suggest that mental diseases are neural network disorders. Neurites and synapses represent the sub-cellular elements organizing these networks, and the molecules that regulate their formation, retraction and adaptive remodeling may contribute to the pathology of mental disorders. Various syndromes may share alterations of functional network leading to symptoms overlapping through clinical diagnoses.
INTRODUCTION
Despite their great diversity of morphology, most vertebrate nerve cells exhibit distinctive polarized structures with a single long axon and a specific dendritic arbor depending on their location. The axon/dendrite identity that influences the synaptic genesis and inputs that each neuron can integrate emerges as a convergent product of specific pattern of growth, branching and retraction and is differentially regulated at multiple points, including the control of the number of primary branches and their mode and frequency of branching by inhibitory (e.g., Sema3, Nogo-A) and permissive [e.g., brain derived neurotrophic factor (BDNF); fibroblast growth factor (FGF)][1-4] factors. Moreover, neurons do not connect indiscriminately between themselves but form an intricate non-random highly selective short [the hippocampal-prefrontal cortex (HIP-PFC) or hippocampal-amygdala-prefrontal cortex circuits][5,6] to long (the corpus callosum that connects the two cerebral hemispheres -facilitating an array of connective function- and influences higher cognition as well) range white matter axon fibers connections[7] producing characteristic networks capable of ensuring proper healthy behavior. The neuronal structure and neurotransmission in these networks, developed by interactions with the environment, are constantly remodeled via “activity-dependent synaptic plasticity”[8] to process, store and transmit relevant information. Accordingly, abnormal changes in the production of neurotrophic factors and permissive or inhibitory guidance cues may induce structural and/or functional abnormalities which may alter information processing and consequently link neuronal network formation/maintenance to mental disease(s). Since psychiatric disorders have overlapping symptoms - e.g., cognitive impairment and emotional dysregulation that can be found in schizophrenia, depression and anxiety disorders it is likely that this similarity is the consequence of disruption in common brain circuits (e.g., hippocampus-prefrontal cortex) over time. In this review, we provide new molecular and cellular insights detailed from gene targeting/genome-wide association data (GWAS)[9,10] that extend the understanding of mental diseases and improve their treatment, to network theories that highlight structural and functional brain connectivity. Structural connectivity corresponds to the anatomical neurites, synapses- connections between neural elements whereas the functional and effective connectivity refers to a statistical dependence between the physiological signals measured in each region and the influence that one region exerts over another respectively. We focus on two brain areas essential in the emotional and cognitive domains: The hippocampal formation (hipF) and the prefrontal cortex. Their functional coupling support multiple functions, including emotion, mood, memory, thinking about the past and future, self and others, which are altered in mental diseases.
MOLECULAR BASIS OF NEURONAL NETWORK GENESIS AND MAINTENANCE
Neurogenesis and neuritogenesis
The generation of neurons (neurogenesis) in various regions of the central nervous system depends on a carefully regulated process of neural progenitor cells proliferation and differentiation. During early development, the neural tube wall constitutes a pseudo-stratified epithelium made of highly polarized neuroepithelial cells. The proper amount of neurons is spatially and temporally controlled by accumulative activities of numerous extra-/intra-cellular factors. At the onset of neurogenesis and initiated by extracellular regulators (Notch, bone morphogenetic proteins, BDNF/TrKB/p75, Wnt/β-catenin/Sonic hedgehog)[11-13] and intracytoplasmic transcription factors (bHLH, Insm1, AP2γ, TRIM32)[14-16], these cells switch their identity and turn into radial glial cells (RG) expressing glutamate-aspartate transporter and brain-lipid binding protein[17]. They generate all diverse intermediate progenitor neurons and glial cells through orchestrations of numerous molecules (Nrg1, Foxg1, Retinoid acid)[18-20] and signaling mechanisms (Jack/Stat, Notch, BMP, FGF)[21-23]. Shortly after, the pro-neural factors (Ngn1/Ngn2/Ascl1, also known as Mash1)[24,25] and transcription factors (Salb2, Sima1Am2, Lhx2)[26-28] activate a generic program of neurogenesis, arrest the division of progenitor cells, suppress the alternative astroglial fate and select the neuronal fate. Subsequently, the newborn neurons extend neurites (neuritogenesis which begins with the concerted accumulation and organization of actin/microtubules) and migrate to appropriate locations. However, due to heterogeneous neuronal phenotypes generated at different times, in different layers and locations, there is considerable heterogeneity in neuritogenesis mechanisms. This makes it difficult to observe in vivo although we found recently that collapsin-response-mediator-protein (CRMP)3[29] has a profound influence on lamellopodia formation, neuritogenesis and dendritic arborization in vertebrate hippocampal neurons: The CRMP3-/- knock-out mice[30] display abnormal functional and structural neural networks associated with a delay in neurite outgrowth and alterations of dendrites and spines but not axon-morphology in hippocampal neurons during development that persist in adults. These alterations affect a subset of hippocampal circuits and hippocampal function: CRMP3-/- mice have a deficit in prepulse inhibition found in several mental diseases and abnormal long term potentiation (LTP). Such a bona fide mouse model is a critical first step towards exploring pathogenic mechanisms. There is increasing evidence that other CRMPs are involved in neurogenesis in the adult dentate gyrus and the olfactory system, and also regulate dendritic/axonal outgrowth[31-33]. Specifically, required for NT3-induced axon outgrowth/branching and linking kinesin to the Sra-1/WAVE cargo complex in axons, CRMP2 can convert established dendrites to axon or induce supernumerary axons[34] while mice lacking individual CRMP1, 4 or 5 present alterations in neuronal differentiation of specific brain areas and the behavior they control[35-37].
Another way to investigate neuritogenesis is using primary hippocampal neurons in culture. Few hours after plating, neurons start to extend minor neurites; then one of the multiple neurites extends rapidly and by morphological transformation generates the axon through stochastic selection; the other neurites mature into dendrites, leading to neuronal polarity. Live cell imaging using time-lapse video microscopy shows that the first two neurites have the highest potential to become axon. Because cultured neurons develop polarity without any presence of exogenous extracellular guidance/neurotropic cues, it has been suggested that an internal polarization program exists involving several intra-neuronal organelles and distinct repertoires of signaling molecules intrinsic to the neuron. Using that model, we reported that whereas neurons from heterozygous CRMP3+/- mice polarized and grew similarly to control wild type (WT), all CRMP3-/- neurons from homozygous CRMP3-/- littermates did not establish neuronal polarity. Such impairment in neurites to progress from stage 1 to stage 2 represents a failure of neurite initiation. Moreover, the correlation between the levels of CRMP3 expression and its activation of L- and N-type of voltage-gated calcium channels suggests that the facilitatory role of CRMP3 on neurite initiation, dendritic development and plasticity may be mediated via Ca2+ influx[38]. Other factors such as neurotrophins, extracellular matrix proteins, attractant/repellent guidance cues and guide-post proteins are considered extrinsic signals. Interactions between these molecules can provide short/long range guidance information or stably change the intrinsic ability of a neuron to extend/retract neurites during development or to engage them into an axono/dendritic differentiation path. It is tempting to suggest that similar players are required to refine neuritogenesis and to make synaptic connections (synaptogenesis) in vivo within the developing brain.
Synaptogenesis and neuronal network genesis
Brain complexity comes from the large diversity and number of neurons and the variety and number of synapses where neurons transfer their electrical and/or chemical signals to other neurons/cells. Electrical and chemical synapses differ in the molecular mechanisms supporting the transmission of information and in their morphological organization[39,40]. Their structure and composition vary across brain regions and their disruption in function and morphology may be involved in many neurological/mental diseases and after trauma. Electrical synapses are also a prerequisite for the chemical synapses formation in mammal brain during development. Within electrical synapses the gap junction channels processed by connexins and pannexins serve as conduits allowing a direct bidirectional communication and passage/exchange of metabolites, intracytoplasmic messengers, and ions between the cytoplasm of two cells (Figure 1B). Within neuronal networks, these electrical synapses provide synchronous electrical activity and field potential oscillations. They mediate an important form of direct intercellular communication and allow rapid transfers of pre-synaptic excitatory electrical impulses to post-synaptic potentials throughout the intercellular gap by generating synchronous oscillations of gamma-frequency (30-70 Hz) rhythms important for field potential oscillation within neuronal networks and necessary for the interplay of neural populations involved in memory processes.
Figure 1.
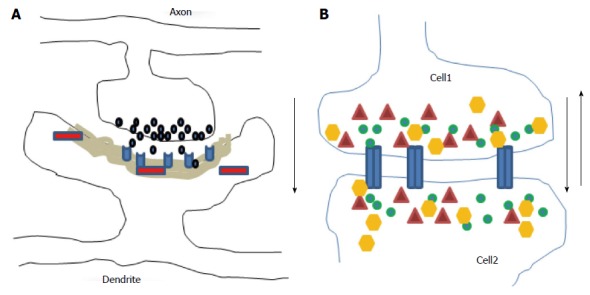
Diagrams illustrate chemical (A) and electrical (B) synapses. At chemical synapses, neurotransmitters (black) released from axonal boutons bind to postsynaptic receptors (light blue) and trigger specific signaling pathways via activation of proteins (red) in postsynaptic cells with prominent postsynaptic densities component (grey area). The information transmission is unidirectional (black arrow). At electrical synapses, gap junction channels (blue) directly connect the two adjacent cells, thus enable the bidirectional passage of electrical currents (black arrows) carried by ions (green), and of small peptides (yellow) or second messengers (dark red).
In contrast, there is no cytoplasmic junction between two cells at the two chemical synaptic subtypes: The excitatory asymmetric (mainly glutamatergic; Figure 1A) type I synapse has marked postsynaptic densities (PSD) while the inhibitory symmetric (mainly GABAergic) type II synapse has no thickened PSD. The genesis of chemical synapses is characterized by an enormous degree of complexity and diversity of protein-protein interactions. Axonal presynaptic boutons of excitatory synapses contain round clear vesicles loaded with the neurotransmitter glutamate and connected with dendritic spines while inhibitory presynaptic boutons contain slightly smaller vesicles and are most abundant at the neuronal soma. In CNS, maturation and stabilization of synaptic structures depend on neurexins and neurolignins, the molecule pairs in the CAM family[41-43], while plasticity which allows an individual to adapt to a rapid changing environment through strengthening, weakening, pruning or adding synaptic connection- is partially dependent on BDNF, ephrin, Wnts, NgR1, semaphorins class 3 and non-coding RNAs[44-48]. The mechanisms contributing to the synaptic plasticity include structural remodeling of the synapse, structural reorganization of presynaptic active zone, postsynaptic density area, protein synthesis, signal transduction pathways, Ca2+ fluxes, kinases activities, neuronal activities, change in transmitters release and receptors trafficking. However, plasticity creates a significant challenge to the intrinsic architecture and integrity of neural networks which is counterbalanced by compensatory regulatory mechanisms for maintaining neuronal homeostasis, leading to a balance between wiring plasticity and stability.
Synapses, the sites where two neurons connect and pass information, are the building blocks of the neuronal networks defined by “a set of elements with time-variant properties that interact with each other” and network dynamics defined by the real-time changes in response to internal and external stimuli. It should be noted however that information is not stored in a chemical form but is processed and retrieved by neuronal networks. The global neuronal network complexity can be defined as a dynamic interconnected functional system characterized by a series of simpler networks organized into increasingly effective local or global complex networks constrained however by the intrinsic structural brain architecture[49]. It has the capacity to support complex thought, action and learned behavior that any single neuron/element of the system would not be able to support alone and its resting-state reflects the stable and intrinsic functional architecture of the brain. Importantly, dysfunction of local networks may spread easily between linked elements, leading to pathological cascades that cause dedifferentiation or trans-neuronal degeneration and encompass large areas of the system. It may also lead to dynamic adjustments and reorganization of the other networks to compensate these changes. Consistent with the view that network organization fundamentally influences brain diseases, many studies including connectomic approach, address the behavioral impairments that arise from network insults or dysfunction and challenge to predict patterns of disease spread and targets of intervention[50,51]. In various mental diseases, neuroimaging observations, diffusion tensor imaging, electroencephalography (EEG) and magnetoencephalography report altered structures and functions of PFC and hipF and deficits in functional integration between these two elements (Figure 2) suggesting overlapping pathogenic mechanisms.
Figure 2.
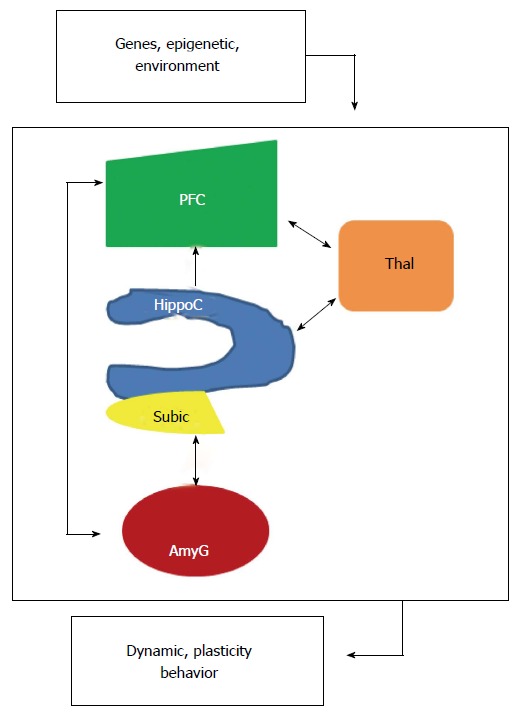
Localized interregional connectivity. Diagram shows pathway connections of hippocampus (blue), amygdala (dark red), thalamus (orange) and prefrontal cortex (green). HippoC-PFC pathway originating from the subiculum and the CA1 of the hippocampus to the PFC is unidirectional, direct in a monosynaptic manner in rodents and primates. HippoC-AmyG pathway shows bidirectional connection between ventral HippoC with AmyG and the pathway AmyG-PFC has also bidirectional connections. Together, both the HippoC and PFC are reciprocally connected with the AmyG and disruption of these pathways, anatomically or functionally may be a common origin of mental diseases. Moreover, there are bidirectional connections between PFC/HippoC with Thalamus. PFC: Prefrontal cortex.
The PFC, which matures later in development than more caudal cortical regions, exerts “top-down” control of many cortical and sub-cortical areas; moreover some of its neuronal subpopulations exhibit complex dendritic arbor. Its development is characterized by growth in early childhood, decrease in adolescence and continued maturation in adulthood. It is well established in human that PFC is involved in language, maintenance of attention, executive functioning, organization of inputs from diverse sensory sources, coordination of goal-directed behavior, socialization and moral decisions. HipF is structurally and functionally heterogeneous. The anterior and posterior parts receive/extend different afferent and efferent connections and play a role in various functions such as learning and memory, stress, spatial and emotional processing. The neuronal projection from the HIP either directly -monosynaptic- or indirectly polysynaptic to the PFC is referred to as the hipocampal-PFC pathways (HIP-PFC). In rats, the direct monosynaptic HIP-PFC pathway originating from the CA1 and the subiculum projects to the anterior cingulated areas of the PFC through fimbria/fornix system. It exhibits activity-dependent synaptic plasticity such as LTP/LTD or depotentiation. Treatment with lidocaine disrupts its performance. However, fine details of the human HIP-PFC are lacking because direct powerful tract-tracing techniques cannot be applied. Within the hipF, neuroanatomical studies show that the ventral CA1 and subiculum also project to the basolateral amygdala (AmyG) which has a critical role in expression of fear and autonomic defense responses whereas recent works highlight the importance of distinct AmyG projections to other structures and their importance in controlling reward/learned behavior. Functional imaging in patients with major depressive disorder (MDD) and bipolar disorder (BP) shows that the rate of resting cerebral blood flow and glucose metabolism in AmyG is elevated and positively correlates with depression severity and relapse. In addition, the dual-projection of hippocampal neurons is crucial for coordinating PFC/AmyG activity during memory retrieval. Meanwhile, AmyG neurons densely arborize within superficial layers of PFC and form synapses with layer II pyramidal neurons. Disruption of the AmyG-PFC pathway increases choice of risky rewards suggesting that it is important for top-down control of emotion, anxiety and fear. Presently, it is well-established that patients suffering from a number of mental diseases, such as schizophrenia, major depression, bipolar and PTSD, display cognitive impairment, have structural abnormalities, disorganized neural networks and aberrant functional coupling within HIP/PFC/AmyG and their pathways[52-56].
NEURONAL NETWORK ABNORMALITIES IN MENTAL DISEASES
Schizophrenia
Schizophrenia is a complex psychiatric disorder with variable symptomatology characterized by hallucination, delusion, anhedonia and cognitive dysfunction. Studies of post-mortem brains provide patterns of abnormalities that reflect failures in early brain development and maturation which can be attributed to alterations in gene expression (NRG1/Akt/Dysbindin-1/Reelin/COMT/DISC1) affecting neuronal differentiation[57-60], gene duplication (25q11-13, 16p11.2, 16p13.1)[61,62] or deletions (2p53, 3q29, 15q13-q14, 15p11.322q11.2)[63,64] . There is also evidence for contribution of multiple different epigenetic events [stress that activates the hypothalamic-pituitary-adrenal (HPA) axis and increases dopamine brain function, viral infection, DNA methylation, histone modification, non-coding RNAs]. Of note, recent proteomic, gene targeting, post-mortem and SNP linkage studies include CRMP1 and 2 in the list of schizophrenia susceptibility genes[65,66]. As schizophrenia typically starts in the late adolescence and as brain development is a continuous process, the remodeling of cortical and hippocampal structures and synaptic connections is thought to be critical. Indeed, patients with schizophrenia show structural anomalies in the HIP and cortical thinning in the PFC associated with aberrant functional coupling -i.e., reduced fractional anisotropy values in the superior longitudinal fasciculus white matter bundle between two areas during resting state or working memory present in both first-episode patients and persons at risk[67]. This scheme suggests that the dysfunction is not a consequence of the disease or treatment but is consistent with abnormal HIP/PFC interaction. Precisely, it has been proposed that deficit in emotional regulation is likely dependent on the dysfunction of HIP, which contributes to aberrant dopaminergic synaptic activities in nucleus accumbens, which in turn influences PFC maladaptive processes leading to delusion and psychosis. To this extent, structural and functional vulnerability and abnormalities of GABAergic, glutamatergic serotoninergic and cholinergic synapses have been reported by several groups[68,69]. As synapses are ultimately linked to neurotransmitter release and their signal transduction and as spine morphology is closely linked to synaptic function, altered spine shape, size and density have multiple functional effects on neuronal networks, and dendritic spine dysfunction may have an etiological role in schizophrenia. Several post-mortem studies reveal altered white matter myelination/projection and a profound reduction in spine density in the PFC of patients while the reduction in spine density found in the auditory cortex could potentially be associated with auditory hallucinations. Other evidence suggest that the heterogeneity of schizophrenia may originate from larger disturbances in neural elements of several interconnected brain circuits and structures[70] including HIP, parahippocampal gyrus, entorhinal cortex, AmyG, superior and transverse temporal gyri, prefrontal and anterior cingulate cortex, and several nuclei of the thalamus. Indeed normal neural structure in morphometric imaging may not guarantee normal function.
Bipolar disorder
Studies of physiopathology in BP have identified brain structural/functional and connectivity alterations associated with the prominent mood swings -i.e., alternating recurrent episodes of mania and depression with psychotic symptoms in some cases. Neuroimaging has convincingly showed that bipolar disorder (BP) is a brain disease involving multiple abnormal brain structures -AmyG, HipF, PFC, thalamus and basal ganglia- and neuronal circuits, particularly the limbic-AmyG-thalamic-cortical pathways interconnected by excitatory glutaminergic projections and the limbic-cortical-striatal-pallidal-thalamic circuits[71]. These two networks share components and regulate AmyG response in complex emotions such as melancholic feeling and neuroendocrine/diurnal rhythms. Functional imaging, which permits direct examination of functional brain structures, find decreased blood flow and metabolism in PFC during depression and a reverse increased metabolism during mania[72]. Additionally, postmortem histopathology shows reductions in cortex volume, glial cell counts, and neuronal size in PFC, AmyG, basal ganglia and dorsal raphe nuclei[73,74]. The reduction of glial cells oligodendroglia and microglia that a play critical role in modulating neurotransmission- provides new insights into possible key CNS cellular abnormalities in BP. Altogether, the altered brain structure/network characteristics suggesting that BP is a developmental disease and the identical-twin concordance rates/adoption studies/family history confirm that it has a strong genetic component. The involvement of multiple genes and their epigenetic (psychosocial and environmental interaction)/epistasis (genes interaction) effects make the clear-cut elucidation of altered risk gene expression particularly challenging, although recently the Human Genome Project has helped to overcome some of these difficulties. Linkage and GWAS have reported many BP risk genes, including ANK3, ZNF804A (important for white matter integrity)[75], NCAN (cortical thickness)[76], TCF4 (ventricular volume)[77], CACNA1C (functional connectivity during executive task. CACNA1C encodes a subunit of the L-type voltage dependent calcium channel[78] that seems to be involved in the dendritic arborization activity of CRMP3 in HIP)[38], BDNF (executive function deficit, neurotrophic factor, plasticity)[79]. Importantly, meta-analyses of the polygenic score profile indicate a large molecular overlap in vulnerability alleles for BP and schizophrenia. In BP, CRMP2 protein levels are decreased in the CA2/CA3 areas and the frontal cortex whereas CRMP4 is decreased in HIP[80].
MDD
MDD is often a recurrent and severe psychiatric disease characterized by decreased density in dendrites and dendritic spines in hippocampus that can be reversed by antidepressant treatments[81]. In the past, most studies have focused on mono-amine system. Another etiological hypothesis proposes that deficiency of neurotrophic factors may mediate depressive symptoms. BDNF is one such factor and there are numerous reports of reduced BDNF in MDD[82]. It has been suggested, in a gene-environment interaction network analysis, that BDNF polymorphism may be involved in MDD. Other evidence which strengthens this hypothesis is provided by studies in BDNF mutant mice[83]. However there are enormous gaps in our understanding of MDD and looking beyond mono-amine and neurotrophic mechanisms to explore the complex neuronal network topologies influence[84] may bring new effective treatment. This notion has received considerable experimental support: (1) it has been shown that the level of CRMP2 important for growth cones formation and neurite arborization is decreased in the brain of patients with depression[85]; (2) recent neuroimaging studies highlight structural alterations in various brain areas of MDD patients[86] -predominately in the HIP and PFC, suggesting overlapping brain abnormalities between the main mental disorders- while the resting-state functional magnetic resonance imaging provides evidence of major change in HIP-PFC circuits[87] responsible for action responses, emotion, sleep, EEG synchronization, attention and memory; (3) the structural/functional abnormalities may contribute to disturbance in mood and cognition in MDD patients and strengthen the hypothesis that MDD is associated with the breakdown of the healthy neuronal networks circuitries; and (4) animal models of depression present similar neuronal dystrophy, reduced synaptic density in PFC and pyramidal cells of the HIP[83].
Post-traumatic stress disorders
After exposure to a traumatic event, e.g., war-related events, physical assault, violence, a small percentage of individuals develop post-traumatic stress disorders (PTSD), characterized by re-experiencing the event with emotional numbing and hyper-arousal symptoms. The identification of structural brain abnormalities, biological and genetic risk in PTSD is required to identify the causal pathways, and inform treatment. Neuroimaging studies of PTSD patients reveal structural and functional alterations within HIP, AmyG, medial frontal cortex and bilateral orbito-frontal cortex[88]. The functional connectivity studies show that PTSD patients exhibit diminished levels of connectivity between the posterior cingulated cortex region and the right superior frontal cortex and the left thalamus during the resting state[89]. Other biological findings detect the dysregulation of HPA axis and release of corticosteroids -critically involved in mediating the deleterious effect of stress including the decrease of dendritic spines density in HIP in line with the decrease of BDNF[90].
Neuropathic pain
Neuropathic pain (NP)[91] is linked either to peripheral nervous system lesions with drastic changes in gene expression pattern, protein interaction network and non-coding RNAs (i.e., likely induced by inflammatory molecules such as histamine, prostaglandins or bradikinin)[92,93] or in relay structures of CNS (i.e., arisen from metabolic disorders, traumatic injury or neurotoxicity)[94]. These changes can persist long after the initial injury (nerve loss, phantom limb). Common causes of NP are acute or chronic trauma, neurotoxins, diabetes, tumor compression, viral infections or side effect of chemotherapy. A systematic approach of NP is based on the characterization of all pain aspects, including emotional, behavioral, psycho-social, anatomical, genetic and molecular genetic factors[95,96]. Neuroimaging studies provide evidence suggesting that NP is associated with structural, functional and neurochemical alterations distributed across multiple brain structures and networks[97]. However, because many environmental factors may interact with genetic polymorphism to influence pain perception, data from proteomic studies to elucidate genetic contribution to NP remain limited and inconsistent although diverse molecules (NMDA/AMPA/ P2X3 ion channel receptors, G-protein-coupled receptors, AnnexinV/CaM/CRMP2 calcium signaling protein, N-type voltage-gated calcium channels CaV2.2, receptor tyrosine kinases, trkB for BDNF, non-coding RNAs)[98-101] can account for changes that arise in pathological pain states.
TREATMENT OF NEURAL NETWORK DEFICITS AND NP
Remodeling neuronal connectivity by transcranial magnetic stimulation
Transcranial magnetic stimulation (TMS) consists of promoting a localized electric current through a localized magnetic field produced by a TMS-coil[102]. It is assumed that TMS and repetitive TMS have an inhibitory or facilitatory effect on neurons and neuronal networks. They can induce plasticity, modulate neurotransmission and increase neuroprotection against oxidative effect via BNDF/TrkB signaling system in the stimulated site and in other structures functionally connected with it[103-105]. These effects associated with magnetic stimulation -low intensity stimulation results in neurite sprouting and increase in synaptic contacts while high intensity stimulation has devastating effects- can be maintained as long as 6 mo after treatment[106]. Furthermore, it has been shown that TMS reduces structural, functional and behavioral abnormalities in ephrin-A2A5-/- mice but do not adversely affect the control WT[107]. Altogether, these observations suggest that TMS treatment may modulate synaptic strength not only locally but at distant sites, modulating the connectivity networks and offering the hope of a focal intervention capable of ameliorating the altered circuitries underlying psychiatric disorders. Indeed clinical trials directed to the treatment of major depression, schizophrenia, bipolar disorders, anxiety disorders, PTSD and neuropathic pain have supported a possible therapeutic effect of TMS alone or in combination with drugs[108]. Although the mechanisms of TMS activity are hypothesized to be based on induction of neuronal firing, certain effects may be due to the cryptochrome (CRY)/photolyase family: Present in all cell nuclei, CRYs that contain magnetosensitive radical pairs may provide the abilities of cells to specifically respond to magnetic field[109,110].
Remodeling neuronal connectivity with selective modulation of BDNF/CRMPs expression
One of the most studied and best characterized neurotrophins in CNS is BDNF. It has received remarkable attention from scientists because it is essential for neurogenesis, neuronal differentiation (including neuritogenesis/dendritic arborization, spine formation and axonogenesis via nonphosphorylated CRMP2), survival, migration, apoptosis, synaptic plasticity (via TrkB and p75NTR activation) and neuronal network formation, and from clinicians because it is required for normal development/functions of the brain and its expression is found decreased in several brain regions in post-mortem studies of patients with neurodegenerative and psychiatric diseases[60,79,81-83,111,112]. In physiological condition, BDNF activity depends on the activation of its downstream intracellular signaling cascades -Ras/MAPK, PLC-γ, PI3K/AKT. An additional level of regulation is provided by the balance between neurotrophic signaling through mature-BDNF/TrkB and apoptotic signaling through pro-BDNF/p75, which determines which connections are maintained within the neuronal network and which neurons are eliminated. Importantly, altered BDNF activity has been reported in brain pathology, particularly in limbic structure, AmyG, orbital and medial PFC and related keys circuits. Antidepressant activity seems to be linked to increasing levels of BDNF. Similar to BDNF, CRMPs express abundantly in the nervous system and while manipulations of their expression by RNAi, gene targeting or overexpression has confirmed their critical role in axono-dendritic growth and collapse, neuronal migration/survival and spinogenesis[29,30,33,34,36], other studies reveal that their expression is also altered in human pathologies including mental disorders[33,85,113-115]. Restoration of altered BDNF/CRMPs activities in these affected areas and/or affective circuits with newer and more refined/targeted immuno-pharmacological agents will likely yield more effective treatments, particularly in treatment-refractory cases, and greater understanding of the mechanisms underlying mental disorders.
NP treatment with CBD3, a peptide derived from CRMP2
Presently, the understanding of the molecular and cellular mechanisms of NP is incomplete, and new concepts are needed to improve its treatment. Genetic and clinical studies have validated N-type voltage-gated calcium channels (CaV2.2) as targets for NP treatment[116-118] but selective blockers have potential serious side effects. Targeting protein interactions to direct channel block has been proposed[119]. This approach has yielded a novel peptide-derived therapeutic prototype for NP relief: A CRMP2-derived peptide (tat-CBD3; Figure 3) that disrupts the CaV2.2/CRMP2 interaction shows anti-nociceptive activity in animal models of neuropathic pain[120]. The relative lack of toxicity provides evidence that CBD3 has therapeutic promise[121]. Recent efforts to optimize the peptide’s efficacy resulted in the generation of a myristoylated version of the peptide; myr-tat-CBD3 -a myristoylated CBD3 peptide harboring a 14-carbon fatty acid, myristate, onto an N-terminal glycine[122]. N-myristoylation is a lipid anchor modification of eukaryotic and viral proteins targeting them irreversibly to locate to the membrane[123-125]. Tethering the CBD3 peptide to the membrane through myristoylation confines its action(s) to the uncoupling of membrane CaV2.2-CRMP2 and blocks Ca2+ influx without affecting the CRMP2 functions mediated by interactions with other cytoplasmic proteins[126].
Figure 3.
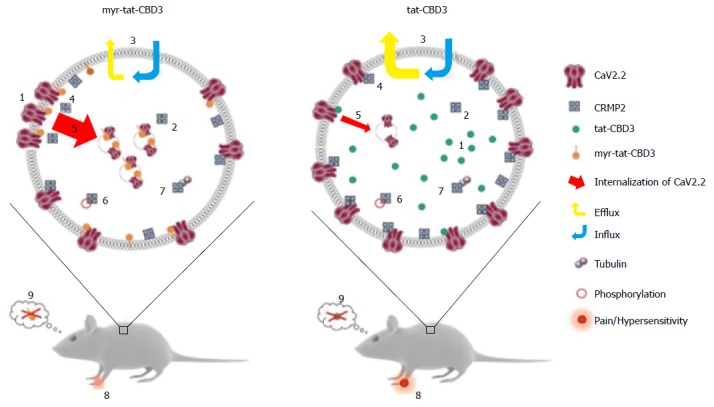
A possible model for N-myristoylated collapsin response mediator protein 2 peptide’s actions on CaV2.2 trafficking and efficacy in neuropathic pain model. Application of an N-myristoylated tat-conjugated CRMP2 peptide (myr-tat-CBD3) results in membrane-delimited “rimming” of the peptide whereas the non-myristoylated version (tat-CBD3) appears to be spatially diffusely distributed in the cell cytoplasm. Analysis of penetration of peptides into GPMVs, which are “blebs” of membrane devoid of organelles and actin cytoskeleton, reveals an unrestricted distribution of the membrane sensitive dye (di-4-ANNEPDHQ) with tat-CBD3, whereas the myristoylated peptide induces a lateral heterogeneity of the fluorescent signal resulting in dye aggregation into micro domains within these model membranes[122]. While CRMP2 has been demonstrated to exist as a tetramer, the oligomeric state of membrane proximal CRMP2 is as yet unknown; however, neither peptide appears to affect CRMP2 oligomerization[123]. Whereas the cells take up both forms of the peptide with similar efficiency, the myristoylated peptide demonstrates a lesser degree of efflux[124]. The apparent increase in retention of myr-tat-CBD3 translates into a superior potency and efficacy in inhibition of evoked calcium influx in sensory neurons presumably via greater uncoupling of CRMP2-CaV2.2 interactions at or juxta-membrane[125]. Increased inhibition of CaV2.2 surface trafficking induced by myr-tat-CBD3 compared with tat-CBD3 may account[126] for the more pronounced restriction of calcium influx imposed by the myristoylated peptide. Cdk5-phosphorylated CRMP2 has been demonstrated to have an enhanced interaction with CaV2.2[129]; however myr-tat-CBD3 does not affect the levels of Cdk5-phosphorylated CRMP2[127], thereby ruling out a role of phosphorylated CRMP2 in regulating calcium influx. CRMP2 binding to tubulin is strengthened by the peptides[130]; the consequences of this are currently unknown. Importantly, where tat-CBD3 is completely ineffective in reversing mechanical hypersensitivity in a rat neuropathic pain model (tibial nerve injury), the myristoylated peptide reverses this hypersensitivity when administered in vivo[122]. Neither peptide elicits any reward-like addictive behaviors. GPMVs: Giant plasma membrane vesicles; CRMP: Collapsin response mediator proteins.
As summarized in Figure 3, the myr-tat-CBD3 peptide is equal or better in its efficacy to tat-CBD3 on reducing pain-related behaviors with pronounced reversal of mechanical hypersensitivity in a postoperative incisional pain model[127] at dose of 0.1 mg/kg in contrast to the lack of effect observed even at 20 mg/kg of tat-CBD3[122]. Moreover sustained relief (> 6 wk) of NP was obtained with an AAV-targeted expression of CBD3 peptide in rat DRG[128]. Both tat-CBD3 and myr-tat-CB3 reduce pain-like behaviors without demonstrating any reward-like potential. These experiments demonstrate the possibility of tailoring molecules that affect membrane targets for specific inhibition of CaV2.2-CRMP2 interactions (Figure 3).
CONCLUSION
Data from various basic science studies together with experimental/clinical studies suggest a correlative causal link between specific neural networks dysfunctions and mental disorders. However, one could argue that (1) a correlation between experimental and clinical observations is a correlative statement but not a causal demonstration; (2) an observed electrical current dysfunction may not support this common view of correlative network specificity because it can result from different network stimulations with many unknown parameters and consequently underspecifying any structural function; and (3) more direct experiments will be needed to consolidate these observations. This approach is critical since it may raise further technical inventions/interventions and challenges such as generating new tools more powerful optogenetic/acousto-optical deflectors approaches in animal models[131,132] to visualize, control and compute simultaneously single neuronal activity and neuronal circuitries, to integrate in parallel behavioral measures for a better treatment of mental diseases, less side-effects associated to newer psychotropic drugs and gene therapy[133-137], and more importantly a better understanding of the physiology and pathology of the human brain in healthy activity and abnormal behavior respectively.
Footnotes
Conflict-of-interest statement: No potential conflict of interest. No financial support.
Open-Access: This article is an open-access article which was selected by an in-house editor and peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution non-commercial license (CC BY-NC 4.0), which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial.
Peer-review started: September 1, 2015
First decision: November 6, 2015
Article in press: January 7, 2016
P- Reviewer: Boto GR, Flyckt L S- Editor: Qi Y L- Editor: A E- Editor: Jiao XK
References
- 1.Schmidt EF, Strittmatter SM. The CRMP family of proteins and their role in Sema3A signaling. Adv Exp Med Biol. 2007;600:1–11. doi: 10.1007/978-0-387-70956-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmandke A, Schmandke A, Schwab ME. Nogo-A: Multiple Roles in CNS Development, Maintenance, and Disease. Neuroscientist. 2014;20:372–386. doi: 10.1177/1073858413516800. [DOI] [PubMed] [Google Scholar]
- 3.Terwisscha van Scheltinga AF, Bakker SC, Kahn RS, Kas MJ. Fibroblast growth factors in neurodevelopment and psychopathology. Neuroscientist. 2013;19:479–494. doi: 10.1177/1073858412472399. [DOI] [PubMed] [Google Scholar]
- 4.Benarroch EE. Brain-derived neurotrophic factor: Regulation, effects, and potential clinical relevance. Neurology. 2015;84:1693–1704. doi: 10.1212/WNL.0000000000001507. [DOI] [PubMed] [Google Scholar]
- 5.Thierry AM, Gioanni Y, Dégénétais E, Glowinski J. Hippocampo-prefrontal cortex pathway: anatomical and electrophysiological characteristics. Hippocampus. 2000;10:411–419. doi: 10.1002/1098-1063(2000)10:4<411::AID-HIPO7>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 6.Ishikawa A, Nakamura S. Ventral hippocampal neurons project axons simultaneously to the medial prefrontal cortex and amygdala in the rat. J Neurophysiol. 2006;96:2134–2138. doi: 10.1152/jn.00069.2006. [DOI] [PubMed] [Google Scholar]
- 7.Filley CM, Brown MS, Onderko K, Ray M, Bennett RE, Berry-Kravis E, Grigsby J. White matter disease and cognitive impairment in FMR1 premutation carriers. Neurology. 2015;84:2146–2152. doi: 10.1212/WNL.0000000000001612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ganguly K, Poo MM. Activity-dependent neural plasticity from bench to bedside. Neuron. 2013;80:729–741. doi: 10.1016/j.neuron.2013.10.028. [DOI] [PubMed] [Google Scholar]
- 9.Shastry BS. Gene disruption in mice: models of development and disease. Mol Cell Biochem. 1998;181:163–179. doi: 10.1023/a:1006865210012. [DOI] [PubMed] [Google Scholar]
- 10.Panagiotou OA, Willer CJ, Hirschhorn JN, Ioannidis JP. The power of meta-analysis in genome-wide association studies. Annu Rev Genomics Hum Genet. 2013;14:441–465. doi: 10.1146/annurev-genom-091212-153520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaiano N, Nye JS, Fishell G. Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron. 2000;26:395–404. doi: 10.1016/s0896-6273(00)81172-1. [DOI] [PubMed] [Google Scholar]
- 12.Paridaen JT, Huttner WB. Neurogenesis during development of the vertebrate central nervous system. EMBO Rep. 2014;15:351–364. doi: 10.1002/embr.201438447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lameu C, Trujillo CA, Schwindt TT, Negraes PD, Pillat MM, Morais KL, Lebrun I, Ulrich H. Interactions between the NO-citrulline cycle and brain-derived neurotrophic factor in differentiation of neural stem cells. J Biol Chem. 2012;287:29690–29701. doi: 10.1074/jbc.M111.338095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ross SE, Greenberg ME, Stiles CD. Basic helix-loop-helix factors in cortical development. Neuron. 2003;39:13–25. doi: 10.1016/s0896-6273(03)00365-9. [DOI] [PubMed] [Google Scholar]
- 15.Pinto L, Drechsel D, Schmid MT, Ninkovic J, Irmler M, Brill MS, Restani L, Gianfranceschi L, Cerri C, Weber SN, et al. AP2gamma regulates basal progenitor fate in a region- and layer-specific manner in the developing cortex. Nat Neurosci. 2009;12:1229–1237. doi: 10.1038/nn.2399. [DOI] [PubMed] [Google Scholar]
- 16.Schwamborn JC, Berezikov E, Knoblich JA. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell. 2009;136:913–925. doi: 10.1016/j.cell.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martynoga B, Drechsel D, Guillemot F. Molecular control of neurogenesis: a view from the mammalian cerebral cortex. Cold Spring Harb Perspect Biol. 2012;4:a008359. doi: 10.1101/cshperspect.a008359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilkinson G, Dennis D, Schuurmans C. Proneural genes in neocortical development. Neuroscience. 2013;253:256–273. doi: 10.1016/j.neuroscience.2013.08.029. [DOI] [PubMed] [Google Scholar]
- 19.Joshi K, Lee S, Lee B, Lee JW, Lee SK. LMO4 controls the balance between excitatory and inhibitory spinal V2 interneurons. Neuron. 2009;61:839–851. doi: 10.1016/j.neuron.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siegenthaler JA, Tremper-Wells BA, Miller MW. Foxg1 haploinsufficiency reduces the population of cortical intermediate progenitor cells: effect of increased p21 expression. Cereb Cortex. 2008;18:1865–1875. doi: 10.1093/cercor/bhm209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pierfelice T, Alberi L, Gaiano N. Notch in the vertebrate nervous system: an old dog with new tricks. Neuron. 2011;69:840–855. doi: 10.1016/j.neuron.2011.02.031. [DOI] [PubMed] [Google Scholar]
- 22.Kamakura S, Oishi K, Yoshimatsu T, Nakafuku M, Masuyama N, Gotoh Y. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat Cell Biol. 2004;6:547–554. doi: 10.1038/ncb1138. [DOI] [PubMed] [Google Scholar]
- 23.Oliveira SL, Pillat MM, Cheffer A, Lameu C, Schwindt TT, Ulrich H. Functions of neurotrophins and growth factors in neurogenesis and brain repair. Cytometry A. 2013;83:76–89. doi: 10.1002/cyto.a.22161. [DOI] [PubMed] [Google Scholar]
- 24.Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- 25.Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Südhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcos-Mondéjar P, Peregrín S, Li JY, Carlsson L, Tole S, López-Bendito G. The lhx2 transcription factor controls thalamocortical axonal guidance by specific regulation of robo1 and robo2 receptors. J Neurosci. 2012;32:4372–4385. doi: 10.1523/JNEUROSCI.5851-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srinivasan K, Leone DP, Bateson RK, Dobreva G, Kohwi Y, Kohwi-Shigematsu T, Grosschedl R, McConnell SK. A network of genetic repression and derepression specifies projection fates in the developing neocortex. Proc Natl Acad Sci USA. 2012;109:19071–19078. doi: 10.1073/pnas.1216793109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schweitzer J, Löhr H, Bonkowsky JL, Hübscher K, Driever W. Sim1a and Arnt2 contribute to hypothalamo-spinal axon guidance by regulating Robo2 activity via a Robo3-dependent mechanism. Development. 2013;140:93–106. doi: 10.1242/dev.087825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quach TT, Wilson SM, Rogemond V, Chounlamountri N, Kolattukudy PE, Martinez S, Khanna M, Belin MF, Khanna R, Honnorat J, et al. Mapping CRMP3 domains involved in dendrite morphogenesis and voltage-gated calcium channel regulation. J Cell Sci. 2013;126:4262–4273. doi: 10.1242/jcs.131409. [DOI] [PubMed] [Google Scholar]
- 30.Quach TT, Massicotte G, Belin MF, Honnorat J, Glasper ER, Devries AC, Jakeman LB, Baudry M, Duchemin AM, Kolattukudy PE. CRMP3 is required for hippocampal CA1 dendritic organization and plasticity. FASEB J. 2008;22:401–409. doi: 10.1096/fj.07-9012com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veyrac A, Giannetti N, Charrier E, Reymond-Marron I, Aguera M, Rogemond V, Honnorat J, Jourdan F. Expression of collapsin response mediator proteins 1, 2 and 5 is differentially regulated in newly generated and mature neurons of the adult olfactory system. Eur J Neurosci. 2005;21:2635–2648. doi: 10.1111/j.1460-9568.2005.04112.x. [DOI] [PubMed] [Google Scholar]
- 32.Crews L, Ruf R, Patrick C, Dumaop W, Trejo-Morales M, Achim CL, Rockenstein E, Masliah E. Phosphorylation of collapsin response mediator protein-2 disrupts neuronal maturation in a model of adult neurogenesis: Implications for neurodegenerative disorders. Mol Neurodegener. 2011;6:67. doi: 10.1186/1750-1326-6-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quach TT, Honnorat J, Kolattukudy PE, Khanna R, Duchemin AM. Collapsin response mediator protein 3 increases the dendritic arborization of hippocampal neurons. Mol Psychiatry. 2015;20:1027. doi: 10.1038/mp.2015.123. [DOI] [PubMed] [Google Scholar]
- 34.Kawano Y, Yoshimura T, Tsuboi D, Kawabata S, Kaneko-Kawano T, Shirataki H, Takenawa T, Kaibuchi K. CRMP-2 is involved in kinesin-1-dependent transport of the Sra-1/WAVE1 complex and axon formation. Mol Cell Biol. 2005;25:9920–9935. doi: 10.1128/MCB.25.22.9920-9935.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamashita N, Takahashi A, Takao K, Yamamoto T, Kolattukudy P, Miyakawa T, Goshima Y. Mice lacking collapsin response mediator protein 1 manifest hyperactivity, impaired learning and memory, and impaired prepulse inhibition. Front Behav Neurosci. 2013;7:216. doi: 10.3389/fnbeh.2013.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niisato E, Nagai J, Yamashita N, Abe T, Kiyonari H, Goshima Y, Ohshima T. CRMP4 suppresses apical dendrite bifurcation of CA1 pyramidal neurons in the mouse hippocampus. Dev Neurobiol. 2012;72:1447–1457. doi: 10.1002/dneu.22007. [DOI] [PubMed] [Google Scholar]
- 37.Yamashita N, Mosinger B, Roy A, Miyazaki M, Ugajin K, Nakamura F, Sasaki Y, Yamaguchi K, Kolattukudy P, Goshima Y. CRMP5 (collapsin response mediator protein 5) regulates dendritic development and synaptic plasticity in the cerebellar Purkinje cells. J Neurosci. 2011;31:1773–1779. doi: 10.1523/JNEUROSCI.5337-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quach TT, Wang Y, Khanna R, Chounlamountri N, Auvergnon N, Honnorat J, Duchemin AM. Effect of CRMP3 expression on dystrophic dendrites of hippocampal neurons. Mol Psychiatry. 2011;16:689–691. doi: 10.1038/mp.2011.6. [DOI] [PubMed] [Google Scholar]
- 39.Harris KM, Weinberg RJ. Ultrastructure of synapses in the mammalian brain. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a005587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hormuzdi SG, Filippov MA, Mitropoulou G, Monyer H, Bruzzone R. Electrical synapses: a dynamic signaling system that shapes the activity of neuronal networks. Biochim Biophys Acta. 2004;1662:113–137. doi: 10.1016/j.bbamem.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 41.Thalhammer A, Cingolani LA. Cell adhesion and homeostatic synaptic plasticity. Neuropharmacology. 2014;78:23–30. doi: 10.1016/j.neuropharm.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 42.Craig AM, Kang Y. Neurexin-neuroligin signaling in synapse development. Curr Opin Neurobiol. 2007;17:43–52. doi: 10.1016/j.conb.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inestrosa NC, Varela-Nallar L. Wnt signalling in neuronal differentiation and development. Cell Tissue Res. 2015;359:215–223. doi: 10.1007/s00441-014-1996-4. [DOI] [PubMed] [Google Scholar]
- 44.Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14:7–23. doi: 10.1038/nrn3379. [DOI] [PubMed] [Google Scholar]
- 45.Yamamoto N, Tamada A, Murakami F. Wiring of the brain by a range of guidance cues. Prog Neurobiol. 2002;68:393–407. doi: 10.1016/s0301-0082(02)00129-6. [DOI] [PubMed] [Google Scholar]
- 46.Thiede-Stan NK, Schwab ME. Attractive and repulsive factors act through multi-subunit receptor complexes to regulate nerve fiber growth. J Cell Sci. 2015;128:2403–2414. doi: 10.1242/jcs.165555. [DOI] [PubMed] [Google Scholar]
- 47.Lerch JK, Kuo F, Motti D, Morris R, Bixby JL, Lemmon VP. Isoform diversity and regulation in peripheral and central neurons revealed through RNA-Seq. PLoS One. 2012;7:e30417. doi: 10.1371/journal.pone.0030417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin J, Yuan Q. Structural homeostasis in the nervous system: a balancing act for wiring plasticity and stability. Front Cell Neurosci. 2014;8:439. doi: 10.3389/fncel.2014.00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuste R. From the neuron doctrine to neural networks. Nat Rev Neurosci. 2015;16:487–497. doi: 10.1038/nrn3962. [DOI] [PubMed] [Google Scholar]
- 50.Goldenberg D, Galván A. The use of functional and effective connectivity techniques to understand the developing brain. Dev Cogn Neurosci. 2015;12:155–164. doi: 10.1016/j.dcn.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fornito A, Zalesky A, Breakspear M. The connectomics of brain disorders. Nat Rev Neurosci. 2015;16:159–172. doi: 10.1038/nrn3901. [DOI] [PubMed] [Google Scholar]
- 52.Penzes P, Buonanno A, Passafaro M, Sala C, Sweet RA. Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. J Neurochem. 2013;126:165–182. doi: 10.1111/jnc.12261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seshadri S, Zeledon M, Sawa A. Synapse-specific contributions in the cortical pathology of schizophrenia. Neurobiol Dis. 2013;53:26–35. doi: 10.1016/j.nbd.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poch C, Campo P. Neocortical-hippocampal dynamics of working memory in healthy and diseased brain states based on functional connectivity. Front Hum Neurosci. 2012;6:36. doi: 10.3389/fnhum.2012.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van den Heuvel MP, Fornito A. Brain networks in schizophrenia. Neuropsychol Rev. 2014;24:32–48. doi: 10.1007/s11065-014-9248-7. [DOI] [PubMed] [Google Scholar]
- 56.Castrén E. Neuronal network plasticity and recovery from depression. JAMA Psychiatry. 2013;70:983–989. doi: 10.1001/jamapsychiatry.2013.1. [DOI] [PubMed] [Google Scholar]
- 57.Gatt JM, Burton KL, Williams LM, Schofield PR. Specific and common genes implicated across major mental disorders: a review of meta-analysis studies. J Psychiatr Res. 2015;60:1–13. doi: 10.1016/j.jpsychires.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 58.Farrell MS, Werge T, Sklar P, Owen MJ, Ophoff RA, O’Donovan MC, Corvin A, Cichon S, Sullivan PF. Evaluating historical candidate genes for schizophrenia. Mol Psychiatry. 2015;20:555–562. doi: 10.1038/mp.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Craddock N, O’Donovan MC, Owen MJ. Genes for schizophrenia and bipolar disorder? Implications for psychiatric nosology. Schizophr Bull. 2006;32:9–16. doi: 10.1093/schbul/sbj033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- 61.Rees E, Walters JT, Chambert KD, O’Dushlaine C, Szatkiewicz J, Richards AL, Georgieva L, Mahoney-Davies G, Legge SE, Moran JL, et al. CNV analysis in a large schizophrenia sample implicates deletions at 16p12.1 and SLC1A1 and duplications at 1p36.33 and CGNL1. Hum Mol Genet. 2014;23:1669–1676. doi: 10.1093/hmg/ddt540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramalingam A, Zhou XG, Fiedler SD, Brawner SJ, Joyce JM, Liu HY, Yu S. 16p13.11 duplication is a risk factor for a wide spectrum of neuropsychiatric disorders. J Hum Genet. 2011;56:541–544. doi: 10.1038/jhg.2011.42. [DOI] [PubMed] [Google Scholar]
- 63.Hosak L, Silhan P, Hosakova J. Genomic copy number variations: A breakthrough in our knowledge on schizophrenia etiology? Neuro Endocrinol Lett. 2012;33:183–190. [PubMed] [Google Scholar]
- 64.Leonard S, Freedman R. Genetics of chromosome 15q13-q14 in schizophrenia. Biol Psychiatry. 2006;60:115–122. doi: 10.1016/j.biopsych.2006.03.054. [DOI] [PubMed] [Google Scholar]
- 65.Bader V, Tomppo L, Trossbach SV, Bradshaw NJ, Prikulis I, Leliveld SR, Lin CY, Ishizuka K, Sawa A, Ramos A, et al. Proteomic, genomic and translational approaches identify CRMP1 for a role in schizophrenia and its underlying traits. Hum Mol Genet. 2012;21:4406–4418. doi: 10.1093/hmg/dds273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu Y, Pham X, Zhang L, Chen PL, Burzynski G, McGaughey DM, He S, McGrath JA, Wolyniec P, Fallin MD, et al. Functional variants in DPYSL2 sequence increase risk of schizophrenia and suggest a link to mTOR signaling. G3 (Bethesda) 2014;5:61–72. doi: 10.1534/g3.114.015636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Benetti S, Mechelli A, Picchioni M, Broome M, Williams S, McGuire P. Functional integration between the posterior hippocampus and prefrontal cortex is impaired in both first episode schizophrenia and the at risk mental state. Brain. 2009;132:2426–2436. doi: 10.1093/brain/awp098. [DOI] [PubMed] [Google Scholar]
- 68.Stephan KE, Friston KJ, Frith CD. Dysconnection in schizophrenia: from abnormal synaptic plasticity to failures of self-monitoring. Schizophr Bull. 2009;35:509–527. doi: 10.1093/schbul/sbn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nikolaus S, Hautzel H, Müller HW. Neurochemical dysfunction in treated and nontreated schizophrenia - a retrospective analysis of in vivo imaging studies. Rev Neurosci. 2014;25:25–96. doi: 10.1515/revneuro-2013-0063. [DOI] [PubMed] [Google Scholar]
- 70.Williamson PC, Allman JM. A framework for interpreting functional networks in schizophrenia. Front Hum Neurosci. 2012;6:184. doi: 10.3389/fnhum.2012.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Savitz J, Drevets WC. Neuroimaging and neuropathological findings in bipolar disorder. Curr Top Behav Neurosci. 2011;5:201–225. doi: 10.1007/7854_2010_68. [DOI] [PubMed] [Google Scholar]
- 72.Strakowski SM, Delbello MP, Adler CM. The functional neuroanatomy of bipolar disorder: a review of neuroimaging findings. Mol Psychiatry. 2005;10:105–116. doi: 10.1038/sj.mp.4001585. [DOI] [PubMed] [Google Scholar]
- 73.Berretta S, Pantazopoulos H, Lange N. Neuron numbers and volume of the amygdala in subjects diagnosed with bipolar disorder or schizophrenia. Biol Psychiatry. 2007;62:884–893. doi: 10.1016/j.biopsych.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 74.Brauch RA, Adnan El-Masri M, Parker JC, El-Mallakh RS. Glial cell number and neuron/glial cell ratios in postmortem brains of bipolar individuals. J Affect Disord. 2006;91:87–90. doi: 10.1016/j.jad.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 75.Ferreira MA, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, Fan J, Kirov G, Perlis RH, Green EK, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Craddock N, Sklar P. Genetics of bipolar disorder. Lancet. 2013;381:1654–1662. doi: 10.1016/S0140-6736(13)60855-7. [DOI] [PubMed] [Google Scholar]
- 77.Bhat S, Dao DT, Terrillion CE, Arad M, Smith RJ, Soldatov NM, Gould TD. CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog Neurobiol. 2012;99:1–14. doi: 10.1016/j.pneurobio.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Berger SM, Bartsch D. The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res. 2014;357:463–476. doi: 10.1007/s00441-014-1936-3. [DOI] [PubMed] [Google Scholar]
- 79.Knable MB, Barci BM, Webster MJ, Meador-Woodruff J, Torrey EF. Molecular abnormalities of the hippocampus in severe psychiatric illness: postmortem findings from the Stanley Neuropathology Consortium. Mol Psychiatry. 2004;9:609–20, 544. doi: 10.1038/sj.mp.4001471. [DOI] [PubMed] [Google Scholar]
- 80.Föcking M, Dicker P, English JA, Schubert KO, Dunn MJ, Cotter DR. Common proteomic changes in the hippocampus in schizophrenia and bipolar disorder and particular evidence for involvement of cornu ammonis regions 2 and 3. Arch Gen Psychiatry. 2011;68:477–488. doi: 10.1001/archgenpsychiatry.2011.43. [DOI] [PubMed] [Google Scholar]
- 81.Castrén E, Rantamäki T. The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Dev Neurobiol. 2010;70:289–297. doi: 10.1002/dneu.20758. [DOI] [PubMed] [Google Scholar]
- 82.Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Magariños AM, Li CJ, Gal Toth J, Bath KG, Jing D, Lee FS, McEwen BS. Effect of brain-derived neurotrophic factor haploinsufficiency on stress-induced remodeling of hippocampal neurons. Hippocampus. 2011;21:253–264. doi: 10.1002/hipo.20744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leistedt SJ, Linkowski P. Brain, networks, depression, and more. Eur Neuropsychopharmacol. 2013;23:55–62. doi: 10.1016/j.euroneuro.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 85.Johnston-Wilson NL, Sims CD, Hofmann JP, Anderson L, Shore AD, Torrey EF, Yolken RH. Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. The Stanley Neuropathology Consortium. Mol Psychiatry. 2000;5:142–149. doi: 10.1038/sj.mp.4000696. [DOI] [PubMed] [Google Scholar]
- 86.Sheline YI. Neuroimaging studies of mood disorder effects on the brain. Biol Psychiatry. 2003;54:338–352. doi: 10.1016/s0006-3223(03)00347-0. [DOI] [PubMed] [Google Scholar]
- 87.Lorenzetti V, Allen NB, Fornito A, Yücel M. Structural brain abnormalities in major depressive disorder: a selective review of recent MRI studies. J Affect Disord. 2009;117:1–17. doi: 10.1016/j.jad.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 88.Qin LD, Wang Z, Sun YW, Wan JQ, Su SS, Zhou Y, Xu JR. A preliminary study of alterations in default network connectivity in post-traumatic stress disorder patients following recent trauma. Brain Res. 2012;1484:50–56. doi: 10.1016/j.brainres.2012.09.029. [DOI] [PubMed] [Google Scholar]
- 89.Shaw ME, Strother SC, McFarlane AC, Morris P, Anderson J, Clark CR, Egan GF. Abnormal functional connectivity in posttraumatic stress disorder. Neuroimage. 2002;15:661–674. doi: 10.1006/nimg.2001.1024. [DOI] [PubMed] [Google Scholar]
- 90.Broekman BF, Olff M, Boer F. The genetic background to PTSD. Neurosci Biobehav Rev. 2007;31:348–362. doi: 10.1016/j.neubiorev.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 91.Treede RD, Jensen TS, Campbell JN, Cruccu G, Dostrovsky JO, Griffin JW, Hansson P, Hughes R, Nurmikko T, Serra J. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology. 2008;70:1630–1635. doi: 10.1212/01.wnl.0000282763.29778.59. [DOI] [PubMed] [Google Scholar]
- 92.Belfer I, Dai F. Phenotyping and genotyping neuropathic pain. Curr Pain Headache Rep. 2010;14:203–212. doi: 10.1007/s11916-010-0110-1. [DOI] [PubMed] [Google Scholar]
- 93.Max MB, Stewart WF. The molecular epidemiology of pain: a new discipline for drug discovery. Nat Rev Drug Discov. 2008;7:647–658. doi: 10.1038/nrd2595. [DOI] [PubMed] [Google Scholar]
- 94.Calvino B, Grilo RM. Central pain control. Joint Bone Spine. 2006;73:10–16. doi: 10.1016/j.jbspin.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 95.Kuner R. Central mechanisms of pathological pain. Nat Med. 2010;16:1258–1266. doi: 10.1038/nm.2231. [DOI] [PubMed] [Google Scholar]
- 96.Price DD. Psychological and neural mechanisms of the affective dimension of pain. Science. 2000;288:1769–1772. doi: 10.1126/science.288.5472.1769. [DOI] [PubMed] [Google Scholar]
- 97.Moisset X, Bouhassira D. Brain imaging of neuropathic pain. Neuroimage. 2007;37 Suppl 1:S80–S88. doi: 10.1016/j.neuroimage.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 98.Kang SK, So HH, Moon YS, Kim CH. Proteomic analysis of injured spinal cord tissue proteins using 2-DE and MALDI-TOF MS. Proteomics. 2006;6:2797–2812. doi: 10.1002/pmic.200500621. [DOI] [PubMed] [Google Scholar]
- 99.Ding Q, Wu Z, Guo Y, Zhao C, Jia Y, Kong F, Chen B, Wang H, Xiong S, Que H, et al. Proteome analysis of up-regulated proteins in the rat spinal cord induced by transection injury. Proteomics. 2006;6:505–518. doi: 10.1002/pmic.200500296. [DOI] [PubMed] [Google Scholar]
- 100.Katano T, Mabuchi T, Okuda-Ashitaka E, Inagaki N, Kinumi T, Ito S. Proteomic identification of a novel isoform of collapsin response mediator protein-2 in spinal nerves peripheral to dorsal root ganglia. Proteomics. 2006;6:6085–6094. doi: 10.1002/pmic.200600300. [DOI] [PubMed] [Google Scholar]
- 101.Alzate O, Hussain SR, Goettl VM, Tewari AK, Madiai F, Stephens RL, Hackshaw KV. Proteomic identification of brainstem cytosolic proteins in a neuropathic pain model. Brain Res Mol Brain Res. 2004;128:193–200. doi: 10.1016/j.molbrainres.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 102.Lisanby SH, Kinnunen LH, Crupain MJ. Applications of TMS to therapy in psychiatry. J Clin Neurophysiol. 2002;19:344–360. doi: 10.1097/00004691-200208000-00007. [DOI] [PubMed] [Google Scholar]
- 103.Chervyakov AV, Chernyavsky AY, Sinitsyn DO, Piradov MA. Possible Mechanisms Underlying the Therapeutic Effects of Transcranial Magnetic Stimulation. Front Hum Neurosci. 2015;9:303. doi: 10.3389/fnhum.2015.00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hasan A, Falkai P, Wobrock T. Transcranial brain stimulation in schizophrenia: targeting cortical excitability, connectivity and plasticity. Curr Med Chem. 2013;20:405–413. [PubMed] [Google Scholar]
- 105.Müller MB, Toschi N, Kresse AE, Post A, Keck ME. Long-term repetitive transcranial magnetic stimulation increases the expression of brain-derived neurotrophic factor and cholecystokinin mRNA, but not neuropeptide tyrosine mRNA in specific areas of rat brain. Neuropsychopharmacology. 2000;23:205–215. doi: 10.1016/S0893-133X(00)00099-3. [DOI] [PubMed] [Google Scholar]
- 106.Spampinato C, Aguglia E, Concerto C, Pennisi M, Lanza G, Bella R, Cantone M, Pennisi G, Kavasidis I, Giordano D. Transcranial magnetic stimulation in the assessment of motor cortex excitability and treatment of drug-resistant major depression. IEEE Trans Neural Syst Rehabil Eng. 2013;21:391–403. doi: 10.1109/TNSRE.2013.2256432. [DOI] [PubMed] [Google Scholar]
- 107.Rodger J, Mo C, Wilks T, Dunlop SA, Sherrard RM. Transcranial pulsed magnetic field stimulation facilitates reorganization of abnormal neural circuits and corrects behavioral deficits without disrupting normal connectivity. FASEB J. 2012;26:1593–1606. doi: 10.1096/fj.11-194878. [DOI] [PubMed] [Google Scholar]
- 108.Ziemann U, Reis J, Schwenkreis P, Rosanova M, Strafella A, Badawy R, Müller-Dahlhaus F. TMS and drugs revisited 2014. Clin Neurophysiol. 2015;126:1847–1868. doi: 10.1016/j.clinph.2014.08.028. [DOI] [PubMed] [Google Scholar]
- 109.Lin C, Todo T. The cryptochromes. Genome Biol. 2005;6:220. doi: 10.1186/gb-2005-6-5-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ritz T, Ahmad M, Mouritsen H, Wiltschko R, Wiltschko W. Photoreceptor-based magnetoreception: optimal design of receptor molecules, cells, and neuronal processing. J R Soc Interface. 2010;7 Suppl 2:S135–S146. doi: 10.1098/rsif.2009.0456.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Autry AE, Monteggia LM. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev. 2012;64:238–258. doi: 10.1124/pr.111.005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Allen SJ, Watson JJ, Dawbarn D. The neurotrophins and their role in Alzheimer’s disease. Curr Neuropharmacol. 2011;9:559–573. doi: 10.2174/157015911798376190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nakata K, Ujike H, Sakai A, Takaki M, Imamura T, Tanaka Y, Kuroda S. The human dihydropyrimidinase-related protein-2 gene on chromosome 8p21 is associated with paranoid-type schizophrenia. Biol Psychiatry. 2003;53:571–576. doi: 10.1016/s0006-3223(02)01729-8. [DOI] [PubMed] [Google Scholar]
- 114.Hensley K, Venkova K, Christov A, Gunning W, Park J. Collapsin response mediator protein-2: an emerging pathologic feature and therapeutic target for neurodisease indications. Mol Neurobiol. 2011;43:180–191. doi: 10.1007/s12035-011-8166-4. [DOI] [PubMed] [Google Scholar]
- 115.Charrier E, Reibel S, Rogemond V, Aguera M, Thomasset N, Honnorat J. Collapsin response mediator proteins (CRMPs): involvement in nervous system development and adult neurodegenerative disorders. Mol Neurobiol. 2003;28:51–64. doi: 10.1385/MN:28:1:51. [DOI] [PubMed] [Google Scholar]
- 116.Kim C, Jun K, Lee T, Kim SS, McEnery MW, Chin H, Kim HL, Park JM, Kim DK, Jung SJ, et al. Altered nociceptive response in mice deficient in the alpha(1B) subunit of the voltage-dependent calcium channel. Mol Cell Neurosci. 2001;18:235–245. doi: 10.1006/mcne.2001.1013. [DOI] [PubMed] [Google Scholar]
- 117.McGivern JG, McDonough SI. Voltage-gated calcium channels as targets for the treatment of chronic pain. Curr Drug Targets CNS Neurol Disord. 2004;3:457–478. doi: 10.2174/1568007043336743. [DOI] [PubMed] [Google Scholar]
- 118.Cao YQ. Voltage-gated calcium channels and pain. Pain. 2006;126:5–9. doi: 10.1016/j.pain.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 119.Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL, Liu N, Xiong W, Ripsch MS, Wang Y, et al. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca²+ channel complex. Nat Med. 2011;17:822–829. doi: 10.1038/nm.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ju W, Li Q, Allette YM, Ripsch MS, White FA, Khanna R. Suppression of pain-related behavior in two distinct rodent models of peripheral neuropathy by a homopolyarginine-conjugated CRMP2 peptide. J Neurochem. 2013;124:869–879. doi: 10.1111/jnc.12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Piekarz AD, Due MR, Khanna M, Wang B, Ripsch MS, Wang R, Meroueh SO, Vasko MR, White FA, Khanna R. CRMP-2 peptide mediated decrease of high and low voltage-activated calcium channels, attenuation of nociceptor excitability, and anti-nociception in a model of AIDS therapy-induced painful peripheral neuropathy. Mol Pain. 2012;8:54. doi: 10.1186/1744-8069-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.François-Moutal L, Wang Y, Moutal A, Cottier KE, Melemedjian OK, Yang X, Wang Y, Ju W, Largent-Milnes TM, Khanna M, et al. A membrane-delimited N-myristoylated CRMP2 peptide aptamer inhibits CaV2.2 trafficking and reverses inflammatory and postoperative pain behaviors. Pain. 2015;156:1247–1264. doi: 10.1097/j.pain.0000000000000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Asada A, Yamamoto N, Gohda M, Saito T, Hayashi N, Hisanaga S. Myristoylation of p39 and p35 is a determinant of cytoplasmic or nuclear localization of active cyclin-dependent kinase 5 complexes. J Neurochem. 2008;106:1325–1336. doi: 10.1111/j.1471-4159.2008.05500.x. [DOI] [PubMed] [Google Scholar]
- 124.Yu G, Felsted RL. Effect of myristoylation on p27 nef subcellular distribution and suppression of HIV-LTR transcription. Virology. 1992;187:46–55. doi: 10.1016/0042-6822(92)90293-x. [DOI] [PubMed] [Google Scholar]
- 125.Bhatnagar RS, Gordon JI. Understanding covalent modifications of proteins by lipids: where cell biology and biophysics mingle. Trends Cell Biol. 1997;7:14–20. doi: 10.1016/S0962-8924(97)10044-7. [DOI] [PubMed] [Google Scholar]
- 126.Khanna R, Wilson SM, Brittain JM, Weimer J, Sultana R, Butterfield A, Hensley K. Opening Pandora’s jar: a primer on the putative roles of CRMP2 in a panoply of neurodegenerative, sensory and motor neuron, and central disorders. Future Neurol. 2012;7:749–771. doi: 10.2217/FNL.12.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brennan TJ, Vandermeulen EP, Gebhart GF. Characterization of a rat model of incisional pain. Pain. 1996;64:493–501. doi: 10.1016/0304-3959(95)01441-1. [DOI] [PubMed] [Google Scholar]
- 128.Fischer G, Pan B, Vilceanu D, Hogan QH, Yu H. Sustained relief of neuropathic pain by AAV-targeted expression of CBD3 peptide in rat dorsal root ganglion. Gene Ther. 2014;21:44–51. doi: 10.1038/gt.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Brittain JM, Wang Y, Eruvwetere O, Khanna R. Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with CaV2.2. FEBS Lett. 2012;586:3813–3818. doi: 10.1016/j.febslet.2012.09.022. [DOI] [PubMed] [Google Scholar]
- 130.Wang LH, Strittmatter SM. Brain CRMP forms heterotetramers similar to liver dihydropyrimidinase. J Neurochem. 1997;69:2261–2269. doi: 10.1046/j.1471-4159.1997.69062261.x. [DOI] [PubMed] [Google Scholar]
- 131.Häusser M. Optogenetics: the age of light. Nat Methods. 2014;11:1012–1014. doi: 10.1038/nmeth.3111. [DOI] [PubMed] [Google Scholar]
- 132.Steinberg EE, Christoffel DJ, Deisseroth K, Malenka RC. Illuminating circuitry relevant to psychiatric disorders with optogenetics. Curr Opin Neurobiol. 2015;30:9–16. doi: 10.1016/j.conb.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jacobs AH, Winkler A, Castro MG, Lowenstein P. Human gene therapy and imaging in neurological diseases. Eur J Nucl Med Mol Imaging. 2005;32 Suppl 2:S358–S383. doi: 10.1007/s00259-005-1960-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lim ST, Airavaara M, Harvey BK. Viral vectors for neurotrophic factor delivery: a gene therapy approach for neurodegenerative diseases of the CNS. Pharmacol Res. 2010;61:14–26. doi: 10.1016/j.phrs.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xu H, Li Z, Si J. Nanocarriers in gene therapy: a review. J Biomed Nanotechnol. 2014;10:3483–3507. doi: 10.1166/jbn.2014.2044. [DOI] [PubMed] [Google Scholar]
- 136.Mulligan RC. Development of gene transfer technology. Hum Gene Ther. 2014;25:995–1002. doi: 10.1089/hum.2014.2543. [DOI] [PubMed] [Google Scholar]
- 137.Rothe M, Schambach A, Biasco L. Safety of gene therapy: new insights to a puzzling case. Curr Gene Ther. 2014;14:429–436. doi: 10.2174/1566523214666140918110905. [DOI] [PubMed] [Google Scholar]