

Abstract
Fatigue occurs frequently in patients with cancer, neurological diseases and chronic inflammatory diseases, but the biological mechanisms that lead to and regulate fatigue are largely unknown. When the innate immune system is activated, heat shock proteins (HSPs) are produced to protect cells. Some extracellular HSPs appear to recognize cellular targets in the brain, and we hypothesize that fatigue may be generated by specific HSPs signalling through neuronal or glial cells in the central nervous system. From a cohort of patients with primary Sjögren’s syndrome, 20 patients with high and 20 patients with low fatigue were selected. Fatigue was evaluated with a fatigue visual analogue scale. Plasma concentrations of HSP32, HSP60, HSP72 and HSP90α were measured and analysed to determine if there were associations with the level of fatigue. Plasma concentrations of HSP90α were significantly higher in patients with high fatigue compared with those with low fatigue, and there was a tendency to higher concentrations of HSP72 in patients with high fatigue compared with patients with low fatigue. There were no differences in concentrations of HSP32 and HSP60 between the high- and low-fatigue groups. Thus, extracellular HSPs, particularly HSP90α, may signal fatigue in chronic inflammation. This supports the hypothesis that fatigue is generated by cellular defence mechanisms.
Keywords: Chronic fatigue, innate immunity, cellular stress, heat shock proteins, autoimmune diseases, Sjögren’s syndrome
Introduction
Fatigue has been described as ‘an overwhelming sense of tiredness, lack of energy and feeling of exhaustion’.1 It is a frequent feature of chronic inflammatory and immunological diseases, cancer, and neurological disorders. Patients often have fatigue so profound that it severely interferes with activities of daily living and leads to longstanding sick leave and disability, resulting in economic burdens to society.
The mechanisms that lead to and regulate fatigue are debated. Depression and socioeconomic burden are important, but increasing evidence points to genetic and molecular mechanisms that are activated during inflammation and cellular stress conditions, and signalled via neuro-immune and oxidative/nitrosative stress pathways.2–4
‘Sickness behaviour’ is a phenomenon observed in animals during infection or ‘danger’, and it is highly conserved during evolution.5 It is characterized by sleepiness; depressive mood; social withdrawal; and loss of grooming, thirst, and appetite. Sickness behaviour constitutes a complex and automated behaviour thought to protect the sick individual from predators. Fatigue in humans has some similarities to sickness behaviour in other animals. Several animal studies have explored the pathways involved in sickness behaviour and demonstrated the fundamental role of IL-1β signalling to the brain.6 Activation of innate immunity cells such as macrophages and granulocytes rapidly leads to increased production of IL-1β. This activates other immune cells to destroy and eliminate the pathogen or the endogenous danger molecules. Simultaneously, IL-1β is actively transported to the brain through several mechanisms; once in the brain, it binds to specific IL-1 receptors.7 This initiates the subconscious sickness behaviour, which persists throughout the immune response. In humans, studies indicate that IL-1β is important in the generation of fatigue.8–10 In chronic inflammatory diseases or other conditions characterized by cellular stresses, IL-1β signalling is continuously active; thus, the fatigue phenomenon is persistent and chronic. Treatment with IL-1 blocking agents improves fatigue in humans.9,10 This supports the hypothesis that IL-1β plays a role in fatigue signalling in humans.
When the innate immune system is activated during infections, pathogens are engulfed and destroyed by highly reactive oxygen and nitrogen species (ROS and RNS, respectively) in phagolysosomes. This is an important part of the body’s first line of defence against pathogens. High levels of ROS/RNS can be detrimental, as they can cause DNA damage and cell death. To protect cellular life, cells have therefore developed strong defence systems composed of a large number of active substances and enzymes that counteract the reactive molecules, maintain redox homeostasis and protect vital cellular functions. Oxidative stress is a term used to describe the situation in which reactive molecules prevail over antioxidant and cellular defences, such as in infectious or chronic inflammatory conditions. Several studies have indicated an association between oxidative stress and fatigue.11–15
Heat shock proteins (HSPs) are highly conserved proteins that serve important protective functions under conditions of oxidative stress and a wide range of other cellular stresses. HSPs constitute a large family of proteins and are classified according to their molecular mass.16 Some HSPs are known for their role in protein folding, where they serve as chaperone molecules. Other HSPs are induced upon cellular stress and are released from the cells. These extracellular HSPs take part in cell-to-cell signalling; it is tempting to speculate that extracellular HSPs are part of a signalling system that coordinates several overarching defence mechanisms, including behavioural strategies for survival. Because of the central role of HSPs in cellular defence and the presence of cellular targets for some HSPs in the brain,17,18 we hypothesize that certain HSPs play a signalling role in fatigue.
Primary Sjögren’s syndrome (pSS) is a chronic autoimmune disease that is characterized by inflammation of exocrine glands and subsequent dryness phenomena.19 Fatigue is common in pSS patients, with a reported prevalence of 30–67%, depending on the fatigue instruments used and the patient cohorts investigated.20,21 It poses a major impact on quality of life, described by patients as an ever-present state, unpredictable, fluctuating, and beyond their own control.22 As observed in other diseases, the severity of fatigue is influenced by pain, depression and sleep disturbances.20,21
As there is no effective drug treatment for pSS, gene activity and molecular interactions are relatively undisturbed by drug treatment compared with many other diseases. Therefore, we chose to investigate the expression of HSPs in a cohort of patients with pSS. To explore the potential role of HSPs in fatigue, we included four different HSPs based on the following criteria: (1) secretion from cells upon stimulation, (2) expression in the brain and the presence of targets on cells in the brain, and (3) previous reports of possible associations with fatigue or fatigue-related mechanisms.
Patients and methods
From a cohort of 72 pSS patients, all of whom fulfilled the American–European Consensus Group criteria for pSS,23 we selected the 20 patients with the highest and the 20 patients with the lowest scores on a fatigue visual analogue scale (fVAS). The patients took part in a study in which they were admitted to Stavanger University Hospital for research purposes only. All examinations, testing and blood sampling were performed under strict standardized conditions, with blood sampling at fixed times during the day.
fVAS is a generic fatigue instrument that has been widely used to measure fatigue in patients with pSS and other diseases.24 It consists of a 100-mm horizontal line with vertical anchoring lines. The description at the left end (0 mm) is ‘no fatigue’, and the description at the right end (100 mm) is ‘fatigue as bad as it can be’. The subjects are asked to draw a vertical line at the point corresponding to their experience of fatigue the last week, and the distance from the left anchor is measured, yielding a numerical score for fatigue.
The Beck Depression Inventory (BDI) was used to assess mood. A BDI score of <13 is normally regarded as no depression, a score of 13–19 represents mild depression and a score of >19 reflects moderate-to-severe depression.
Blood samples
Routine haematological and biochemical tests were performed at the hospital’s routine laboratory. Antinuclear antibodies and antibodies to SSA/Ro and SSB/La were analysed with the QUANTA Lite ENA 6 kit (Inova Diagnostics, San Diego, CA, USA). Positive results were confirmed by QUANTA Lite SS-A and SS-B. The clinical characteristics of the patients are provided in Table 1.
Table 1.
Selected clinical variables for patients with pSS with high and low fVAS scores.
| Variables | High fatigue (n = 20) | Low fatigue (n = 20) | P-Value |
|---|---|---|---|
| Age, yr [range] | 58 [32–79] | 59 [36–87] | 1.00 |
| Duration, yr [range] | 9.1 [0.8–14.8] | 4.1 [1.8–11.0] | 0.04 |
| Female sex (%) | 16 (80) | 18 (90) | 0.82 |
| Anti-SSA/SSB (%) | 18 (90) | 14 (70) | 0.31 |
| CRP [mg/l), median [range] | 3 [0–13] | 0 [0–8] | <0.01 |
| BDI scores, median [range] | 13 [5–38] | 6 [0–18] | <0.01 |
| fVAS scores, median [range] | 88 [76–96] | 20 [3–44] | <0.01 |
| Immunosupressive drugs (%) | 9 (45) | 8 (40) | |
| Corticosteroids (%) | 2 (10) | 0 | |
| Antimalarials (%) | 5 (25) | 3 (15) | |
| Corticosteroids and antimalarials (%) | 3 (15) | 3 (15) | |
| Corticosteroids and azathioprine | 1 | 1 | |
| Cyclophosphamide | 0 | 1 |
SSA: Sjögren’s-syndrome-related antigen A (Ro); SSB: Sjögren’s-syndrome-related antigen B (La); CRP: C-reactive protein.
Samples to be analysed for HSPs were collected in EDTA tubes on ice, centrifuged at 2500 g at 4℃ for 15 min, aliquoted and stored at −80℃ until analyses. Plasma concentrations of HSP32, HSP60, HSP72 and HSP90α were measured with commercial ELISA kits (Enzo Life Sciences, Farmingdale, NY, USA). Samples were thawed on ice and diluted 1:3 (HSP32), 1:2 (HSP60), 1:4 (HSP72) and 1:20 (HSP90α) with assay buffer. All samples were assayed in duplicate and analysed according to the manufacturer’s recommendations. HSP concentrations were read as absorbance at 450 nm for HSP32, HSP60 and HSP90α, and 495 nm for HSP72 on a Synergy H1 plate reader (BioTek, Bad Friedrichshall, Germany). Coefficients of variation (CV) between duplicates were <15% for HSP32 and HSP90α and <20% for HSP60 and HSP72. Intra-assay variability (Enzo Life Sciences) was <10% for the HSP32, HSP60 and HSP90 kits (n = 10). For the HSP72 kit, intra-assay variability was <10% for high concentrations (0.5 ng/ml) and 15% for low concentrations (0.14 ng/ml) (n = 20).
Statistics
Owing to the non-normal distribution of the data, results are presented as median and range and Mann–Whitney U-test was used for comparisons of continuous variables in the two patient groups. Logistic regression was used to analyse associations between fatigue and HSP concentrations and other relevant patient data. Relevant variables were first tested in a univariable logistic regression model, and only variables with a P-value < 0.2 were added to the multivariable logistic regression model. Stepwise backward and forward model selection was used to choose the final multivariable model. Goodness of fit was examined by the Hosmer–Lemeshow test. For all analyses, significance was set to P < 0.05. SPSS 22 and RStudio 0.98.1102 (with R 3.1.2) were used for statistical analysis and generation of graphs.
Ethics approval and patients consent
The study was performed according to the Declaration of Helsinki and approved by the Regional Ethics Committee West (2010/1455). All participants signed a legal consent form and were free to refuse any specific part of the examination.
Results
Plasma concentrations of the four different HSPs in the high- and low-fatigue groups are illustrated in Figure 1. HSP90α plasma concentrations were considerably higher in patients with high vs. low fatigue: 40.8 (20.0–105.0) vs. 29.9 (4.9–73.3) ng/ml (P = 0.02). For HSP72 there was a tendency to higher concentration in the high- vs. low-fatigue group, not reaching statistical significance: 1.14 (0.32–12.32) vs. 0.32 (0.32–15.41) ng/ml (P = 0.06). For HSP32 and HSP60, there were no significant differences between the groups with high and low fatigue: HSP32, 3.02 (0.67–8.25) vs. 3.08 (0.0–7.63) ng/ml (P = 0.48); HSP60, 3.0 (3.0–5.0) vs. 3.0 (3.0–210.0) ng/ml (P = 0.25). Because of the higher number of standards needed for HSP72 and HSP90α analyses, one sample from each of these two groups (n = 4) were excluded owing to the limited number of wells on the plates. Twelve out of the 156 samples (7.7%) analysed had CVs that were too high for duplicates and were thus excluded from analysis (HSP72 = 6; HSP60 = 5; and HSP90α = 1).
Figure 1.
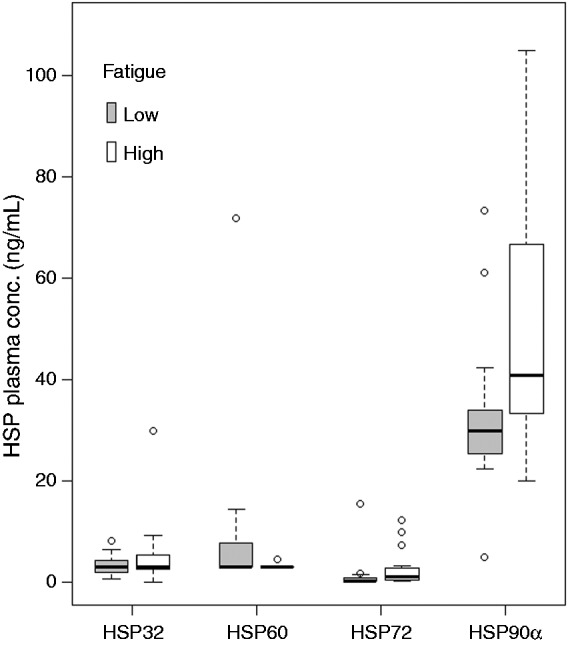
HSP plasma concentrations compared with levels of fatigue. HSP90α concentration differed between high- and low-fatigue groups (P = 0.02). The difference in HSP72 concentrations between high- and low-fatigue groups was close to significant (P = 0.06). There were no associations between fatigue level and plasma concentrations for HSP32 and HSP60. For ease of presentation, one data point for HSP60 (210 ng/ml) in a low-fatigue patient was not included in the Figure.
To investigate whether relevant variables other than HSP90α influenced the fVAS scores, a multivariable logistic regression model was fitted, with high- and low-fatigue groups as the dependent variable and HSP90α, BDI, age, sex, disease duration, C-reactive protein levels and the presence of anti-SSA/SSB Abs as independent variables. In both backward and forward selection, only HSP90α and BDI remained in the final model (Table 2). The Hosmer–Lemeshow test for goodness of fit demonstrated a good model fit for the final model (P = 0.71).
Table 2.
Logistic regression model for association of HSP90α and relevant variables with high and low fatigue.
| Variables | OR | CI (95%) | P-Value |
|---|---|---|---|
| HSP90α | 1.12 | 1.02–1.24 | 0.02 |
| BDI | 1.55 | 1.09–2.21 | 0.02 |
Variables not in the final model: age, duration, sex, anti-SSA/SSB, and CRP.
OR: odds ratio; CI: confidence interval.
To further investigate the influence of depression, patients were dichotomized into one group with BDI scores <13 and a second group with BDI scores ≥13. Plasma concentrations of HSP32, HSP60, HSP72 and HSP90α were then compared between these groups. There were no differences in HSP concentrations between the two BDI groups (Figure 2).
Figure 2.

HSP plasma concentrations vs. BDI score. Patients were dichotomized into one group with no depression (BDI < 13) and one group with mild-to-severe depression (BDI ≥ 13). Plasma concentrations of HSP32, HSP60, HSP72 and HSP90α were then compared between the two depression groups. No differences in BDI score and plasma levels of the respective HSPs were observed between the two groups. For ease of presentation, one data point for HSP60 (210 ng/ml) in a low-fatigue patient was not included in the figure.
Discussion
We found that plasma levels of HSP90α were significantly higher in pSS patients with high fatigue compared with those with low fatigue. In addition, there was a close to significant increase in HSP72 levels in patients with high vs. low fatigue. The influence of HSP90α was considerable, as the odds ratio for high vs. low fatigue was 1.12, indicating that an increase of 1 ng/ml in the HSP90α concentration increased the odds of being in the high-fatigue group by 12%. The influence of depression was even stronger than for HSP90α, as a rise of 1 in the BDI score increased the odds of being in the high-fatigue group by 55%. No differences in HSP32 and HSP60 levels were observed between the high- and low-fatigue groups.
The influence of depression on fatigue, as revealed in the multivariate model, was expected. It is well known that affective states can have a strong impact on fatigue.25,26 However, this association is a complex issue, and it is important to realize that questionnaires used for assessing fatigue and depression often have similar wording. This may lead to circular reasoning and false conclusions regarding their relationship. Moreover, there are indications that depression and fatigue may be signalled through more or less shared molecular pathways via the IL-1 system.27,28 Of note in this regard is the lack of difference in HSP90α concentrations between the groups with and without depression. This suggests that HSP90α itself has no direct effect on the depressive state.
Our findings point to the possibility of a mechanism in which extracellular HSP90α and, to a lesser degree, HSP72 signal to the brain and induce a state of fatigue. This implies that the HSPs have to cross the blood–brain barrier (BBB). Recombinant HSP72 has been demonstrated to cross the BBB in ischaemic brains.29 Whether this is true for HSP90α is unknown, but it raises the question of whether HSPs can be transported across the BBB under normal conditions or only under certain conditions, such as inflammation. Specialized areas of the BBB, known as the circumventricular organs, have no functional BBB because of fenestrated capillaries, and they permit the passage of small molecules across the BBB.30 The circumventricular organs could represent a route for HSP trafficking and signalling into the brain.
Another possible transport option through the BBB is via exosomes. Exosomes are nanovesicles that contain RNA, microRNA and intracellular-derived proteins. Exosomes participate in intercellular signalling by being actively taken up and then releasing their contents.31–33 Exosomes constitute a major secretory pathway for HSPs and can cross the BBB.34–37
A third possibility is that HSP90α and HSP72 are influenced by a common factor that more directly influences fatigue. If this is the case, levels of HSP90α and HSP72 may simply vary in response to this factor.
Finally, it could be that HSPs act as damage associated molecular proteins (DAMPs) together with redox-derived DAMPs on innate immunity cells by activating their TLRs, thus inducing the fatigue phenomenon.38
Specific receptors for HSPs have not been identified in neuronal or glial brain cells. However, TLR4, which is known to be activated by bacterial surface LPS, is a receptor expressed on both neurons and microglial cells. Interestingly, with respect to fatigue, administration of LPS to animals induces sickness behaviour, while simultaneous blocking of the TLR4 signalling pathway by interfering peptides prevents the behaviour.39 In addition to LPS, HSP90α and HSP72 are among the endogenous molecules that activate TLR4.18,40
In cell cultures, HSP32, HSP72 and HSP90α activate microglial cells via TLR4 and lead to cytokine production by activation of p38 MAPK and NF-κB.41 In the context of fatigue generation, it is therefore possible that HSPs in humans interact with TLR4 on microglia and produce IL-1, which is a known inducer of sickness behaviour.6 Another possibility is a direct influence on cerebral neurons through HSP interactions with TLR4 on the surface of the neurons. The presence of TLR4 on cerebral neurons, with co-localization of HSP70, has previously been demonstrated in vivo.42
We did not find any association between fatigue and levels of HSP60 or HSP32. A recent study found antibodies against epitopes of bacterial and human HSP60 in patients with myalgic encephalomyelitis;43 however, to our knowledge, there are no reports regarding blood levels of HSP60 protein and fatigue.
This study has some limitations. It could be argued that we should have used other fatigue measuring instruments, such as multidimensional or disease-specific instruments. However, more than 200 different fatigue instruments exist today, and it is difficult to argue the superiority of one over the other. Because it is well known that VAS show good responsiveness to change over time, and owing to of our long-term experience with the fVAS, we regarded this as the most optimal choice for this study.
Also, it has previously been documented in pSS that a number of other factors influence fatigue, such as muscular and joint pain, mental depression and poor sleep.20,21 The severity of fatigue in the individual patient is therefore a complex phenomenon, modulated by several cofactors, and cannot be attributed to one single player.
In the HSP60 and HSP72 assays, some samples had signals close to and under the lower limit of detection, resulting in high variation between duplicate measures. Five HSP60 samples and six HSP72 samples were consequently removed from analyses. Serum might have been a better sample matrix for HSP60 and HSP72, but we choose to use plasma because we wanted to employ rapid centrifugation and aliquoting of samples at low temperature to avoid degradation of analytes.
Importantly, to be valid, our findings need to be replicated in other and even larger cohorts of pSS patients, and also in cohorts of patients with other diseases.
The strengths of our study are the relatively large groups of well-characterized patients, who were matched in age and nearly matched in sex and were not undergoing drug treatment that could potentially interfere with analysis.
In conclusion, extracellular HSP90α and, to a lesser degree, HSP72 may represent a mechanism by which fatigue is signalled to the brain under conditions characterized by cellular stress, such as chronic inflammatory diseases. These counteractive and down-regulatory processes of inflammation may explain why the severity of fatigue is seldom reported to be a function of disease activity or inflammatory markers.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Kjetil Bårdsen was supported by the Western Norway Regional Health Authority (WNRHA, ‘Helse Vest’), grant number 911775.
References
- 1.Krupp LB, Pollina DA. Mechanisms and management of fatigue in progressive neurological disorders. Curr Opin Neurol 1996; 9: 456–460. [DOI] [PubMed] [Google Scholar]
- 2.Norheim KB, Jonsson G, Omdal R. Biological mechanisms of chronic fatigue. Rheumatology (Oxford) 2011; 50: 1009–1018. [DOI] [PubMed] [Google Scholar]
- 3.Morris G, Berk M, Walder K, et al. Central pathways causing fatigue in neuro-inflammatory and autoimmune illnesses. BMC Med 2015; 13: 28–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maes M. Inflammatory and oxidative and nitrosative stress pathways underpinning chronic fatigue, somatization and psychosomatic symptoms. Curr Opin Psychiatry 2009; 22: 75–83. [DOI] [PubMed] [Google Scholar]
- 5.Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev 1988; 12: 123–37. [DOI] [PubMed] [Google Scholar]
- 6.Dantzer R, Heijnen CJ, Kavelaars A, et al. The neuroimmune basis of fatigue. Trends Neurosci 2014; 37: 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith DE, Lipsky BP, Russell C, et al. A central nervous system-restricted isoform of the interleukin-1 receptor accessory protein modulates neuronal responses to interleukin-1. Immunity 2009; 30: 817–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harboe E, Tjensvoll AB, Vefring HK, et al. Fatigue in primary Sjögren’s syndrome—a link to sickness behaviour in animals? Brain Behav Immun 2009; 23: 1104–8. [DOI] [PubMed] [Google Scholar]
- 9.Norheim KB, Harboe E, Gøransson LG, et al. Interleukin-1 inhibition and fatigue in primary Sjögren’s syndrome—a double blind, randomised clinical trial. PLoS One 2012; 7: e30123–e30123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Omdal R, Gunnarsson R. The effect of interleukin-1 blockade on fatigue in rheumatoid arthritis—a pilot study. Rheumatol Int 2005; 25: 481–484. [DOI] [PubMed] [Google Scholar]
- 11.Avalos I, Chung CP, Oeser A, et al. Oxidative stress in systemic lupus erythematosus: relationship to disease activity and symptoms. Lupus 2007; 16: 195–200. [DOI] [PubMed] [Google Scholar]
- 12.Kennedy G, Spence Va, McLaren M, et al. Oxidative stress levels are raised in chronic fatigue syndrome and are associated with clinical symptoms. Free Radic Biol Med 2005; 39: 584–589. [DOI] [PubMed] [Google Scholar]
- 13.Brkic S, Tomic S, Maric D, et al. Lipid peroxidation is elevated in female patients with chronic fatigue syndrome. Med Sci Monit 2010; 16: CR628–R632. [PubMed] [Google Scholar]
- 14.Segal B, Thomas W, Zhu X, et al. Oxidative stress and fatigue in systemic lupus erythematosus. Lupus 2012; 21: 984–992. [DOI] [PubMed] [Google Scholar]
- 15.Chung CP, Titova D, Oeser A, et al. Oxidative stress in fibromyalgia and its relationship to symptoms. Clin Rheumatol 2009; 28: 435–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stetler RA, Gan Y, Zhang W, et al. Heat shock proteins: cellular and molecular mechanisms in the central nervous system. Prog Neurobiol 2010; 92: 184–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basu S, Binder RJ, Ramalingam T, et al. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 2001; 14: 303–313. [DOI] [PubMed] [Google Scholar]
- 18.Triantafilou M, Triantafilou K. Heat-shock protein 70 and heat-shock protein 90 associate with Toll-like receptor 4 in response to bacterial lipopolysaccharide. Biochem Soc Trans 2004; 32: 636–639. [DOI] [PubMed] [Google Scholar]
- 19.Jonsson R, Vogelsang P, Volchenkov R, et al. The complexity of Sjögren’s syndrome: novel aspects on pathogenesis. Immunol Lett 2011; 141: 1–9. [DOI] [PubMed] [Google Scholar]
- 20.Karageorgas T, Fragioudaki S, Nezos A, et al. Fatigue in primary Sjogren’s syndrome: clinical, laboratory, psychometric and biological associations. Arthritis Care Res (Hoboken) 2016; 68: 123–131. [DOI] [PubMed] [Google Scholar]
- 21.Segal B, Thomas W, Rogers T, et al. Prevalence, severity, and predictors of fatigue in subjects with primary Sjögren’s syndrome. Arthritis Rheum 2008; 59: 1780–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mengshoel AM, Norheim KB, Omdal R. Primary Sjögren’s syndrome: fatigue is an ever-present, fluctuating, and uncontrollable lack of energy. Arthritis Care Res (Hoboken) 2014; 66: 1227–1232. [DOI] [PubMed] [Google Scholar]
- 23.Vitali C, Bombardieri S, Jonsson R, et al. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis 2002; 61: 554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolfe F. Fatigue assessments in rheumatoid arthritis: comparative performance of visual analog scales and longer fatigue questionnaires in 7760 patients. J Rheumatol 2004; 31: 1896–1902. [PubMed] [Google Scholar]
- 25.Arnold LM. Understanding fatigue in major depressive disorder and other medical disorders. Psychosomatics 2008; 49: 185–190. [DOI] [PubMed] [Google Scholar]
- 26.Omdal R, Waterloo K, Koldingsnes W, et al. Fatigue in patients with systemic lupus erythematosus: the psychosocial aspects. J Rheumatol 2003; 30: 283–287. [PubMed] [Google Scholar]
- 27.Goshen I, Kreisel T, Ben-Menachem-Zidon O, et al. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry 2008; 13: 717–728. [DOI] [PubMed] [Google Scholar]
- 28.Lawson MA, McCusker RH, Kelley KW. Interleukin-1 beta converting enzyme is necessary for development of depression-like behavior following intracerebroventricular administration of lipopolysaccharide to mice. J Neuroinflammation 2013; 10: 54–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shevtsov MA, Nikolaev BP, Yakovleva LY, et al. Neurotherapeutic activity of the recombinant heat shock protein Hsp70 in a model of focal cerebral ischemia in rats. Drug Des Devel Ther 2014; 8: 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benarroch EE. Circumventricular organs: receptive and homeostatic functions and clinical implications. Neurology 2011; 77: 1198–1204. [DOI] [PubMed] [Google Scholar]
- 31.Svensson KJ, Christianson HC, Wittrup A, et al. Exosome uptake depends on ERK1/2-heat shock protein 27 signaling and lipid raft-mediated endocytosis negatively regulated by caveolin-1. J Biol Chem 2013; 288: 17713–17724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antonucci F, Turola E, Riganti L, et al. Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO J 2012; 31: 1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chivet M, Javalet C, Laulagnier K, et al. Exosomes secreted by cortical neurons upon glutamatergic synapse activation specifically interact with neurons. J Extracell Vesicles 2014; 3: 24722–24722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hegmans JPJJ, Bard MPL, Hemmes A, et al. Proteomic analysis of exosomes secreted by human mesothelioma cells. Am J Pathol 2004; 164: 1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lancaster GI, Febbraio MA. Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J Biol Chem 2005; 280: 23349–23355. [DOI] [PubMed] [Google Scholar]
- 36.Merendino AM, Bucchieri F, Campanella C, et al. Hsp60 is actively secreted by human tumor cells. PLoS One 2010; 5: e9247–e9247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alvarez-Erviti L, Seow Y, Yin H, et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol 2011; 29: 341–345. [DOI] [PubMed] [Google Scholar]
- 38.Lucas K, Morris G, Anderson G, et al. The Toll-like receptor radical cycle pathway: a new drug target in immune-related chronic fatigue. CNS Neurol Disord Drug Targets 2015; 14: 838–854. [DOI] [PubMed] [Google Scholar]
- 39.Hines DJ, Choi HB, Hines RM, et al. Prevention of LPS-induced microglia activation, cytokine production and sickness behavior with TLR4 receptor interfering peptides. PLoS One 2013; 8: e60388–e60388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asea A, Rehli M, Kabingu E, et al. Novel signal transduction pathway utilized by extracellular HSP70. Role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem 2002; 277: 15028–15034. [DOI] [PubMed] [Google Scholar]
- 41.Kakimura J-I, Kitamura Y, Takata K, et al. Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J 16: 601–603. [DOI] [PubMed] [Google Scholar]
- 42.Ohara K, Shimizu K, Matsuura S, et al. Toll-like receptor 4 signaling in trigeminal ganglion neurons contributes tongue-referred pain associated with tooth pulp inflammation. J Neuroinflammation 2013; 10: 139–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elfaitouri A, Herrmann B, Bölin-Wiener A, et al. Epitopes of microbial and human heat shock protein 60 and their recognition in myalgic encephalomyelitis. PLoS One 2013; 8: e81155–e81155. [DOI] [PMC free article] [PubMed] [Google Scholar]