

Abstract
In the recently characterized Type IX Secretion System (T9SS), the conserved C-terminal domain (CTD) in secreted proteins functions as an outer membrane translocation signal for export of virulence factors to the cell surface in the Gram-negative Bacteroidetes phylum. In the periodontal pathogen Porphyromonas gingivalis, the CTD is cleaved off by PorU sortase in a sequence-independent manner, and anionic lipopolysaccharide (A-LPS) is attached to many translocated proteins, thus anchoring them to the bacterial surface. Here, we solved the atomic structure of the CTD of gingipain B (RgpB) from P. gingivalis, alone and together with a preceding immunoglobulin-superfamily domain (IgSF). The CTD was found to possess a typical Ig-like fold encompassing seven antiparallel β-strands organized in two β-sheets, packed into a β-sandwich structure that can spontaneously dimerise through C-terminal strand swapping. Small angle X-ray scattering (SAXS) revealed no fixed orientation of the CTD with respect to the IgSF. By introducing insertion or substitution of residues within the inter-domain linker in the native protein, we were able to show that despite the region being unstructured, it nevertheless is resistant to general proteolysis. These data suggest structural motifs located in the two adjacent Ig-like domains dictate the processing of CTDs by the T9SS secretion pathway.
Protein secretion onto the cell surface or into the extracellular milieu is essential for the survival of living organisms. In the case of diderm bacteria, secreted proteins must traverse two protein-impermeable lipid barriers: the inner- and outer-membranes (IM and OM, respectively), which are integral parts of the cell envelope in Gram-negative bacteria. To achieve this, diderm bacteria have evolved eight unique Types (I to VIII) of Secretion System (TxSS) to translocate cargo proteins to the cell surface, including surface appendages1. Cargo proteins either translocate across both cell envelopes directly from the cytoplasm (T1SS, T3SS, T4SS, and T6SS), or are first exported across the IM into the periplasm via signaling through an N-terminal signal peptide to the Sec translocon or twin-arginine translocation (Tat) systems and then transported through the outer membrane (T2SS, T5SS, T7SS, and T8SS). Subsequently, the cargo proteins become components of surface superstructures or surface proteins by remaining attached to the bacterial cell surface, or are released in a soluble form into the environment.
With the exception of T1SS and T2SS, which are widely distributed among prokaryotes, all other types of secretion systems are limited to proteobacteria with scattered occurrence in a few evolutionary lineages of bacteria2. This is the case for Porphyromonas gingivalis, a bacterium belonging to the Bacteroidetes phylum recognized as a major periodontal pathogen3. The virulence of P. gingivalis is dependent on secreted cysteine proteases called gingipains (RgpA, RgpB, and Kgp) that are essential for corruption of the host immune system and acquisition of nutrients and heme – a vital growth factor and source of black pigmentation of colonies on blood agar4. Gingipains are synthesized with a typical N-terminal signal peptide targeting them to the Sec translocon5,6 and are exported through the OM via a novel secretion system referred to as “Por Secretion System; PorSS” or following the naming convention, Type IX Secretion System (T9SS)7. Currently, the T9SS is composed of several OM, periplasmic, and IM proteins whereby knockout of any of these proteins leads to a gingipain-null, non-pigmented mutant phenotype in which inactive gingipains accumulate in the periplasm7,8,9,10,11,12.
Translocation of gingipains from the periplasm across the OM is dependent on a conserved C-terminal domain (CTD), which appears to be important for secretion of the proteins13 and in particular, truncation of the last few C-terminal residues of this domain leads to accumulation of gingipains in the periplasm14. Subsequently, the T9SS targeting signal was demonstrated to reside within the last 22 residues at the C-terminus of the CTD15. During gingipain translocation across the OM, the CTD is cleaved off by PorU, an essential component of the T9SS that acts as a sorting protease16, and anionic-LPS (A-LPS) is then covalently attached for surface anchorage to the OM17. In the case of RgpB, covalent attachment of A-LPS depends on the length of a linker sequence between the CTD and the preceding immunoglobulin-superfamily domain (IgSF), and occurs concurrently with the proteolytic cleavage of the CTD18.
Apart from gingipains, 27 other proteins in P. gingivalis possess CTDs, several of which were shown to be post-translationally modified during secretion via T9SS19. Together, they form an electron-dense layer of proteins highly enriched in gingipains on the P. gingivalis cell surface20. The Tannerella forsythia surface proteins TsfA and TsfB are secreted and processed in a similar manner (CTD cleavage and glucan attachment) via a T9SS, forming the semi-crystalline S-layer on the bacterial surface21,22. Several other CTD-bearing proteins, including KLIKK proteases23, are also secreted by T. forsythia via T9SS24,25. Collectively, S-layer and secreted CTD proteins regulate the virulence potential of T. forsythia, which together with P. gingivalis and Treponema denticola, are grouped into the so-called “red complex” organisms26. Red complex pathogens are instrumental in causing dysbiosis in the dental microbiota, which eventually leads to the development of chronic periodontitis in susceptible individuals27.
T9SS is not limited to periodontal pathogens and is widespread among members of the Bacteroidetes phylum with gliding motility27. In Cytophaga hutchinsonii and Flavobacterium jonsoniae, T9SS is required for secretion of cell surface gliding motility adhesins and cell surface-associated enzymes required for the digestion of crystalline cellulose and chitin28,29,30,31,32. As in periodontal pathogens, all proteins secreted by gliding Bacteroidetes via T9SS contain a CTD encompassing approximately 60 to 100 amino acid residues that is proteolytically cleaved during or after translocation across the OM19.
A fast growing body of evidence indicates that T9SS is used by diverse members of the Bacteroidetes phylum to secrete CTD-bearing proteins. Despite limited sequence identity, CTDs share some sequential motifs that maybe important in its function. However, not much else is known about the structure of this domain. To elucidate the role of the CTD in RgpB secretion and posttranslational modification, we solved the crystal structure of the CTD alone and in tandem with the preceding IgSF domain. This, and the known structure of mature RgpB in complex with the N-terminal prodomain, allowed us to reconstruct the structure of the RgpB zymogen: the catalytic domain followed by tandem of Ig-like folded domains. Furthermore, small angle X-ray scattering (SAXS) revealed no fixed reciprocal orientation of the IgSF and CTD domains, which are connected by an unstructured linker. Finally, the propensity of the CTD to dimerize may represent a mechanism involved in domain recognition by T9SS machinery.
Results
Structure of the rIgSF-CTD tandem
Whereas the structures of mature RgpB alone and in the inhibitory complex with the N-terminal pro-domain are available33,34, there is no structural data available on the CTD of proRgpB or any other T9SS-translocated protein. This domain is conserved in all proteins secreted by T9SS to signal their translocation across the outer membrane. To cast light on the structure-function relationship of the CTD, we recombinantly expressed this domain alone or together with the preceding IgSF domain (rIgSF-CTD) as GST fusion proteins carrying a Prescission cleavage site. In addition, our previous report described the strange behavior of non-glycosylated RgpB production in the native organism when the junction between the IgSF and CTD was lengthened by insertion of a hexapeptide motif18, to investigate this phenomenon, we also produced recombinant IgSF-CTD with insertion or substitution of hexapeptide motifs in the linker region for analysis.
Despite extensive efforts, we were unable to crystallize the wild-type version of recombinant protein containing the wild-type IgSF-CTD (residues T577–K736; for residue numbering, see UniProt database entry P95493). However, diffraction quality crystals were obtained using mutants of the same construct, which included an inserted hexahistidine after residues S664 or I665 (recombinant products r664i6H and r665i6H, respectively), or by substitution of six consecutive linker residues (I665ADVAN670) with a factor Xa cleavage motif (IEGRAA; r665sXa). The three structures were solved and refined with diffraction data to 1.9 Å, 2.4 Å, and 2.1 Å resolution, respectively (Table S1). Each variant of rIgSF-CTD formed nearly identical crystal structures (RMSD of 0.760 Å for r665i6H and 0.327 Å for r665sXa, with respect to r664i6H for 298 aligned residues). Thus, we performed our analysis with the structure refined to highest resolution (r664i6H).
The recombinant r664i6H product (Table S1) assembled into a monoclinic lattice, producing crystals belonging to monoclinic space group P21. The asymmetric unit contained two molecules, each corresponding to a r664i6H tandem with approximate dimensions of 55 Å × 33 Å × 20 Å (Fig. 1A). In each tandem, the IgSF and CTD domains showed an orthogonal arrangement, with their longest axes tilted by 90 degrees with respect to each other. Regions with defined electron density included the residues V579-G662 (IgSF) and K672–K736 (CTD). The 9-residue linker (T663-D671) between the immunoglobulin-like subdomain and the CTD did not yield visible electron density, indicating that this fragment was disordered in the crystals. The hexahistidine tag was inserted in this disordered area.
Figure 1. Structure and topology diagram of the rIgSF-CTD tandem and structural superposition of the IgSF and CTD domains calculated using the Secondary Structure Matching (SSM) algorithm66.

(A) Overview of the IgSF (cyan)-CTD (yellow) tandem. The structure is elongated, with both domains packing in a perpendicular fashion. The shape of the IgSF closely resembles a β-barrel, whereas the CTD is more a regular β-sandwich. (B) Topology and schematic diagrams of, respectively, IgSF (left) and CTD (right). Although both domains have an Ig-like fold, significant differences are observed between sheet composition. Whereas the N-terminal sheet of each domain is antiparallel in both cases, the C-terminal sheet differs, being mixed in IgSF. In IgSF, strands sA-sA’ and, to a lower extent, sD, are establish interactions with both sheets of the barrel, thus generating a higher curvature in the surface of the sheets. (C) IgSF (cyan, sA–sG) and CTD (yellow, s1–s7) can be superimposed with an RMSD of 2.1 Å for 63 aligned residues, thus confirming that both fragments have an Ig-like fold. However, main differences occur at the first strand (sA–sA’ for IgSF, s1 for the CTD) and in the curvature of the sheets (less prominent for the CTD). (D) Structure-based sequence alignment. Despite low sequence identity, the number, length and type of secondary structures reveals that both are Ig-like domains.
The IgSF domain consists of a seven-stranded β-barrel composed of two β-sheets (sA-sG, with the first strand split into two). The antiparallel smaller N-terminal sheet is initiated with a short strand, sA (Q584-T586), which transitions to a connecting loop leading to strand sA’, which becomes a peripheral, parallel strand in the larger C-terminal 5-stranded β-sheet (Fig. 1A,B). Strand sA’ then folds back to connect with sB, the central strand of the N-terminal sheet. sB progresses via a loop to sC, which forms a β-hairpin and then continues to sD within the C-terminal sheet. This creates the inner core of the Greek-key motif. Turns after sD and sE, with the latter forming a long loop to reach sF, complete the three-stranded, N-terminal sheet. The sF strand is connected by a β-hairpin motif with sG, which completes the IgSF domain. No disulfide bridges are observed within this domain, which is assembled into a cylindrical β-barrel due to the interactions between the lateral strands of both sheets. This is clear at the sA-sA’ strand, which is split and interacts with the two β-sheets, and also at the sD strand, although to a lower extent. Together, these interactions generate a clamping force able to increase the curvature of the outer surface of the barrel.
The overall fold of the rCTD distantly resembles that of the IgSF domain, and both domains superimpose with an RMSD of 2.11 Å for 63 aligned residues (Fig. 1C), despite sharing a sequence identity below 20% (Fig. 1D). As the IgSF domain, the CTD is composed of two almost completely parallel to each other β-sheets that are formed by three and four antiparallel strands (Fig. 1A). This differs with respect to the IgSF domain, in which the overall shape somewhat resembles a flattened ellipsoid. In the CTD, all β-strands are of similar length and no significant interactions occur between the lateral strands of each sheet, thus generating a compact, sandwich-like structure instead of a β-barrel. In comparison to the IgSF domain, the β-turns/hairpins and connecting loops are shorter in the CTD, and the sheets are more regular and clearly identifiable. Nevertheless, the structure also follows a Greek-key topology typical of Ig-like folded domains (Fig. 1A,B, right panel).
The CTD has maximal dimensions of 20 Å × 32 Å × 15 Å. It starts with strand s1, which, after a β-hairpin, continues to strand s2, followed by a loop that crosses to the other side of the sandwich, becoming s3 of the C-terminal sheet (Fig. 1B, right panel). The s3 strand makes a β-hairpin-like loop and progresses to strand s4, thus constituting the inner motif of the Greek-key, similar to the IgSF domain. The s3–s4 loop contains D696, in which the side chain forms double hydrogen bonds with the α-nitrogens of G699 and R700 of the polypeptide backbone (that in this area runs orthogonally to s3), generating a curved hairpin resembling a spoon (Fig. 1A). At the tip of this spoon structure, a methionine protrudes from the surface. A similar structure occurs in the loop that connects s4 of the C-terminal sheet with s5, the last strand of the N-terminal sheet. Within this loop, double hydrogen bounds between N707 and the α-nitrogens of S687 and A690 produce a structure that resembles a highly curved antiparallel, double-stranded sheet. The long loop joins s5 and s6 within the C-terminal sheet, then the polypeptide chain connects via a β-turn with s7, the last and longest strand of the domain, leading to the end of the protein. The two hairpin motifs (strands s1–s2 and s6–s7) are exposed in opposed sides along the longest axis of the CTD.
It can be speculated that the unusual conformation of the s3–s4 and s4–s5 loops, along with conservation of D696 followed by methionine or branched aliphatic amino acids (valine, leucine and isoleucine) in T9SS cargo proteins (Fig. S1) might be involved in the function of the CTD in domain sorting proteins for secretion via T9SS. Nevertheless, mutation of D696 in RgpB does not affect gingipain secretion or posttranslational modification18.
Structural similarity searches using the CTD domain identified the human surface antigens CD48 (PDB 2DRU; DALI Z-score = 8.5; RMSD = 2.1 Å; length of alignment = 64; sequence identity = 13%), CD58 (1CCZ; 8.4; 2.1; 64; 13), CD2 (1HNG; 8.3; 2.1; 64; 13), and CD4 (1CID; 8.2; 2.7; 63; 16), all of which possess an Ig-like fold. This result confirms again that the CTD belongs to the Ig-like superfamily, making the IgSF-CTD arrangement a tandem of Ig-like domains.
Small-angle X-ray scattering (SAXS) structure of rIgSF-CTD tandem
Small-angle X-ray scattering (SAXS) data from recombinant rIgSF-CTD and the r664i6H variant (Fig. 2) indicate that in solution the two domains are connected by a flexible linker. The small-angle scattering for both constructs indicate they are pure monomers in solution (see Methods) and thus the SAXS data are suitable for structural analysis (Table S2). Inverse Fourier transform of I(q) versus q to yield the density-weighted atom-pair length distribution (P(r) versus r (Fig. 2B) shows an asymmetric profile with a peak at ∼20 Å, a shoulder around 50 Å and a maximum dimension (dmax) ∼100 Å, consistent with an elongated, two-domain structure. As might be expected, the Rg value for the wild type construct (25.2 ± 0.1 Å) is somewhat smaller than that for the r664i6H variant (26.0 ± 0.2 Å) containing the hexahistidine insertion, but there appears little difference in dmax values (97–98 Å) and the overall P(r) profile is very similar, with some small variation above r values of 60 Å.
Figure 2. Small angle X-ray scattering (SAXS) of rIgSF-CTD.

(A) SAXS data for wild-type rIgSF-CTD (black symbols) and the r664i6H variant (red symbols) with the ensemble optimisation model fit shown as lines in the reverse colours. (B) Atom pair distribution, P(r) versus r, calculated as the Fourier transform of the I(q) versus q profiles in A, with the same colour coding. (C) Ensemble optimisation modelling (EOM) selected representative structures aligned via the IgSF domains (magenta) with the small flexible tip of N-terminal residues and the linker in yellow (surface representation only), showing the range of positions for the CTD domains within the wild-type rIgSF-CTD construct; the CTD domains coloured differently for each conformation within the set. (D) Overlay of EOM selected sets for wild-type rIgSF-CTD (cyan CTD domains) and r664i6H variant (orange CTD domains) to show the range of flexibility of the CTD with respect to the aligned IgSF domain (magenta).
Modelling against the scattering data for each of the two constructs using dummy residues to define a volume occupied by each structure shows an extended asymmetric shape (Fig. S2) consistent with the expected two domains and the P(r) analysis. Atomistic models were created to test against the scattering data using the two structured domains from the crystal structure (IgSF residues 579–662 and CTD residues 672–736) and a linker of undefined structure for the wild type (residues 663–671) and r664i6H variant (residues 663–671 plus the hexahistidine insertion). An optimised average best-fit structure was generated (using the program BUNCH) that yielded a χ2 value for the model fits to the scattering data of 1.3 (Table S2) and models that showed few or no interactions between the domains (Fig. S2). Multiple runs of BUNCH produced similar fits, with varying orientations of the IgSF and CTD domains for both structures.
SAXS data yield structural information on the time and ensemble average conformation in solution, and to test the possibility that the domain linker region might be flexible in solution, an ensemble optimisation modelling (EOM) approach was used with the same two domain structures and flexible linkers. The EOM calculations for both constructs gave a significantly improved fit to the data with a χ2 values of 1 (Table S2) and an excellent fit to the data over the measured q-range (Fig. 2A). Examination of the Rg and dmax population distributions for the two sets of ensembles (Fig. S3) shows a weighting toward more compact structures with Rg values ∼2–3 Å smaller than the average values, but a significant population of more extended conformations are also observed. Further, the hexahistidine insertion in the r664i6H variant provides for greater range of Rg and dmax values in the ensemble; by ∼4 Å in Rg and >10 Å for dmax. Figure 2C shows the selected representative example structures used to define the ensemble in the wild-type rIgSF-CTD construct. The scattering data are consistent with the hexahistidine insertion in the r664i6H construct having extended the overall length of native linker between the IgSF and CTD domains and thus facilitating a wider sampling of conformational space in the EOM results (Fig. 2D), perhaps enabling the inter-domain contacts that aided in crystallisation of this variant.
Structure of a domain-swapped rCTD dimer
In addition to the rIgSF-CTD tandem of the Ig-like folded domains, we solved the structure of the rCTD alone. Crystals were obtained from a fragment encompassing residues S664–K736, which crystallized in the hexagonal space group, P65 (Table S1). The structure was solved to 1.9 Å resolution (Table S1) and the Fourier map enabled us to trace residues K672–K736. The asymmetric unit contained a dimer of rCTD molecules (approximately 75 Å × 21 Å × 14 Å) in a side-to-side arrangement, with the monomers related to each other by a twofold axis. The interface between the monomers had a total surface area of 1,440 Å, accounting for 28% of the total accessible surface area of the dimer. Superimposition of one monomer with the corresponding domain in the rIgSF-CTD tandem revealed that the conformations of strands s1–s6 (corresponding to the 55 N-terminal residues) of the rCTD were very similar (Fig. 3). In both structures, these strands constitute a parallel sandwich of two three-stranded β-sheets with the fully conserved inner Greek-key topology, including the same β-turns/hairpins and connecting loops (Fig. 3B). However, a significant difference was observed between the polypeptide chain encompassing residues V717–V735 corresponding to the s6-s7 loop and the last strand (s7) of the CTD. In this structure, strand 7 was swapped between the two monomers and constituted the primary interface between them. Thus, in this conformation, the s6-s7 loop, instead of a turn, adopts an extended conformation along the s6 main axis, bringing s7 to interact with the neighboring monomer. This change rotates the position of E725 side chain by almost 180 degrees, and repositions T724, which is able to establish both main and side chain H-bonding interactions with the backbone of K727 from the neighboring monomer, thus stabilizing this new conformation (Fig. 3C). The equivalent region from the neighboring monomer then occupies the space generated by this movement in an identical manner. This domain swap, as a consequence, generates an antiparallel β-sheet between strands s6 and s7, as occurs in the absence of swapping, that has the same backbone and side chain set of interactions (see Fig. 1A), with the only differences being the monomer that provides s7 and the conformation of the s6-s7 loop. Minor variations occur on the surface side chains of T730 and R721 due to the difference in content and packing of the crystals.
Figure 3. The rCTD structure.
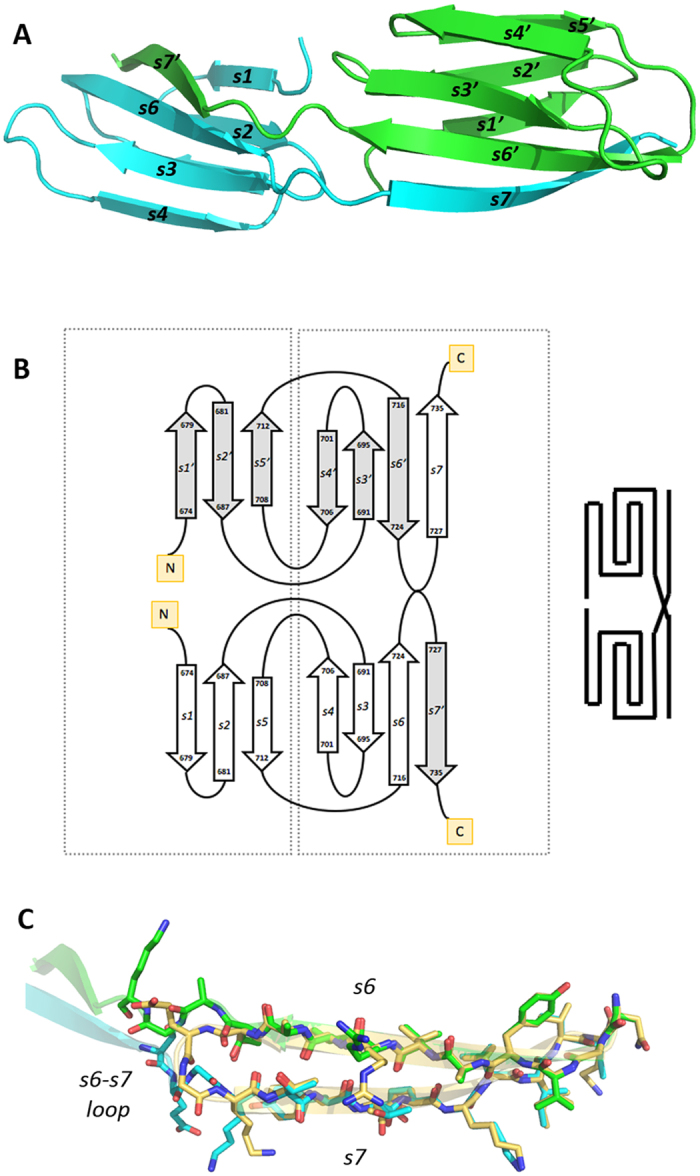
(A) Purified rCTD yielded a dimeric structure (chains in cyan and green), in which both domains interact head to back. The C-terminal strand does not fold back, running antiparalel to the penultimate strand but rather runs in extended conformation and provides the last strand of the symmetric partner. (B) Topology diagram showing the domain swap of the C-terminal strands. (C) Stick model showing the superposition of strands s6 and s7 in the CTD of the IgSF-CTD tandem structure (carbon atoms in gold) and in the dimeric standalone CTD structure (strand s6 of one monomer in green, strand s7 of the other monomer in cyan). Except for the respective linkers to the preceding strands s6, the two s7 strands nicely fit on top of each other.
Probing rIgSF-CTD and its variants by limited proteolysis
Limited proteolysis is a technique commonly used to determine domain composition and probe for structural differences induced by physiological and/or pathological modifications36. We applied this method to elucidate differences in the conformation of the linker between the IgSF and CTD domains, which may explain the failure of the native rIgSF-CTD form to crystallize. To this end, we probed r664i6H and native rIgSF-CTD by limited proteolysis using V8 protease, Lys-C endopeptidase, and human neutrophil elastase (HNE) (Fig. 4A,B). In both cases, the same peptide bonds within the IgSF-like domains were cleaved, but with different kinetics. Apart from E599#V, E634#S, and K627#A cleavages (“#” indicates hydrolyzed peptide bonds) away from the IgSF-CTD junction, all other major primary cleavages were mapped to a loop between strands s6 and s7 and directly at the end of s7 (Fig. 4C). Proteolysis of Lys-Asp within strand s7 and cleavage of Thr-Ser peptide bonds by Lys-C peptidase and HNE, respectively, appear to be secondary to upstream cleavages at Lys-Val, Val-Gly, and Val-Thr. Interestingly, no proteolysis occurred directly within the flexible linker loop, despite the presence of several peptide bonds (Ile-Ala, Ala-Asp, Val-Ala, and Ala-Asn) that should be susceptible to proteolysis by HNE.
Figure 4. Probing the structure of the wild-type rIgSF-CTD and the r665i6H variant by limited proteolysis.

Both proteins were incubated with V8 protease (V8), Lys-C endopeptidase (Lys-C) and human neutrophil elastase (HNE) at (A) different substrate:enzyme molar ratios (concentration-dependent proteolysis) for 1 h or (B) at a constant 100:1 substrate:enzyme ratio for different time intervals (time-dependent proteolysis). Boiling with SDS-PAGE reducing sample buffer terminated reaction and samples were subjected to SDS-PAGE, transferred onto PVDF membrane for N-terminal sequencing (sequences next to each panel). (C) Identified primary cleavage sites within the linker region are indicated by solid arrows: red for elastase, blue for Lys-C and green for V8 protease. Open arrows indicate secondary cleavages by the respective enzymes. Polypeptide sequence comprising the IgSF domain is in blue font; the linker region between IgSF and CTD domains is in lowercase and inserted 6×His is in red. Horizontal arrows below the sequence indicate β-strands in the IgSF-CTD tertiary structure.
Since the first round of probing by limited proteolysis did not reveal any cleavage events within the inter-domain linker, we constructed and analyzed three additional variants of rIgSF-CTD for susceptibility to cleavage by porcine pancreatic elastase (PPE) and factor Xa: substitution of residues I665ADVAN670 with hexahistidine residues in r665s6H; and insertion of hexahistidine or Xa cleavage sequence (IEGRAA) after residue Gly662 in recombinant proteins r662i6H and r662iXa, respectively (Fig. 5D). Interestingly, the protein variants with insertions elongating the inter-domain linker were cleaved faster by PPE than those for which substitution altered the sequence but not the length of the linker region (Fig. 5A,D). Primary cleavage by PPE occurred at the end of strand s6 (Val-Gly) followed by hydrolysis of the Ile-Lys peptide bond. All cleavages downstream of strand s7 were secondary (see Fig. 5A, dashed arrows) and most likely represent truncation of CTD. Again, despite the presence of many peptide bonds potentially susceptible to proteolysis by PPE (Ile-Ala, Ala-Ala, Ala-Asp, Val-Ala, Ala-Asn) in the junction region of the native IgSF-CTD protein, none were initially cleaved by PPE. In addition, the same bonds shifted downstream by insertion of 6×His (r662i6H) or the factor Xa cleavage motif (IEGRAA; in construct r662iXa) were also resistant to proteolysis by PPE. Furthermore, potential PPE cleavage sites (Ala-Ala, Ala-Asp, and Ala-Thr) within the IEGRAA sequence in r662sXa and r664iXa were secondary PPE cleavage sites in proteins with the insertion of the sequence, but not in the protein in which it was introduced by substitution. The lack of cleavage observed within the linker by elastase is clearly not due to inaccessibility of this loop to proteolytic enzymes, as proteins bearing the IEGRAA motif by insertion/substitution were efficiently cleaved at the Arg-Ala peptide bond by fXa (Fig. 5B,D). Interestingly, however, there was a clear difference in the kinetics of cleavage events, with r662iXa being far more susceptible to proteolysis than r665sXa (Fig. 5C).
Figure 5. Probing the structure of insertion/substitution variants of IgSF-CTD by limited proteolysis.

IgSF-CTD variants were incubated with (panels A and B) porcine pancreatic elastase (PPE) or (panel C) factor Xa (fXa) at different substrate:enzyme molar ratios (concentration-dependent proteolysis) for 1 h or at a constant 100:1 substrate:enzyme ratio for different time intervals (time-dependent proteolysis). Boiling with SDS-PAGE reducing sample buffer terminated reaction and samples were subjected to SDS-PAGE, transferred onto PVDF membrane for N-terminal sequencing (sequences next to each panel). (D) Identified primary cleavage sites within the linker region are indicated by solid arrows: red for PPE and black for fXa. Open and broken red arrows indicate secondary and minor cleavage sites for PPE. Polypeptide sequence comprising the IgSF domain is in blue font; the linker region between IgSF and CTD domains is in lowercase with inserted/substituted sequences in red font. Horizontal arrows below the sequence indicate β-strands in the IgSF-CTD tertiary structure.
Taken together, these results demonstrate that insertion and substitution within the connecting linker do not affect domain structure or inter-domain arrangement, and suggest that the linker is resistant to proteolysis despite the presence of peptide bonds potentially susceptible to cleavage by HNE and PPE. Instead, these two proteases very efficiently cleave the native and mutated rIgSF-CTD upstream within a loop between s6 and s7. Conversely, the intra-domain loop in mutants bearing the IEGRAA motif was highly susceptible to proteolysis by fXa, a protease with unique specificity37.
Probing full-length proRgp for proteolytic release of CTD
In stark contrast to the wild-type W83 strain which retains anionic-LPS conjugated RgpB on the OM, P. gingivalis mutants expressing RgpB with insertion of a hexahistidine-tag or an 8-residue StrepII© tag after residue G662 in the junction of IgSF-CTD secrete the RgpB into the culture medium in a soluble, non-glycosylated form18. Conversely, replacement of residues S664–N670 with Ala-6×His yielded a mutant with wild-type phenotype. The effect of these and other mutations in the linker region between the IgSF and CTD domains on RgpB secretory phenotype suggests that the length of the junction is critical for RgpB glycosylation18. This observation was confirmed presently using IEGRAA as the motif for insertion after residue G662 of RgpB in P. gingivalis (mutant RgpB662iXa6H) resulting in secretion of soluble, non-glycosylated RgpB or substitution of residues I665–N670 (RgpB665sXa6H) with IEGRAA resulting in glycosylated, membrane-bound RgpB (Fig. S4).
Based on subtle but clear differences in the kinetics of cleavage of IgSF-CTD carrying an insertion vs. a substitution, we hypothesize that the rate of proteolysis of the CTD during the secretion process is responsible for the lack of glycosylation of insertion-carrying RgpB. To further investigate this possibility, we purified full-length, C-terminally hexahistidine-tagged proRgpB in which the IEGRAA motif was either inserted in the IgSF-CTD linker (RgpB662iXa6H) or replaced residues I665–N670 with the motif (RgpB665sXa6H). The mutated RgpB variants were expressed in the ΔporN secretion-deficient mutant of P. gingivalis W83 to allow accumulation of full-length proRgpB in the periplasm and for purification by affinity chromatography on Ni2+-Sepharose. The proRgp variants were then subjected to concentration- (Fig. 6A) and time-dependent (Fig. 6B) digestion with fXa. Interestingly, while the proRgpB from RgpB662iXa6H was susceptible to proteolysis by fXa, proRgpB from RgpB665sXa6H was completely resistant even after 72 h incubation at a ratio of 10:1 with fXa (Fig. 6B, lower panel). Further, although most of proRgpB from RgpB662iXa6H was cleaved very rapidly (at a substrate:fXa ratio of 10:1), the remaining protein resisted proteolysis, regardless of incubation time. This suggests that proRgpB from RgpB662iXa6H occurs in two different forms: one resistant to proteolysis by fXa and the other susceptible. Cumulatively, our findings suggest that the rate of proteolytic cleavage of the IgSF-CTD linker region may influence the process of glucan attachment to the translocating RgpB with rapid cleavage resulting in the non-glycosylated form of the protein.
Figure 6. Probing of full-length proRgpB variants expressed in the native organism bearing factor Xa cleavage motif (IEGRAA) in the junction between the IgSF and CTD domains.

(A) Concentration-dependent and (B) time-dependent limited proteolysis with factor Xa. (A) 1.67 μM of proRgpB with insertion (RgpB662iXa6H) or substitution (RgpB665sXa6H) of residues in the linker region with Xa cleavage motif were incubated either alone or with fXa at the molar ratio 50:1, 100:1 and 1,000:1 at 37 °C for 3 h (upper panels) or 15 h (lower panels). The proteolysis was assessed by SDS-PAGE. Ctrl, control of proRgpB variant incubated alone. (B) ProRgpB variants were incubated at 37 °C with fXa at 10:1 ratio for up to 72 h. At indicated time intervals, samples were withdrawn and analyzed by SDS-PAGE for proteolytic degradation.
Confirmation of CTD dimerization phenomenon
To confirm that the rCTD subjected to crystallization was indeed a stable dimer apparently formed by strand swapping, we exposed freshly extracted, serially diluted rCTD to chemical crosslinker, glutaraldehyde, followed by SDS-PAGE analysis for the presence of the dimer. This method is commonly used to assess protein oligomeric status38,39. As shown in Fig. 7A, regardless of the dilution, even at as low protein concentration as 0.1 mg ml−1, the clear presence of dimer was observed after cross-linking. To exclude the possibility that the observed dimer of rCTD after glutaraldehyde treatment was an artifact, rCTD and r664i6H at the same concentration (0.1 mg ml−1) were exposed to different glutaraldehyde concentrations (0.1, 0.05, 0.01%). Dimer formation was observed only in the rCTD sample (data not shown). Keeping in mind that cross-linking is never 100% efficient and the fact that only one form of rCTD was detected through gel filtration as represented by a single, symmetrical peak at 10.2 kDa (Fig. 7B), we interpret these data to be consistent with the rCTD existing predominantly as a dimeric form (theoretical molecular mass of 15.8 kDa) in solution.
Figure 7. Soluble rCTD is a dimer in equilibrium and the CTD cleaved off natively expressed proRgpB spontaneously dimerizes.

(A) rCTD at 1, 0.5 and 0.1 mg ml−1 was treated with glutaraldehyde and analysed by SDS-PAGE. (B) Recombinant CTD (rCTD) (red), proRgpB662iXa (black), and proRgpB662iXa preincubated with fXa (blue) were subjected to size exclusion chromatography on a Superdex 75 10/300 GL column equilibrated with 50 mM Tris, 150 mM NaCl, 2.5 mM CaCl2, 0.02% NaN3 pH 7.5 (C) Indicated fractions of resolved proteins were analysed by Western blot using anti-rCTD antibodies to reveal the CTD content in each analysed fraction.
Considering the structural similarities in the swapped area in the homodimer, formation of heterodimers with the IgSF domain, or even with another Ig-like domain, is conceivable, as this occurs in several other proteins with Ig-like folds35. Therefore, to explore this energetically affordable domain swapping as possible part of the mechanism of recognition and/or translocation of T9SS cargo proteins, we investigated propensity of CTD released from proRgpB produced in the native organism to dimerize upon cleavage. To this end, full-length, purified proRgpB bearing the factor Xa cleavable motif inserted in the linker between IgSF and CTD (proRgpB662iXa) was incubated with Xa, the digest was resolved on a gel filtration column and selected fractions were analyzed for the presence of the CTD. As shown in Fig. 7, both the elution profile (Fig. 7B) and the WB signal intensity profile (Fig. 7C) of the CTD released from proRgpB662iXa overlap very nicely with the profiles of the rCTD dimer. Moreover, when the CTD released from proRgpB662iXa was crosslinked with glutaraldehyde, the dimer band could also be detected (Fig. 7C). These observations that the CTDs can spontaneously swap their C-terminal β-strand may represent an important feature of the CTD which contributes to the recognition of cargo protein secretion by T9SS.
Discussion
IgSF protein domains are widely distributed in living organisms with a general function related to protein-protein interactions, such as protein binding or molecular recognition processes40. Here, we show for the first time that the OM translocation signal, a conserved CTD of RgpB, has a typical Ig-like fold encompassing seven antiparallel β-strands organized in two β-sheets that are packed against each other in a β-sandwich (Fig. 1). Significantly, structure-based alignment of 32 CTDs identified or predicted in P. gingivalis (Fig. S1) shows that the (predicted) β-strands overlap perfectly with the conservative motifs identified through the structure of RgpB CTD: motif A is composed of strands 1 and 2, motif B contains strands 3 and 4, and motifs C,D, and E encompass strands s5, s6, and s7, respectively13,14. In β-strands, hydrophobic and hydrophilic residues occur in an alternating pattern, with hydrophobic side chains pointing towards the interior of the molecule. This pattern, especially in strands s2, s3, s5, and s6, is strongly conserved (Fig. S1). The structurally equivalent buried hydrophobic residues of these strands form a ring of hydrophobic interactions in the core of the domain and are engaged in folding of IgSF domains41. In addition, similar conserved motifs are also present in CTDs derived from other proteins secreted via T9SS by disparate Gram-negative bacteria: T. forsythia, Prevotella intermedia, and Parabacteroides distasonis19. This strongly implies that in addition to P. gingivalis, the CTD domains of other Bacteroidetes T9SS cargo proteins also share the common Ig-like fold despite having a negligible degree of identity with respect to the primary structure.
The C-terminus of the CTD has been reported to contain the export signal that targets the cargo protein for OM export through the T9SS system14. Indeed, fusing the last 22 residues of CTD from HPB35 – which overlap perfectly with the last two β-strands of the CTD – to a green fluorescent protein (GFP), resulted in its secretion and glycosylation in P. gingivalis15. Of note, this C-terminal export signal is the same region of the protein that strand swapping occurs in our rCTD dimer. Collectively, the data suggest that the hairpin motifs formed by the two highly conserved β-strands in the CTD C-terminus comprise the primary T9SS recognition signal.
In RgpB, the IgSF domain directly follows the catalytic caspase-like catalytic domain. The IgSF structure in the mature RgpB33, in complex with the N-terminal prodomain of RgpB34, and in the present report adopts a nearly identical conformation. Taking as a reference the IgSF domain (579–661) from the latter structure, RMSDs of 0.456 Å (mature RgpB, 83 aligned residues) and 0.453 Å (proRgpB, 82 aligned residues) was calculated, demonstrating that neither the presence of the preceding catalytic domain nor the succeeding CTD induces any conformational change in the IgSF domain. This is clearly visible by the almost perfect superimposition of IgSF in RgpB and in IgSF-CTD structures (Fig. 8A). Therefore, by superpositioning of the N-terminal prodomain:RgpB complex onto the IgSF-CTD tandem, we constructed a model corresponding to the periplasmic form of RgpB, including the N-terminal prodomain, the catalytic domain, the IgSF and CTD domains, prior to entering the T9SS secretory pathway (Fig. 8B). This structural model suggests that the IgSF domain may act as a spacer or insulator between the catalytic and CTD domains. To date, the IgSF domains in T9SS secreted proteins have been structurally characterized in Kgp42, RgpB33,34, and PPAD43 are found to be similar to one another.
Figure 8. Superimposition of the IgSF domains in the presence of other domains in RgpB, and a model of the full-length latent proRgpB.

(A) The IgSF domain of r664i6H (cyan) superimposes perfectly with the IgSF domain of mature RgpB (PDB id. 1CVR, in red) and RgpB-N-terminal prodomain complex (PDB id. 4IEF, in orange) and shows significant structural conservation with related IgSF sequences from Kgp (PDB id. 4RBM, in green) and PPAD (PDB id. 4YT9, in grey). (B) Surface representation of the chimeric, multi-domain model of the latent full-length proRgpB, transiently present in the periplasm during secretion, including the N-terminal prodomain (green), the catalytic domain (purple), the IgSF domain (cyan) and the CTD (yellow).
In the crystal structure of the rIgSF-CTD tandem of Ig-like folded domains, the interdomain linker was disordered, and was therefore expected to be highly susceptible to proteolysis. Contrary to this expectation, in both the native protein and recombinant proteins modified by insertions or substitutions, the wild-type linker was resistant to proteolysis (Figs 4 and 5). All primary cleavages occurred within the loop connecting β-strands 6 and 7 regardless of the linker modification. From these results, it can be inferred that the linker sequence, although lacking secondary structure by crystallography or SAXS of the recombinant rIgSF-CTD, the junction is impervious to proteolytic attack, suggestive of some kind of structural barrier. SAXS on proRgpB may be more informative in this regard and is currently being explored in our laboratories. The exception was cleavage by factor Xa of the linker engineered to contain an inserted hexapeptide motif (IEGRAA), which is recognized by this very specific protease. In this case, the protein containing the IEGRAA insertion was processed more rapidly than rIgSF-CTD carrying an IEGRAA substitution. Cleavage by fXa likely results from the ability of this protease to interact simultaneously and specifically with several consecutive residues upstream of a hydrolyzed peptide bond37, thus making it amenable to cleavage. It can be expected that linker rearrangement resulting from interaction of the CTD with the secretory machinery facilitates linker cleavage by PorU sortase. Interestingly, however, fXa was unable to release the CTD from the full-length proRgpB in which the IEGRAA motif was substituted for native residues. By contrast, full-length proRgpB in which the motif was inserted into the linker was susceptible to proteolysis by fXa. This indicates that elongation of the IgSF-CTD interdomain linker results in enhanced susceptibility of the linker region to proteolytic processing.
Finally, the observed propensity of the CTD to form dimers by β-strand swapping may also contribute to recognition of this domain by the T9SS apparatus for OM translocation and A-LPS modification of cargo proteins. The fact that the integrity of the last β-strand of the CTD is essential for secretion of RgpB14 lends some credit to this hypothesis, which is also supported by domain/strand swapping events observed previously in Ig-domains closely structurally related to CTD: CD244 and CD445. Furthermore, β-sheet complementation in the Ig-like domains observed in subunit-subunit and chaperone-subunit interactions functions as the mechanism of bacterial pili assembly by the chaperone-usher secretion pathway (T7SS)46. Therefore, it is likely that a somewhat similar mechanism may be involved in the secretion and modification of proteins secreted via T9SS in Bacteroidetes. This possibility is being currently tested in our laboratories.
Methods
Bacterial strains and general growth conditions
Porphyromonas gingivalis wild-type W83 and mutants were grown in enriched tryptic soy broth medium (eTSB; per litre: 30 g trypticase soy broth, 5 g yeast extract, 5 mg haemin, pH 7.5 and supplemented with 0.5 g L-cysteine and 2 mg menadione) or on blood eTSB agar (eTSB medium + 15 g agar per litre and supplemented with 4% defibrinated sheep’s blood) at 37 °C in an anaerobic chamber (Don Whitley Scientific, UK) with an atmosphere of 90% N2, 5% CO2 and 5% H2. E. coli strain DH5α was used for all plasmid construction work and was grown in Luria–Bertani medium and agar. For antibiotic selection in E. coli, ampicillin was used at 100 μg ml−1. For P. gingivalis growth selection on solid media erythromycin was used at 5 μg ml−1and tetracycline at 1 μg ml−1.
Recombinant protein expression and purification
The coding sequence of CTD alone (bases 1987–2208) and IgSF-CTD (bases 1729–2208) of rgpB (PG0506; NCBI Reference Sequence: WP_010956050.1) were amplified from P. gingivalis W83 genomic DNA by PCR using Accuprime Pfx DNA polymerase (Invitrogen, CA, USA) and the following forward primers: RgpBCterBamHIF and RgpBIgBamHIF, and a common reverse primer: RgpBCterXhoIR (Table S3). Amplicons were purified and cloned initially into the pGEX-5T expression vector47 using the BamHI/XhoI sites to give rise to expression plasmids pRCter-B and pRIg-A, respectively. These vectors have a thrombin cleavage site for removal of the GST tag but we have found thrombin to be promiscuous and cleaved the rIgSF-CTD product at multiple sites. Thus, SLIM site-direct mutagenesis was used to replace the thrombin site of pRIg-A to Prescission cleavage site (using primers RThr2PreFA, RThr2PreFB, GThr2PreRA, GThr2PreRB) to give pRIgP-B. Subsequently, the SLIM method was also used to mutate the wild-type construct of IgSF-CTD in pRIgP-B to produce mutant recombinant proteins with 6×His insertion (r662i6H, r664i6H, r665iH) and substitution of linker residues (r665s6H) or insertion of a factor Xa cleavage site (Ile-Glu-Gly-Arg-Ala-Ala) (r662iXa, r664Xa) and substitution of linker residues (r662sXa, r665sXa). The constructs were confirmed by DNA sequencing and they were transformed into E. coli strain BL21 (DE3) (New England Biolabs, Ipswich, MA, USA) under the control of the lac promoter. The resulting recombinant products included an N-terminal GST tag, a PreScission protease cleavage site, followed by the cloned protein.
Transformed E. coli was grown in Luria-Bertani (LB) media at 37 °C to approximately OD600 0.75 and expression of recombinant proteins were induced by the addition of 0.1 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG). After 16 h of cultivation at room temperature, cells were harvested by centrifugation (6000 × g, 15 min, 4 °C) and resuspended in PBS supplemented with 5 mM EDTA and 0.02% NaN3 (15 ml per pellet from 1 L of culture) and subsequently lysed by sonication (cycle of 30 × 0.5 s pulses at amplitude of 70% per pellet from 1 L of culture) using Branson Sonifier Digital 450 (Branson Ultrasonics, Danbury, CT, USA). Cell lysates were clarified by centrifugation (40 000 × g, 40 min, 4 °C), filtered through a 0.45-μm syringe filter, and applied at 4 °C onto a glutathione-Sepharose 4 Fast Flow column (bed volume 5 ml) equilibrated with PBS supplemented with 5 mM EDTA and 0.02% NaN3. After washing, 10 ml of equilibration/washing buffer containing 100 μl of PreScission protease stock solution (1 U/ml) was applied and the column was incubated for 40 h at 4 °C before elution. The final purification was accomplished by size exclusion chromatography on calibrated Superdex S75 column using ÄCTA Purifier System (GE Healthcare Life Sciences). Protein concentration in the final preparation was determined by measurement of absorbance at 280 nm using Nanodrop (NanoDrop Products, Wilmington, DE, USA) and BCA assay (Thermo Scientific Pierce). Purity of obtained protein was verified by SDS-PAGE electrophoresis.
Crystallization and structural analysis
For all aforementioned rIgSF-CTD constructs, crystallization assays were performed using the sitting drop vapor diffusion method, at both 20 °C and 4 °C in steady temperature crystal farms (Bruker). Briefly, reservoir solutions were prepared with a Tecan robot, whereas crystallization nanodrops of 300 nl were dispensed by a Cartesian robot (Genomic Solutions) in 96 × 2-well MRC plates (Innovadyne), at the joint IBMB-IRB High-Throughput Crystallography Platform at Barcelona Science Park. Crystallization hits were improved and scaled up to the microliter range by mixing 1 microliter of protein and reservoir solutions in 24-well Cryschem crystallization dishes (Hampton Research). The rCTD was crystallized using analogous procedures except that crystals were obtained at Małopolska Centre of Biotechnology of the Jagiellonian University.
Suitable crystals for X-ray diffraction were obtained using protein concentrations of 10, 15, 8 and 6 mg/ml for protein variants rCTD, r664i6H, r665sXa and r665i6H, respectively, in 5 mM Tris, 50 mM NaCl, pH 8.0 (for rCTD) and 50 mM Tris, 5 mM EDTA, pH 8.0 (for remaining constructs). The corresponding reservoir solutions were 25% PEG 3350, 0.2 M Li2SO4, 0.1 M Bis-Tris pH 5.5 (4 °C, for rCTD), 30% PEG 4000, 0.2 M Ammonium Sulfate (with 10% PEG 300 used as additive for r664i6H, and without for r665sXa), and 30% PEG 8000, 0.2 M Li2SO4, 0.1 M NaOAc pH 4.5 (for r665i6H). For cryoprotection, reservoir solutions further containing 15% (v/v) (r664i6H) and 20% (v/v) glycerol (r665sXa and r665i6H) were used. No extra cryo-protection was used for the rCTD crystals.
Complete diffraction data sets were collected from liquid N2 flash cryo-cooled crystals at 100 K (provided by an Oxford Cryosystems 700 series cryostream) at beam lines ID29 (r664i6H) and ID23-2 (r665sXa and r665i6H), respectively on an ADSC Quantum Q315r CCD detector and a Dectris PILATUS 6M pixel detector, of the European Synchrotron Radiation Facility (Grenoble, France) within the “Block Allocation Group Barcelona”. rCTD crystals were measured employing a rotating anode copper source (MicroMax-007 HF, Rigaku) attached to a Rigaku R-AXIS IV++ detector at the Małopolska Centre for Biotechnology of the Jagiellonian University in Kraków.
The diffraction data were processed with programs XDS48 (r664i6H), MOSFLM49 (r665sXa, r665i6H and rCTD dimer), and SCALA50 within the CCP4 suite of programs (Collaborative Computational Project, 1994) (Table S1).
The structure of r664i6H was solved by molecular replacement with program PHASER51 using the IgSF domain (residues 348–432) of RgpB from PDB entry 1CVR as searching model. The map obtained was used as input for electron density modification using the ARP/wARP software52. The resulting maps were used to manually complete the model using program COOT53, which alternated with crystallographic refinement (including TLS refinement) with programs PHENIX54, BUSTER55 and REFMAC556.
The structures of r665sXa and rCTD dimer were solved by molecular replacement with PHASER using the r664i6H structure as searching model and were completed using iterative manual building and restrained refinement cycles as aforementioned. Through an equivalent process, we could obtain a similar model for r665i6H mutant, but it was finally not further refined because it proved indistinguishable from r664i6H and r665sXa and had poor refinement statistics.
SAXS data acquisition, analysis and modelling
SAXS data were measured at the Australian Synchrotron on the SAXS/WAXS beamline57 with the data collection parameters presented in Table S2. Data were reduced to I(q) versus q (I(q) = 4πsinθ/λ and 2θ is the angle between the incident and scattered X-rays, λ their wavelength) using the software scatterBrain [ http://www. synchrotron. org.au/aussyncbeamlines/saxswaxs/software-saxswaxs]. Intensities were placed on an absolute scale using the known scattering from H2O. All samples were prepared as described above under Recombinant protein expression and purification. SAXS measurements were performed on two consecutive elution fractions from size exclusion chromatography (representing the front half and back half of the peak profile, termed fractions A and B) after dilution to a starting concentration of 1.5 mg/mL according to BCA assays. SAXS data were collected on a concentration series representing 100%, 80%, 60%, 40%, 20% and 10% of the starting concentration of each of the four samples (wild type rIgSF-CTD and the r664i6H fractions A and B). All dilutions were made in gel filtration buffer.
A solvent scattering blank was measured on the gel filtration buffer alone. The scattering profiles for wild type rIgSF-CTD and the r664i6H variant were obtained by subtraction of the corresponding solvent blank scattering from the samples (protein + solvent) scattering. There was no discernable concentration dependence to the scattering profiles, nor any difference in the highest concentration measurement from fractions A and B from either wild type rIgSF-CTD or the r664i6H variant. These highest concentration measurements were therefore averaged to improve signal to noise and used for all further analysis. Molecular weight (MW) estimates for the proteins were made using the method of Orthaber et. al.58. Values for contrast and partial specific volumes were determined using the program MULCh59. SAXS data analysis and modeling used the ATSAS program package60, with the specific programs used are specified in the Table S2 along with the data ranges used to determine the given structural parameters.
Both constructs were shown to be monodisperse in solution by virtue of the linear log I(q) versus q2 plot (Guinier plot, Fig. S5), good agreement between Rg values determined by Guinier or P(r) analysis (Table S2) and molecular weight estimates that are within 10% of the expected values from the forward scattering intensity (I(0)) and from shape reconstructions (modelled using DAMMIF) (Table S2).
DAMMIF modelling calculations were submitted to ATSAS on-line, with P1 symmetry for 20 independent runs that yielded a set of similar shapes for each construct with NSD values all significantly less than 1 as follows (standard deviations in parentheses): rIgSF-CTD (wild type) 0.689(0.032), r664i6H 0.551(0.045). BUNCH modelling used default parameters with the structures for the IgSF (residues 579–662) and CTD (residues 672–736) domains from the crystal structure (PDB code 5AG8), with no structure assigned to the respective linkers for the wild type (residues 663–671 inclusive) and for the r664i6H variant (residues 663–671 inclusive plus the hexahistidine insert) so that the domains were free to adopt positions that optimised the fit to the scattering data. Ensemble Optimisation Modelling (EOM) used the same domain structures and flexible linkers and was run using the ATSAS on-line server with default parameters.
Plasmid construction for porN-deletion mutant
Two 1-kb flanking regions 5′ and 3′ to the porN gene (The Institute for Genomic Research [TIGR] accession no. PG0291 in P. gingivalis strain W83; PGN1673 in P. gingivalis strain ATCC33277), encoding an essential component of T9SS7, were amplified by PCR with PG266FrANdeIF + PG266FrASmaIR and PG266FrBXbaIF + PG266FrBPstIR primer pairs using Accuprime Pfx DNA polymerase (Invitrogen, CA) for cloning into pUC19 plasmid (New England Biolabs Inc., MA). An erythromycin resistance cassette ermF-ermAM was amplified (using ermFAMXmaIF and ermFAMXbaIR primers) from plasmid pVA219861 and ligated into the modified pUC19 vector above to give the final suicide plasmid, p266AeB-C, for homologous recombination into the W83 genome to give rise to the ΔPorN strain as described below. Correct sequence and orientation of the DNA inserts in the plasmid were confirmed by sequencing.
Plasmid construction for rgpB mutagenesis studies
Creation of suicide plasmids carrying the desired rgpB mutations for homologous recombination into P. gingivalis was performed as previously described14. Initially, the erythromycin-resistance cassette (ermF-ermAM) in the plasmid pURgpB–E constructed for use in RgpB mutagenic studies14 was replaced by a tetracycline-resistance gene tetQ, amplified from plasmid pNFD13-262 using the primer pair tetQXmaIF and tetQSalIR. Using a modified SLIM method of mutagenesis63, a coding sequence for 6×His tag was inserted at the 3′ end of the rgpB gene, to create the master plasmid p6HRgpBt-A. The same methodology was applied to modify the master plasmid with insertion of a coding sequence for factor Xa cleavage site (IEGRAA) at base position 1987 in rgpB gene to create the RgpB662iXa6H mutant or replacement of bases 1993–2010 in rgpB with the coding sequence for IEGRAA to create the RgpB665sXa6H mutant. All primers are listed in Table S3. DNA sequencing of the pertinent region of mutated plasmids confirmed the correct mutation.
Generation of isogenic mutants via homologous recombination
Electroporation of the suicide plasmid constructs into wild-type P. gingivalis W83 allowed for chromosomal integration of the mutated regions into the P. gingivalis genome via a double homologous recombination event as described previously14. Resistant clones were selected using antibiotic selective media. Southern blotting with digoxigenin-labeled ermAM-ermF or tetQ gene probes was used to confirm the presence of only one crossover site in the genome in each mutant (ΔPorN, RgpB665sXa6H, and RgpB662iXa6H, Table 1). Deletion and site-directed mutagenic mutants were further verified by DNA sequencing of the pertinent regions of the genome. The ΔPorN strain was then used to generate double mutant strains (ΔPorNRgpB665sXa6H and ΔPorNRgpB662iXa6H, Table 1) expressing the modified rgpB gene in the background of non-functional T9SS system, which retains unprocessed progingipains in the periplasm7.
Table 1. P. gingivalis strains and mutants used in this study.
| Strain | Relevant genotype | Source |
|---|---|---|
| W83 | Wild-type | Ref. strain |
| ΔPorN | porN(Ermr) | This study |
| RgpB662iXa6H | rgpBp.G662_T663insIEGRAA; K736insHHHHHH(tetQr) | This study |
| RgpB665sXa6H | rgpBp.A666E;D667G;V668R;N670A; K736insHHHHHH(tetQr) | This study |
| ΔPorN/RgpB662iXa6H | porN(Ermr) rgpBp.G662_T663insIEGRAA; K736insHHHHHH(tetQr) | This study |
| ΔPorN/RgpB665sXa6H | porN(Ermr) rgpBp.A666E;D667G;V668R;N670A; K736insHHHHHH(tetQr) | This study |
Purification of His-tagged modified full-length proRgpB
Strains ΔPorNRgpB665sXa6H and ΔPorNRgpB662iXa6H were grown to a stationary phase (OD600 = 1.5) and bacterial cells collected by centrifugation (8,000 × g, 30 min) from 8 L culture. Cells were washed and resuspended in Ni2+-Sepharose binding buffer (20 mM sodium phosphate buffer, 500 mM NaCl, 20 mM imidazole, pH 7.4, supplemented with 0.02% NaN3 and 1.5 mM 4,4-dithiodipyridine (Sigma-Aldrich)) and lysed by sonication. Insoluble cell debris were removed by ultracentrifugation (100,000 × g, 60 min, 4 °C) and clarified supernatant was applied onto a column of pre-equilibrated Ni2+-Sepharose 6 Fast Flow matrix (1.5 ml) (GE Healthcare). After extensive washing, bound proteins were eluted with binding buffer supplemented with 500 mM imidazole. Final purification of proRgpB variants was accomplished by size exclusion chromatography on Superdex 75 column equilibrated with 20 mM Tris, 50 mM NaCl, 5 mM CaCl2, 0.02% NaN3, pH 7.5 using the ÄCTA Purifier system. Fractions containing proRgpB were pooled together, concentrated by ultrafiltration (30 kDa cut-off membrane) and dialyzed against 20 mM BisTris, 150 mm NaCl, 5 mM CaCl2, pH 6.8 with 0.02% NaN3. Protein concentration of the final samples was determined by BCA Assay (Sigma, St Louis, MO, USA).
Protein cross-linking with glutaraldehyde
The proteins: rCTD, r664i6H and CTD released from proRgpB662iXa at concentrations 1, 0.5 and 0.1 mg ml−1 were incubated in PBS with different concentrations of glutaraldehyde ranging form 0.1% to 0.01% for 5 min at 37 °C and then reaction was stopped by addition of Tris to reach final concentration of 100 mM. Next, the samples were subjected to SDS-PAGE and Western blot analysis as described below.
SDS-PAGE and Western blot analysis
Samples were first boiled in non-reducing SDS-PAGE sample buffer containing 2 mM TLCK for 5 min to inactivate all gingipains prior to the addition of 1% β-mercaptoethanol and boiled for a further 5 min for complete denaturation. Samples were centrifuged briefly at 13,000 × g, 1 min to remove particulates and the supernatant separated on SDS-PAGE and gels were stained with Coomassie Brilliant Blue. For Western blot analysis resolved proteins were subsequently electrotransferred onto 0.22-μm-pore-size nitrocellulose membranes and blocked in 2% (w:v) BSA/PBS solution overnight. RgpB was detected using a 1:2000 dilution of anti-RgpB mouse mAb 18E6 in TTBS (20 mM Tris, 500 mM NaCl, pH 7.5 supplemented with 0.1% Tween 20) for 3 h. Membranes were washed four times with TTBS before being probed for 2 h with a 1:2000 dilution of an alkaline phosphatase-conjugated rabbit anti-mouse polyclonal secondary antibody (Dako Cytomation, Denmark). Development was carried out using the AP Conjugate Substrate kit as per manufacturer’s instructions (Bio-Rad Lab., CA, USA).
The rCTD and CTD released from proRgpB662iXa were detected using a 1:13,500 dilution (1 μg ml−1) of anti-CTD rabbit polyclonal antibody in 5% (w/v) skim milk/TTBS solution. Membranes were washed four times with TTBS before being probed for 1 h with a 1:40,000 dilution of an HRP-conjugated goat anti-rabbit polyclonal secondary antibody (Sigma-Aldrich). Development was carried out using the ECL Western blotting substrate kit according to manufacturer’s instructions (Pierce, Thermo Scientific).
Limited proteolysis investigations
Different rIgSF-CTD variants were incubated with tested proteases in 20 mM Tris, 50 mM NaCl, 5 mM CaCl2, pH 7.6 (bovine factor Xa (Sigma-Aldrich, St. Louis, MO, USA)), 25 mM Tris, 1 mM EDTA, 0.02% NaN3, pH 8.0 (Lys-C endopeptidase and V8 protease (Promega, Fitchburg, WI, USA)) or 0.1 M Tris, 150 mM NaCl, 5 mM EDTA, 0.05% Tween 20, 0.02% NaN3, pH 7.5 (porcine pancreatic elastase (Promega)) and human neutrophil elastase (BioCentrum, Krakow, Poland)) for up to 72 h at different weight ratios. Similarly, the full-length proRgpB forms (RgpB665sXa6H and RgpB662iXa6H) were incubated with factor Xa. At specific time points, aliquots were withdrawn for SDS-PAGE analysis and determination of a cleavage site by N-terminal sequence analysis.
N-terminal sequence analysis
Samples were resolved on 10% SDS-PAGE gels (T/C ratio, 33:1) and electrotransferred in 10 mm CAPS, 10% methanol, pH 11 in a Semi-Dry Transfer Cell onto a PVDF membrane. Protein bands were visualized by Coomassie Brilliant Blue R-250 staining, excised, and analyzed by automated Edman degradation using a Procise 494HT amino acid sequencer (Applied Biosystems, Carlsbad, CA, USA).
Miscellaneous
Figures were prepared with PYMOL (The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC.). Structure similarities were determined with the DALI program64. Experimental model validation was performed with MolProbity65. The final coordinates of the rIgSF-CTD mutants r664i6H and r665sXa and of the rCTD dimer are available from the Protein Data Bank (PDB codes 5AG8, 5AG9, and 5HFS respectively).
Additional Information
How to cite this article: de Diego, I. et al. The outer-membrane export signal of Porphyromonas gingivalis type IX secretion system (T9SS) is a conserved C-terminal β-sandwich domain. Sci. Rep. 6, 23123; doi: 10.1038/srep23123 (2016).
Supplementary Material
Acknowledgments
This study was financially supported in part by grants from European, US American, Polish, Spanish, and Catalan agencies (UMO-2012/04/A/NZ1/00051, UMO-2012/05/B/NZ6/00581, UMO-2013/08/W/NZ1/00696, UMO-2011/01/D/NZ1/01169, 2975/7.PR/13/2014/2, NIH NIDCR DE09761; FP7-PEOPLE-2011-ITN-290246 “RAPID”; FP7-HEALTH-2012-306029-2 “TRIGGER”; BFU2012-32862; BIO2013-49320-EXP; MDM-2014-0435; 1306/MOB/IV/2015/0 (“Mobilność Plus” MK) and 2014SGR9). The Department of Structural Biology of IBMB is a “María de Maeztu” Unit of Excellence from the Ministry of Economy and Competitiveness. Funding for data collection was provided in part by ESRF. The Faculty of Biochemistry, Biophysics and Biotechnology of the Jagiellonian University at Kraków (Poland) is a partner of the Leading National Research Center (KNOW) supported by the Polish Ministry of Science and Higher Education. SAXS experiments were performed using the SAXS/WAXS beamline at the Australian Synchrotron with the assistance of Dr. Nigel Kirby.
Footnotes
Author Contributions K.A.N., J.P. and F.X.G. conceived the work; I.D.M. solved the crystal structure; M.K., L.K., P.G., B.S., M.N., Z.N., B.P., J.A.H. and J.G. carried out the protein work, D.M. and K.A.N. were responsible for molecular biology manipulations; I.B.T. identified proteins; A.H.K. and J.T. solved the SAXS structure; I.D.M., M.K., J.J.E., A.H.K., J.T., G.D., F.X.G., K.A.N. and J.P. analysed the data; I.D.M., M.K., J.T., F.X.G., K.A.N. and J.P. wrote the paper.
References
- Desvaux M., Hébraud M., Talon R. & Henderson I. R. Outer membrane translocation: numerical protein secretion nomenclature in question in mycobacteria. Trends Microbiol. 17, 338–340 (2009). [DOI] [PubMed] [Google Scholar]
- Martínez-García P. M., Ramos C. & Rodríguez-Palenzuela P. T346Hunter: A Novel Web-Based Tool for the Prediction of Type III, Type IV and Type VI Secretion Systems in Bacterial Genomes. PLoS One 10: e0119317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysak J. et al. Porphyromonas gingivalis: major periodontopathic pathogen overview. J Immunol Res 2014: 476068 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick R. E., Wijeyewickrema L. C. & Pike R. N. The gingipains: scissors and glue of the periodontal pathogen, Porphyromonas gingivalis. Future Microbiol. 4, 471–487 (2009). [DOI] [PubMed] [Google Scholar]
- Pavloff N. et al. Molecular cloning and characterization of Porphyromonas gingivalis lysine-specific gingipain. A new member of an emerging family of pathogenic bacterial cysteine proteinases. J. Biol. Chem. 272, 1595–1600 (1997). [DOI] [PubMed] [Google Scholar]
- Pavloff N. et al. Molecular cloning and structural characterization of the Arg-gingipain proteinase of Porphyromonas gingivalis. Biosynthesis as a proteinase-adhesin polyprotein. J. Biol. Chem. 270, 1007–1010 (1995). [DOI] [PubMed] [Google Scholar]
- Sato K. et al. A protein secretion system linked to bacteroidete gliding motility and pathogenesis. Proc. Natl. Acad. Sci. USA 107, 276–281 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K. et al. Identification of a new membrane-associated protein that influences transport/maturation of gingipains and adhesins of Porphyromonas gingivalis. J Biol Chem 280, 8668–8677 (2005). [DOI] [PubMed] [Google Scholar]
- Saiki K. & Konishi K. Identification of a Porphyromonas gingivalis novel protein sov required for the secretion of gingipains. Microbiol. Immunol. 51, 483–491 (2007). [DOI] [PubMed] [Google Scholar]
- Saiki K. & Konishi K. Identification of a novel Porphyromonas gingivalis outer membrane protein, PG534, required for the production of active gingipains. FEMS Microbiol. Lett. 310, 168–174 (2010). [DOI] [PubMed] [Google Scholar]
- Saiki K. & Konishi K. Porphyromonas gingivalis C-terminal signal peptidase PG0026 and HagA interact with outer membrane protein PG27/LptO. Mol Oral Microbiol 29, 32–44 (2014). [DOI] [PubMed] [Google Scholar]
- Ishiguro I., Saiki K. & Konishi K. PG27 is a novel membrane protein essential for a Porphyromonas gingivalis protease secretion system. FEMS Microbiol. Lett. 292, 261–267 (2009). [DOI] [PubMed] [Google Scholar]
- Seers C. A. et al. The RgpB C-terminal domain has a role in attachment of RgpB to the outer membrane and belongs to a novel C-terminal-domain family found in Porphyromonas gingivalis. J. Bacteriol. 188, 6376–6386 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen K. A., Travis J. & Potempa J. Does the importance of the C-terminal residues in the maturation of RgpB from Porphyromonas gingivalis reveal a novel mechanism for protein export in a subgroup of Gram-Negative bacteria? J Bacteriol 189, 833–843 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji M. et al. Por secretion system-dependent secretion and glycosylation of Porphyromonas gingivalis hemin-binding protein 35. PLoS One 6, e21372 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorasia D. G. et al. Porphyromonas gingivalis Type IX Secretion Substrates Are Cleaved and Modified by a Sortase-Like Mechanism. PLoS Pathog. 11, e1005152 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paramonov N. et al. Structural analysis of a novel anionic polysaccharide from Porphyromonas gingivalis strain W50 related to Arg-gingipain glycans. Mol. Microbiol. 58, 847–863 (2005). [DOI] [PubMed] [Google Scholar]
- Zhou X. Y., Gao J. L., Hunter N., Potempa J. & Nguyen K. A. Sequence-independent processing site of the C-terminal domain (CTD) influences maturation of the RgpB protease from Porphyromonas gingivalis. Mol. Microbiol. 89, 903–917 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veith P. D. et al. Protein substrates of a novel secretion system are numerous in the Bacteroidetes phylum and have in common a cleavable C-terminal secretion signal, extensive post-translational modification, and cell-surface attachment. J. Proteome Res. 12, 4449–4461 (2013). [DOI] [PubMed] [Google Scholar]
- Chen Y. Y. et al. The outer membrane protein LptO is essential for the O-deacylation of LPS and the co-ordinated secretion and attachment of A-LPS and CTD proteins in Porphyromonas gingivalis. Mol. Microbiol. 79, 1380–1401 (2011). [DOI] [PubMed] [Google Scholar]
- Tomek M. B. et al. The S-layer proteins of Tannerella forsythia are secreted via a type IX secretion system that is decoupled from protein O-glycosylation. Mol. Oral Microbiol. 29, 307–320 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita Y. et al. Lack of a surface layer in Tannerella forsythia mutants deficient in the type IX secretion system. Microbiology 160, 2295–2303 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek M. et al. KLIKK proteases of Tannerella forsythia: putative virulence factors with a unique domain structure. Front. Microbiol. 6, 312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich V. et al. Outer membrane vesicles of Tannerella forsythia: biogenesis, composition, and virulence. Mol. Oral Microbiol. 30, 451–473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veith P. D. et al. Tannerella forsythia outer membrane vesicles are enriched with substrates of the type IX secretion system and TonB-dependent receptors. J. Proteome Res. 14, 5355–5366 (2015). [DOI] [PubMed] [Google Scholar]
- Socransky S. S., Haffajee A. D., Cugini M. A., Smith C. & Kent R. L. Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25, 134–144 (1998). [DOI] [PubMed] [Google Scholar]
- McBride M. J. & Zhu Y. Gliding motility and Por secretion system genes are widespread among members of the phylum bacteroidetes. J. Bacteriol. 195, 270–278 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava A., Johnston J. J., van Baaren J. M. & McBride M. J. Flavobacterium johnsoniae GldK, GldL, GldM, and SprA are required for secretion of the cell surface gliding motility adhesins SprB and RemA. J. Bacteriol. 195, 3201–3212 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes R. G., Samarasam M. N., Van Groll E. J. & McBride M. J. Mutations in Flavobacterium johnsoniae sprE result in defects in gliding motility and protein secretion. J. Bacteriol. 193, 5322–5327 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y. & McBride M. J. Deletion of the Cytophaga hutchinsonii type IX secretion system gene sprP results in defects in gliding motility and cellulose utilization. Appl. Microbiol. Biotechnol. 98, 763–775 (2014). [DOI] [PubMed] [Google Scholar]
- Kharade S. S. & McBride M. J. Flavobacterium johnsoniae chitinase ChiA is required for chitin utilization and is secreted by the type IX secretion system. J. Bacteriol. 196, 961–970 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharade S. S. & McBride M. J. Flavobacterium johnsoniae PorV is required for secretion of a subset of proteins targeted to the type IX secretion system. J. Bacteriol. 197, 147–158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichinger A. et al. Crystal structure of gingipain R: an Arg-specific bacterial cysteine proteinase with a caspase-like fold. EMBO J. 18, 5453–5462 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Diego I. et al. Porphyromonas gingivalis virulence factor gingipain RgpB shows a unique zymogenic mechanism for cysteine peptidases. J. Biol. Chem. 288, 14287–14296 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remaut H. & Waksman G. Protein-protein interaction through beta-strand addition. Trends Biochem. Sci. 31, 436–444 (2006). [DOI] [PubMed] [Google Scholar]
- Fontana A. et al. Probing protein structure by limited proteolysis. Acta Biochim. Pol. 51, 299–321 (2004). [PubMed] [Google Scholar]
- Schilling O., Auf dem Keller U. & Overall C. M. Factor Xa subsite mapping by proteome-derived peptide libraries improved using WebPICS, a resource for proteomic identification of cleavage sites. Biol. Chem. 392, 1031–1037 (2011). [DOI] [PubMed] [Google Scholar]
- Migneault I., Dartiguenave C., Bertrand M. J. & Waldron K. C. Glutaraldehyde: behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. Biotechniques. 37, 790–802 (2004). [DOI] [PubMed] [Google Scholar]
- Friedman P. N., Chen X., Bargonetti J. & Prives C. The p53 protein is an unusually shaped tetramer that binds directly to DNA. Proc Natl Acad Sci USA 90, 3319–23 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodelón G., Palomino C. & Fernández L. Á. Immunoglobulin domains in Escherichia coli and other enterobacteria: from pathogenesis to applications in antibody technologies. FEMS Microbiol. Rev, 37, 204–250 (2013). [DOI] [PubMed] [Google Scholar]
- Lappalainen I., Hurley M. G. & Clarke J. Plasticity within the obligatory folding nucleus of an immunoglobulin-like domain. J. Mol. Biol. 375, 547–559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Diego I. et al. Structure and mechanism of cysteine peptidase gingipain K (Kgp), a major virulence factor of Porphyromonas gingivalis in periodontitis. J. Biol. Chem. 289, 32291–32302 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulas T. et al. Structure and mechanism of a bacterial host-protein citrullinating virulence factor, Porphyromonas gingivalis peptidylarginine deiminase. Sci. Rep. 5, 11969 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones E. Y., Davis S. J., Williams A. F., Harlos K. & Stuart D. I. Crystal structure at 2.8 Å resolution of a soluble form of the cell adhesion molecule CD2. Nature 360, 232–239 (1992). [DOI] [PubMed] [Google Scholar]
- Brady R. L. et al. Crystal structure of domains 3 and 4 of rat CD4: relation to the NH2-terminal domains. Science 260, 979–983 (1993). [DOI] [PubMed] [Google Scholar]
- Kline K. A., Dodson K. W., Caparon M. G. & Hultgren S. J. A tale of two pili: assembly and function of pili in bacteria. Trends Microbiol. 18, 224–232 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold H. et al. Plasmid pGEX-5T: an alternative system for expression and purification of recombinant proteins. Biotechnol. Lett. 14, 245–250 (1992). [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye T. G., Kontogiannis L., Johnson O., Powell H. R. & Leslie A. G. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 67, 282–292 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrakis A., Harkiolaki M., Wilson K. S. & Lamzin V. S. ARP/wARP and molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 57, 1445–1450 (2001). [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W. G. & Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart O. S. et al. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D Biol. Crystallogr. 68, 368–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby N. M. et al. A low-background-intensity focusing small-angle X-ray scattering undulator beamline. J. Appl. Cryst. 46, 1670–1680 (2013). [Google Scholar]
- Orthaber D., Bergmann A. & Glatter O. SAXS experiments on absolute scale with Kratky systems using water as a secondary standard. J. Appl. Cryst. 33, 218–225 (2000). [Google Scholar]
- Whitten A. E., Cai S. & Trewhella J. MULCh: modules for the analysis of small-angle neutron contrast variation data from biomolecular assemblies. J. Appl. Cryst. 41, 222–226 (2008). [Google Scholar]
- Petoukhov M. V. et al. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Cryst. 45, 342–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher H. M. et al. Virulence of a Porphyromonas gingivalis W83 mutant defective in the prtH gene. Infect. Immun. 63, 1521–1528 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolich M. P., Shoemaker N. B. & Salyers A. A. A Bacteroides tetracycline resistance gene represents a new class of ribosome protection tetracycline resistance. Antimicrob. Agents Chemother. 36, 1005–1012 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu J., March P. E., Lee R. & Tillett D. Site-directed, Ligase-Independent Mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 32, e174 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L. & Sander C. Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 20, 478–480 (1995). [DOI] [PubMed] [Google Scholar]
- Chen V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E. & Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.