

Abstract
The known angiotensin II (AngII) physiological effect of aldosterone synthesis and secretion induction, a steroid hormone that contributes to the pathology of postmyocardial infarction (MI) heart failure (HF), is mediated by both Gq/11 proteins and β‐arrestins, both of which couple to the AngII type 1 receptors (AT1Rs) of adrenocortical zona glomerulosa (AZG) cells. Over the past several years, AngII analogs with increased selectivity (“bias”) toward β‐arrestin‐dependent signaling at the AT1R have been designed and described, starting with SII, the gold‐standard β‐arrestin‐”biased” AngII analog. In this study, we examined the relative potencies of an extensive series of AngII peptide analogs at relative activation of G proteins versus β‐arrestins by the AT1R. The major structural difference of these peptides from SII was their varied substitutions at position 5, rather than position 4 of native AngII. Three of them were found biased for β‐arrestin activation and extremely potent at stimulating aldosterone secretion in AZG cells in vitro, much more potent than SII in that regard. Finally, the most potent of these three ([Sar1, Cys(Et)5, Leu8]‐AngII, CORET) was further examined in post‐MI rats progressing to HF and overexpressing adrenal β‐arrestin1 in vivo. Consistent with the in vitro studies, CORET was found to exacerbate the post‐MI hyperaldosteronism, and, consequently, cardiac function of the post‐MI animals in vivo. Finally, our data suggest that increasing the size of position 5 of the AngII peptide sequence results in directly proportional increases in AT1R‐dependent β‐arrestin activation. These findings provide important insights for AT1R pharmacology and future AngII‐targeted drug development.
Keywords: Aldosterone, angiotensin II, angiotensin II type 1 receptor, biased ligand, structure–activity relationship (SAR), β‐arrestin; post‐MI HF
Abbreviations
- AngII
angiotensin II
- ARB
angiotensin receptor blocker
- AT1R
angiotensin II type 1 receptor
- AZG
adrenocortical zona glomerulosa
- GPCR
G protein‐coupled receptor
- GRK
GPCR‐kinase
- HF
heart failure
- MI
myocardial infarction
- SAR
structure–activity relationship
- StAR
steroidogenic acute regulatory protein
Introduction
Aldosterone is a cardio‐toxic mineralocorticoid hormone, whose circulating levels are significantly elevated in postmyocardial infarction (MI) chronic heart failure (HF), accelerating disease progression (Weber 2001). Its major site of systemic synthesis and secretion is the adrenocortical zona glomerulosa (AZG) cells, which endogenously express AngII type 1 receptors (AT1Rs) (Rainey et al. 2004). Elevated serum K+ levels or AngII binding to the AZG AT1Rs can trigger aldosterone production and release into the systemic circulation (Ganguly and Davis 1994). The AT1R is primarily a G protein‐coupled receptor (GPCR) stimulating aldosterone turnover through Gq/11 proteins (De Gasparo et al. 2000). However, its activation by AngII can also elicit G protein‐independent signaling through β‐arrestins, scaffolding signal transducers originally discovered as terminators of GPCR signaling (Luttrell and Gesty‐Palmer 2010). We have previously shown that AngII‐bound AT1Rs in AZG cells can stimulate aldosterone production via both G proteins and β‐arrestin1, and the latter leads to post‐MI HF‐related hyperaldosteronism (Lymperopoulos et al. 2009, 2011). In fact, β ‐arrestin1 is absolutely crucial for the development of post‐MI hyperaldosteronism, since the circulating aldosterone levels of β‐arrestin1 knockout mice post‐MI appear normal (Bathgate‐Siryk et al. 2014). What's more, this adrenal β‐arrestin1‐dependent signaling pathway to aldosterone biosynthesis may underlie the “aldosterone breakthrough” phenomenon observed with the clinical use of angiotensin receptor blockers (ARBs), that is AT1R‐selective antagonist drugs (Bomback and Klemmer 2007; Lymperopoulos et al. 2014; Dabul et al. 2015).
Although adrenal β ‐arrestin1 activated by the AT1R exerts the cardio‐toxic effect of hyperaldosteronism, in other tissues, such as the heart itself, it may exert beneficial effects (Lymperopoulos and Bathgate 2013). Therefore, in an effort to come up with better cardiovascular therapeutics, several AngII analog peptides have been developed and characterized over the past few years that presumably only activate β‐arrestins at the AT1R while leaving G proteins unaffected (Violin et al. 2013). This was spurred by the discovery of SII ([Sar1, Ile4, Ile8]‐AngII) as being such a β‐arrestin‐”biased” analog more than a decade ago (Holloway et al. 2002). At present, several other, much more potent than SII, β‐arrestin‐”biased” analogs have been produced and described, with one of them, TRV120027 ([Sar1, D‐Ala8]‐AngII), currently under evaluation in early phase clinical trials for acute HF (Violin et al. 2010; Soergel et al. 2013). In this study, we synthesized and tested a series of such β‐arrestin‐”biased” AngII peptide analogs by using the sequence of SII as a template. Thus, compared to the natural octapeptide AngII, we kept the presence of sarcosine (N‐methyl‐glycine) (instead of aspartate) at position 1 and we also substituted the phenylalanine at position 8 with leucine in all of these compounds. However, we chose to replace the isoleucine of position 5 with varied substitutions, instead of replacing the tyrosine of position 4 (as in SII). Based on in vitro assays, one of the analogs turned out to be particularly potent at stimulating β‐arrestin‐dependent aldosterone production and was further evaluated in vivo in post‐MI animals progressing to HF. We were also able to make some inferences on the effect of the substitution of Ile5 of the AngII peptide sequence on AT1R‐ β‐arrestin coupling by comparing the in vitro potencies of the various analogs we synthesized.
Materials and Methods
Materials
All peptides were custom‐made by Pierce Biotechnology (Rockford, IL), except for AngII, SII, CORSML, CORML, and CORET, which were made in Dr. Cordopatis's lab (Univ. of Patras, Patras, Greece). All the peptides used in this study were sequence‐verified via mass spectrometry and purified to >95% purity via HPLC.
DiscoveRx assay
The PathHunter™ β‐Arrestin® assay monitors the activation of a GPCR in a homogenous, nonimaging assay format using a proprietary complementation technology (Fig. S1), developed by DiscoveRx (Fremont, CA) (McGuinness et al. 2009; Dabul et al. 2015). Briefly, the assay utilizes an enzyme fragment complementation reaction with β‐galactosidase (β‐Gal) as the functional reporter. The enzyme is split into two complementary portions expressed as fusion proteins in the cell. The enzyme acceptor (EA) is fused to β‐arrestin and the ProLink donor peptide is fused to the GPCR of interest, in this case, human AT1R overexpressed in Chinese Hamster Ovary (CHO) cells. Upon GPCR stimulation, β‐arrestin is recruited to the receptor for desensitization, bringing the two fragments of β‐Gal together and allowing complementation to occur. This generates an active enzyme that can convert a chemiluminescent substrate to generate an output signal detectable on a standard microplate reader.
CellKey assay
This assay utilizes cellular dielectric spectroscopy (CDS) to detect a range of whole‐cell responses in a label‐free manner. CellKey is a CDS‐based instrument that detects GPCR activation in an automated microplate format (Peters et al. 2007; Dabul et al. 2015). Agonist and antagonist activity can be accurately quantified under conditions of low receptor expression. In this study, human AT1R‐overexpressing human embryonic kidney (HEK)293 cells were used in the CellKey assay system (see also Fig. S2A–B). Briefly, cells were incubated with the peptide of interest for 15 min at room temperature and the change in impedance was recorded to measure degree of AT1R activation.
H295R cell culture and transfections
The human AZG cell line H295R was purchased from American Type Culture Collection (Manassas, VA) and cultured as previously described (Lymperopoulos et al. 2009). Transfection was performed with adenovirus encoding for full length, wild‐type rat β‐arrestin1, as previously described (Lymperopoulos et al. 2009). The construction of this adenovirus has also been described previously (Lymperopoulos et al. 2009).
Plasma and in vitro aldosterone secretion measurements
In vitro aldosterone secretion in the culture medium of H295R cells as well as rat plasma aldosterone levels were determined by EIA (Aldosterone EIA kit, ALPCO Diagnostics, Salem, NH), as described (Lymperopoulos et al. 2009).
Western blotting
Western blots to assess protein levels of StAR (sc‐25806, SantaCruz Biotechnology, Santa Cruz, CA), β ‐arrestin1/2 (sc‐28869, SantaCruz), and GAPDH (MAB374; Chemicon, Temecula, CA) were done using protein extracts from treated H295R cells, as described previously (Lymperopoulos et al. 2009). Immunoblots were revealed by enhanced chemiluminescence (ECL, Life Technologies, Grand Island, NY) and visualized in the FluorChem E Digital Darkroom (Protein Simple, San Jose, CA) (Salazar et al. 2013). Densitometry was performed with the AlphaView software (Protein Simple, San Jose, CA, USA) in the linear range of signal detection (on nonsaturated bands).
Experimental animals and surgical procedures
The animals in this study were handled according to animal welfare regulations and protocols approved by the authors’ Institutional Review (IACUC) Boards and complied with the ARRIVE guidelines. MI was performed in adult male Sprague–Dawley rats using the cryo‐infarct method, as previously described (Lymperopoulos et al. 2011). Adrenal‐specific in vivo gene delivery, at 2 weeks post‐MI, was done essentially as described (Lymperopoulos et al. 2011), via direct delivery of adenovirus encoding full length, wild‐type rat β‐arrestin1 in both adrenal glands (Lymperopoulos et al. 2008). The CORET peptide (100 μg/kg i.p., dose calculated based on EC50 for aldosterone secretion in vitro) or saline was administered daily for seven consecutive days, starting on the day of the adenoviral adrenal‐specific gene delivery (2 weeks post‐MI). Overexpression of adrenal β‐arrestin1 was confirmed via Western blotting and was comparable among animals of the same group (data not shown). All animals were fed exactly the same (low salt) diet throughout the study.
Echocardiography and in vivo hemodynamics
Transthoracic echocardiography was performed with a linear 30‐MHz transducer (VeVo 770 High Resolution Imaging System, VisualSonics, Toronto, ON, Canada), as described previously (Lymperopoulos et al. 2011, 2014). Three independent echocardiographic measurements were taken in both modes. In vivo hemodynamic analysis was also performed as previously described (Lymperopoulos et al. 2011), with a 1.4‐Fr pressure‐conductance catheter (Millar Instruments, Houston, TX) to record cardiac hemodynamics and a polyethylene‐50 catheter inserted into the right external jugular vein for isoproterenol infusions (maximal dose of isoproterenol: 333 ng/kg of body weight).
Statistical analyses
Data are generally expressed as mean + SEM. Unpaired two‐tailed Student's t test, chi‐square test, and one‐ or two‐way ANOVA with Bonferroni or Tukey's tests were generally performed for statistical comparisons, unless otherwise indicated. For multigroup statistical comparisons, Tukey's multiple comparison test using SAS version 8.2 software (Cary, NC, USA) was used (unless otherwise indicated). For all tests, a P value of <0.05 was considered to be significant.
Results
CORET potently stimulates β‐arrestin coupling to the AT1R
In a first set of experiments, we synthesized and tested three AngII peptide analogs: CORML: [Met5, Leu8]‐AngII, CORSML: [Sar1, Met5, Leu8]‐AngII, and CORET: [Sar1, Cys(Et)5, Leu8]‐AngII, and tested them in the PathHunter (DiscoveRx) and in the CellKey assay systems, that is, in CHO and HEK293 cells, respectively, overexpressing the human AT1R (see Figs S1 and 2 for more details). CORET was found particularly potent at inducing β‐arrestin coupling to the AT1R, only ~4 times less potent than the natural agonist AngII in this particular assay (Fig. 1, see also Table 1). Importantly, all three analogs were much more (~20–fold) potent than SII, the prototypic β‐arrestin‐”biased” AngII analog peptide, at stimulating AT1R‐ β‐arrestin coupling in this assay system (Fig. 1B, see also Table 1). Finally, as shown in Figure 1C, all three compounds, plus the analog [Sar1, Cys5, Leu8]‐AngII (“Cys”), were less potent than AngII but more potent than SII at β‐arrestin stimulation in the CellKey assay system (see also Table 1). As far as efficacy is concerned, all compounds in the CellKey assay showed less maximal effect than AngII but higher than SII (~75% of AngII maximal effect on average vs. only ~40% for SII, Fig. 1C and Table 1). Finally, none of our analogs displayed inverse agonist or allosteric modulator properties at the AT1R (data not shown).
Figure 1.
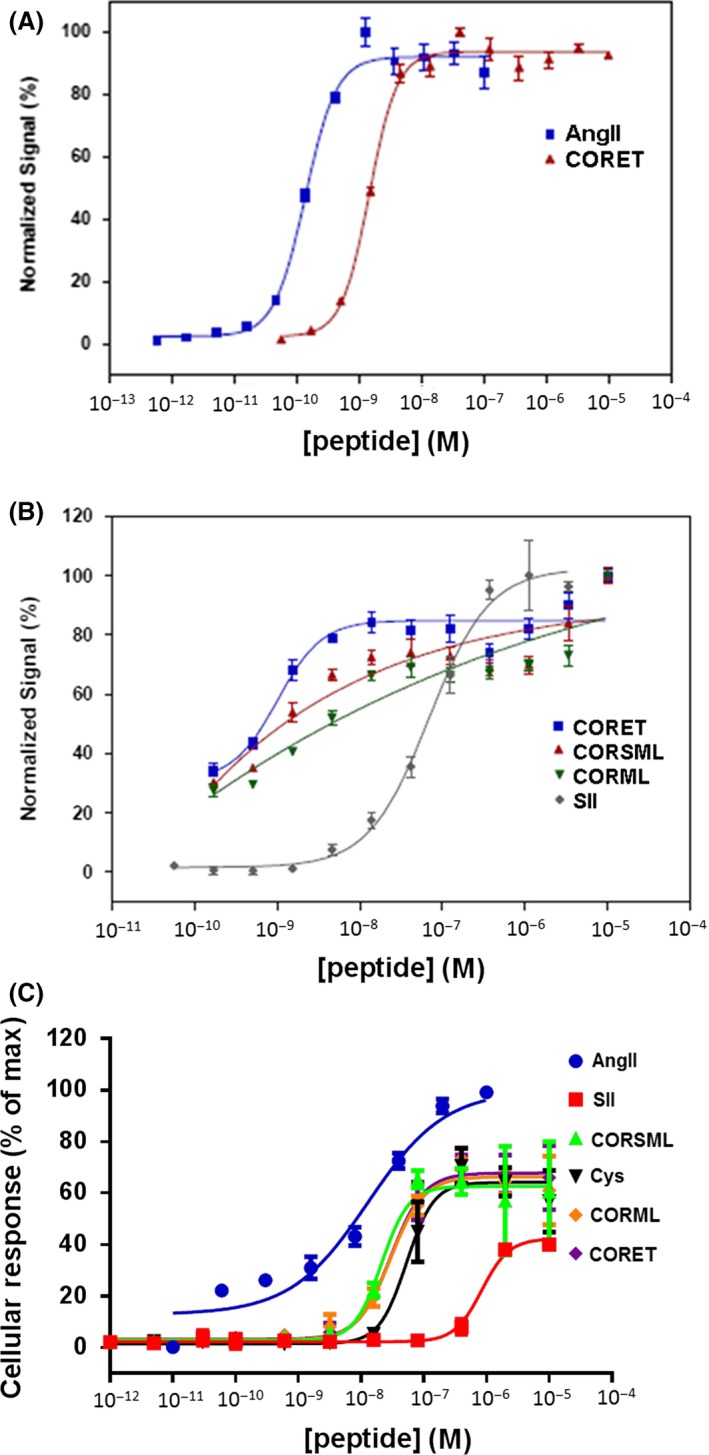
Comparison of potencies of CRSML, CORML, and CORET at stimulating AT1R‐ β‐arrestin coupling. (A) Comparison of CORET versus AngII (DiscoveRx assay). (B) Comparison of CORET, CORSML, and CORML versus SII (DiscoveRx assay). (C) Concentration–response curves for AT1R‐β‐arrestin binding stimulation, as derived from the CellKey assay (Cys: Sar1, Cys5, Leu8‐AngII). n = 5 independent determinations for each peptide in both assays (two‐way ANOVA with Bonferroni test).
Table 1.
Maximal effect (Max. eff., as % of 1 nmol/L AngII ± SEM) and half‐maximal effective concentration (EC50 ± SEM) values for AT1R‐elicited β‐arrestin activation in vitro calculated by the CellKey assay system; for AT1R‐elicited β‐arrestin activation in vitro calculated by the PathHunter assay system; and for AT1R‐elicited β‐arrestin‐dependent aldosterone secretion in H295R cells
| AngII | SII | CORML | CORSML | CORET | PRO | BUT | PEN | |
|---|---|---|---|---|---|---|---|---|
| EC50 (nmol/L) (CellKey) | 0.1 ± 0.06 | 10 ± 5.2 | 0.6 ± 3.2 | 0.5 ± 2.2 | 0.4 ± 2.3 | 6 ± 4.3 | 3 ± 2.3 | 1 ± 3.3 |
| Max. eff. (%) (CellKey) | 100 | 40 ± 6 | 75 ± 10 | 77 ± 11 | 81 ± 4 | 86 ± 6 | 85 ± 6 | 80 ± 5 |
| EC50 (nmol/L) (PathHunter) | 0.1 ± 0.04 | 11 ± 6.1 | 0.6 ± 4.2 | 0.7 ± 3.7 | 0.5 ± 3.3 | 4 ± 3.0 | 2 ± 4.4 | 0.4 ± 5.3 |
| Max. eff. (%) (PathHunter) | 100 | 42 ± 7 | 73 ± 8 | 76 ± 7 | 82 ± 5 | 83 ± 6 | 83 ± 7 | 80 ± 7 |
| EC50 (nmol/L) (Aldosterone) | 6 ± 4.5 | 103± 98.3 | 101 ± 11.3 | 95 ± 12.5 | 55 ± 21.2 | N.D. | N.D. | N.D. |
| Max. eff. (%) (Aldosterone) | 100 | 35 ± 6 | 55 ± 9 | 57 ± 8 | 80 ± 6 | N.D. | N.D. | N.D. |
n = 5 independent determinations per treatment (two‐way ANOVA with Tukey's multiple comparison test).
N.D, Not Determined.
CORET potently induces aldosterone turnover in AZG cells in vitro
Next, we examined what the above findings mean for the physiological effect of AT1R‐induced, β‐arrestin‐dependent aldosterone production (Fig. 2A). β‐arrestin1 has been found, similarly to G protein‐dependent signaling by the activated AT1R in AZG cells, to stimulate aldosterone synthesis and secretion by upregulating Steroidogenic Acute Regulatory protein (StAR), the enzyme that catalyzes the rate‐limiting step of this biosynthetic process (mitochondrial cholesterol uptake) (Fig. 2A) (Rainey et al. 2004; Lymperopoulos et al. 2009). Thus, using the human AZG cell line H295R, transfected to overexpress β‐arrestin1 (Lymperopoulos et al. 2009), and in vitro aldosterone secretion as the readout, we found again that all three peptides (CORML, CORSML, CORET) are more potent and efficacious aldosterone secretion inducers than SII in vitro, with CORET being the most potent and efficient out of the three (Table 1 and Fig. 2B). Compared to AngII, the peptides were less potent (Table 1), as expected, since AngII stimulates aldosterone secretion via both G proteins and β‐arrestin1, whereas these peptides only via β‐arrestin1. Notably, for the very same reason, no effect on aldosterone secretion was seen with any peptide analog (including SII) when endogenous β‐arrestin1 was blocked via overexpression of a dominant negative mutant (Fig. S3A–B).
Figure 2.
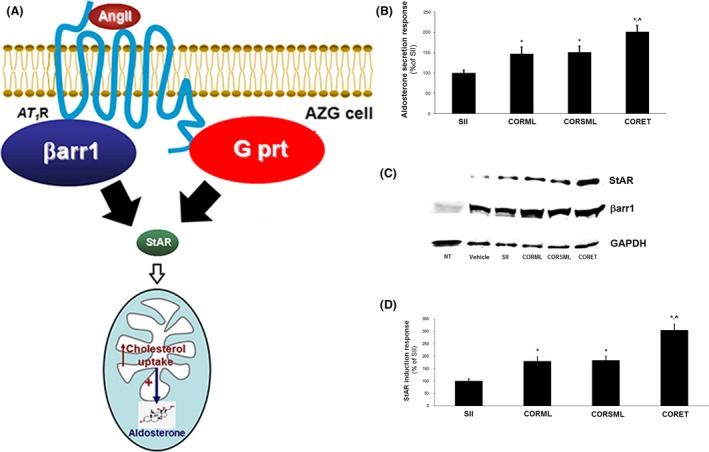
AngII analog peptides and aldosterone turnover in vitro. (A) Schematic representation of the AT1R‐elicited pathways mediated by G proteins (G prt) and β‐arrestin1 (β arr1) converging on aldosterone synthesis and secretion in adrenocortical zona glomerulosa (AZG) cells. (B) In vitro aldosterone secretion from H295R cells overexpressing β‐arrestin1 and stimulated for 4 h with 100 nmol/L CORSML, CORML, or CORET. Data are presented as % of the secretion induced by 10 μmol/L SII. *P < 0.05, versus SII, ^P < 0.05, versus. CORML or CORSML, n = 5 independent experiments/treatment. (C, D) Western blotting for StAR protein levels in the same cells treated as in (B). Representative blots of five independent experiments are shown in (C), including blots from nontransfected (NT) H295R cells for β‐arrestin1 (β arr1) to confirm its overexpression in the transfected cells, and for GAPDH (glyceraldehyde 3‐phosphate dehydrogenase) as loading control, and the StAR protein induction (as % of the SII response), as derived by densitometric quantification, is shown in (D). *P < 0.05, versus SII, ^P < 0.05, versus CORML or CORSML, n = 5 independent experiments/treatment (two‐way ANOVA & chi‐square test with Tukey's multiple comparison test).
Consistent with the findings on aldosterone secretion, all three peptides were again more efficacious stimulators of StAR protein upregulation than SII, with CORET being the most potent of the three again (Fig. 2C‐D), indicating that also synthesis of aldosterone is more elevated with any of the three peptides (and especially with CORET) than it is with SII. β‐arrestin1 overexpression in these cells was confirmed by Western blotting (Fig. 2C) and again, no effects on StAR protein induction were seen when endogenous β‐arrestin1 was blocked (data not shown), as expected.
CORET exacerbates post‐MI hyperaldosteronism and cardiac function in vivo
Since CORET was the most potent AngII analog at stimulating β‐arrestins and β‐arrestin1‐dependent aldosterone production in vitro, and adrenal β‐arrestin1‐dependent hyperaldosteronism plays an important role in experimental post‐MI HF progression (Lymperopoulos et al. 2011), we next sought to examine the in vivo effects of CORET in post‐MI rats progressing to HF and overexpressing β‐arrestin1 specifically in their adrenals (Lymperopoulos et al. 2011). We treated these animals with daily intraperitoneal injections of CORET (or saline) for seven consecutive days starting at 2 weeks post‐MI and at the end of the week‐long treatment period (3 weeks post‐MI), we measured circulating aldosterone levels and cardiac function. Consistent with a potent β‐arrestin stimulation at the adrenal AT1R in vivo, circulating aldosterone levels of the CORET‐treated animals were significantly elevated compared to control, saline‐trated post‐MI animals (Fig. 3A). Importantly, this elevation was on top of the hyperaldosteronism normally present in the animals due to their post‐MI progression to HF (compare with Sham in Fig. 3A), indicating worsening of the post‐MI hyperaldosteronism by the CORET treatment. Finally, this hyperaldosteronism deterioration was reflected on cardiac function of the treated post‐MI animals, as both ejection fraction (Fig. 3B) and isoproterenol (a standard cardiac positive inotrope)‐stimulated contractility (Fig. 3C) were significantly worsened by CORET, compared to control saline‐treated post‐MI animals. Taken together, these results strongly suggest that CORET accelerates post‐MI HF progression by stimulating adrenal β‐arrestin1‐mediated aldosterone elevation.
Figure 3.
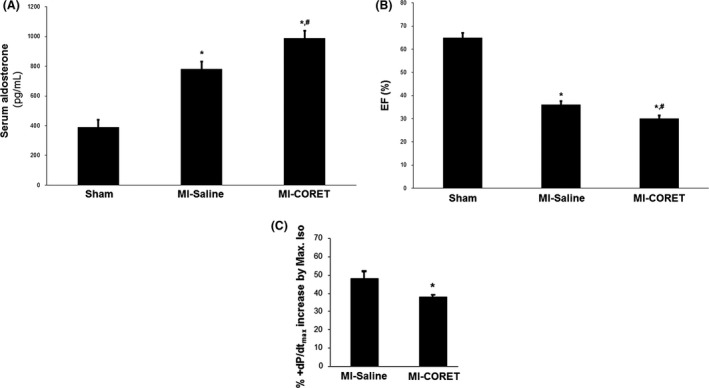
CORET in post‐MI HF in vivo. (A) Circulating aldosterone levels in 3‐week post‐MI rats, overexpressing β‐arrestin1 in their adrenals, and treated for seven consecutive days either with saline (MI‐Saline) or with daily i.p. injections of CORET. Aldosterone levels in age‐matched, sham‐operated, healthy, and untreated (and with normal adrenal β‐arrestin1 expression levels) animals are also included for comparison. *P < 0.05, versus Sham, # P < 0.05, versus MI‐Saline, n = 5 rats/group. (B) Ejection fraction (EF %) of the same rat groups at the end of the 7‐day treatment. *P < 0.05, versus Sham, # P < 0.05, versus MI‐Saline, n = 5 rats/group. (C) Increases in cardiac contractile function in the post‐MI groups at the end of the 7‐day treatment, as inferred by the % increase in +dP/dtmax that a maximal dose of isoproterenol (Max. Iso) induces. *P < 0.05, versus. MI‐Saline, n = 5 rats/group (two‐way ANOVA & chi‐square test with Tukey's multiple comparison test).
AngII Ile5 and AT1R‐ β‐arrestin coupling
Since the most striking structural difference between SII and CORET is substituted Tyr4 versus substituted Ile5 of the AngII sequence (respectively), we posited that maybe this is the reason for the enhanced β‐arrestin agonism at the AT1R displayed by CORET. Thus, we focused on the effect of varying the substitution at this position (Ile5) of the AngII sequence on the potency of β‐arrestin agonism at the AT1R. To this end, we synthesized a series of analogs with Cys5 substitutions of increasing side‐chain size, while keeping the Sar1 and Leu8 substitutions steady (Fig. 4A). We tested all of these compounds in vitro for β‐arrestin agonism potency and efficacy at the human AT1R against the natural (non‐”biased”) agonist AngII and the β‐arrestin‐”biased” SII, via both the DiscoveRx (Fig. 4B) and the CellKey (Fig. 4C) assay systems. As shown in Table 1, increasing the linear size of the Cys5 alkyl side‐chain results in a directly proportional increase in potency (EC50 decrease) for β‐arrestin agonism, while maintaining a high level of efficacy (>80% of the AngII's maximal effect). This trend is preserved even in the presence of a residue as “bulky” as Cys(n‐pentyl) at position 5 of the AngII sequence (Table 1). As far as SII is concerned, consistent with all of our experiments above, it was found much less potent or efficacious than any of the Cys5‐substituted analogs we tested in this study: ~76 times less potent than AngII versus only ~3–4 times less potency than AngII for all of the compounds; only 40% of AngII's maximal effect versus >80% of AngII's maximal effect for all of the compounds (Table 1). These findings suggest a profound effect of AngII's Ile5 aliphatic side‐chain substitution on β‐arrestin activation by the AT1R.
Figure 4.
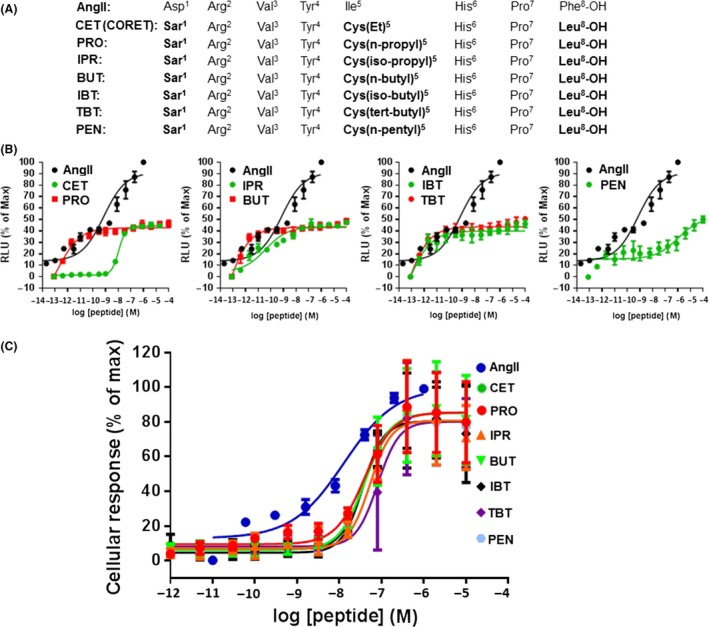
AngII's Ile5 substitution and AT1R‐elicited β‐arrestin activation. (A) The amino acid sequences of all of the peptides studied. The substitutions differing from AngII are highlighted in bold. (B) Concentration‐response curves for stimulation of AT1R‐β‐arrestin binding, based on the DiscoveRx assay. (C) Concentration–response curves for stimulation of AT1R‐β‐arrestin binding based on the CellKey assay. See also Table 1. n = 5 independent determinations for each analog in both assays (two‐way ANOVA with Tukey's multiple comparison test).
Discussion and Conclusions
In this study, we synthesized a series of β‐arrestin‐”biased” AngII Ile5‐substituted peptide analogs and examined their effects on AT1R‐β‐arrestin recruitment, as well as their impact on the AT1R physiological effect of aldosterone production in AZG cells, which is known to be mediated by β‐arrestin1 (Lymperopoulos et al. 2009). We uncovered two main findings: first, one of these peptides, [Sar1, Cys(Et)5, Leu8]‐AngII (CORET), is a potent β‐arrestin agonist at the human AT1R, stimulating aldosterone synthesis and secretion in vitro more robustly than SII, and it also exacerbates post‐MI HF hyperaldosteronism accelerating cardiac performance decline in vivo. Since a large number of AngII β‐arrestin‐”biased” analogs have been developed over the past several years in an effort to come up with safer and more effective cardiovascular therapeutics over the currently available traditional and “unbiased” AT1R ligands (Violin et al. 2013), the possibility of causing excessive aldosterone secretion from the adrenal gland, which can complicate post‐MI HF therapy, has to be taken into account when designing such β‐arrestin‐”biased” AT1R ligands.
The other major finding of this study is that increasing the size of the alkyl side‐chain of Ile5 of the AngII peptide sequence results in proportionally more robust β‐arrestin recruitment to the AT1R. A number of previous studies on structure–activity relationships (SAR) of the AngII molecule with regards to AT1R “bias” toward β‐arrestin versus G protein signaling have indicated that replacement of the aromatic side‐chain of Phe8, absolutely critical for full Gq protein agonism at the AT1R, is necessary to remove the G protein‐activating capacity of the ligand, while maintaining the ability to activate β‐arrestins (Miura et al. 1999; Nikiforovich et al. 2006; Zimmerman et al. 2012; Balakumar and Jagadeesh 2014). Additionally, Asp1 is known to affect only binding (affinity) to the AT1R and not efficacy and, in fact, substitution with Sar1 actually enhances receptor binding (Wilkes et al. 2002). Finally, Tyr4 appears also essential for full Gq protein agonism but does not seem to affect AT1R phosphorylation (Miura et al. 1999; Holloway et al. 2002; Nikiforovich et al. 2006), a crucial prerequisite event for subsequent β‐arrestin binding (Qian et al. 2001). Ile5 does not seem to affect activity toward G proteins but its side‐chain plays a role in maintaining the proper (agonist) conformation of AngII when bound to the AT1R, that is, it contributes to the proper positioning of the two agonist “switches” (Tyr4 & Phe8) of the AngII molecule (Tzakos et al. 2004; Nikiforovich et al. 2006). With regards to the effects on β‐arrestin agonism at the AT1R, virtually nothing is presently known about the Ile5 position; however, it is interesting to note that some of the purported β‐arrestin‐”biased” analogs with beneficial effects in the heart, currently under investigation for therapy of HF and of dilated cardiomyopathy (DCM), have this Ile5 substituted (e.g., to Lys5 in TRV120023), whereas others retain it (e.g., TRV120027) (Violin et al. 2010). Our present data indicate that this position, although not critical for β‐arrestin agonism at the AT1R like the Phe8 position is, can still significantly and positively modulate the potency of the β‐arrestin‐”biased” AT1R ligand. Of course, this awaits confirmation in larger SAR studies and with the crystallization of the full AT1R protein, including the β‐arrestin‐interacting regions. Unfortunately, the recently solved human AT1R crystal structure (Zhang et al. 2015) was missing the full C‐terminal region of the receptor, which the β‐arrestins interact with (Balakumar and Jagadeesh 2014), and thus, could not provide any information in that regard. Of course, the process of the AT1R‐β‐arrestin interaction and its ultimate effects in the cell is very complex and multifactorial, involving, not only the activating ligand, but also GPCR‐kinase (GRK)‐dependent receptor phosphorylation (Qian et al. 2001), which particular β‐arrestin isoform (β‐arrestin1 or ‐2) binds the receptor and the conformational changes the β‐arrestin molecule itself undergoes upon binding to the AT1R (Shukla et al. 2008, 2013), and even the duration of this interaction, that is, the stability of the AT1R‐ β‐arrestin complex in endosomal compartments (Zimmerman et al. 2012). Combined differences in all of these molecular determinants are quite likely to be responsible for the fact that biased AT1R ligand‐activated β‐arrestin‐dependent signaling appears beneficial, at least in the heart, whereas β‐arrestins do not seem capable of providing any functional or structural benefit to the heart when they are activated by the AngII‐bound AT1R. Nevertheless, it becomes clear from the above that simply varying the structure of an AT1R ligand hardly suffices to “bias” the resultant AT1R‐ β‐arrestin complex toward a specific cellular effect or to even predict the effect on AT1R‐elicited signal transduction.
In conclusion, our present findings suggest that adrenal β‐arrestin‐dependent aldosterone production is a potential side‐effect of AT1R β‐arrestin‐”biased” ligands that needs to be taken into account, especially if the ligand is considered a drug candidate for a cardiovascular indication. In addition, we conclude that the substitution of the Ile5 position of AngII, so far largely “overlooked” in β‐arrestin “bias” studies, may play an important role in stimulating the interaction of AT1R with β‐arrestins. Both of these findings may have important implications for AngII pharmacology and for future AT1R‐targeted drug development.
Conflict of Interest
None declared.
Supporting information
Figure S1. Schematic illustration of the DiscoveRx PathHunterTM β‐Arrestin assay.
Figure S2. Schematic illustration of the CellKey assay protocol employed in this study.
Figure S3. (A) In vitro aldosterone secretion from H295R cells overexpressing a dominant negative mutant β‐arrestin1 and stimulated for 4 h with 100 nmol/L CORSML, CORML, or CORET. Data are presented as % of the secretion induced by 10 μmol/L SII. *P < 0.05, versus SII, ^P < 0.05, versus CORML or CORSML, n = 5 independent experiments/treatment. No significant differences were observed among treatments.
Acknowledgments
This work was supported in part by a Scientist Development Grant from the American Heart Association (AHA #09SDG2010138, National Center) and a Nova Southeastern University's President's Faculty Research and Development Grant (PFRDG) (both to A.L.). The authors declare no competing financial or any other interests.
Valero T. R., Sturchler E., Jafferjee M., Rengo G., Magafa V., Cordopatis P., McDonald P., Koch W. J., Lymperopoulos A..Structure–activity relationship study of Angiotensin II analogs in terms of β ‐arrestin‐dependent signaling to aldosterone production, Pharma Res Per, 4(2), 2016, e00226, doi: 10.1002/prp2.226
References
- Balakumar P, Jagadeesh G (2014). Structural determinants for binding, activation, and functional selectivity of the angiotensin AT1 receptor. J Mol Endocrinol 53: R71–R92. [DOI] [PubMed] [Google Scholar]
- Bathgate‐Siryk A, Dabul S, Pandya K, Walklett K, Rengo G, Cannavo A, et al. (2014). Negative impact of β ‐arrestin‐1 on post‐myocardial infarction heart failure via cardiac and adrenal‐dependent neurohormonal mechanisms. Hypertension 63: 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomback AS, Klemmer PJ (2007). The incidence and implications of aldosterone breakthrough. Nat Clin Pract Nephrol 3: 486–492. [DOI] [PubMed] [Google Scholar]
- Dabul S, Bathgate‐Siryk A, Valero TR, Jafferjee M, Sturchler E, McDonald P, et al. (2015). Suppression of adrenal β arrestin1‐dependent aldosterone production by ARBs: head‐to‐head comparison. Sci Rep 5: 8116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T (2000). International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 52: 415–472. [PubMed] [Google Scholar]
- Ganguly A, Davis JS (1994). Role of calcium and other mediators in aldosterone secretion from the adrenal glomerulosa cells. Pharmacol Rev 46: 417–447. [PubMed] [Google Scholar]
- Holloway AC, Qian H, Pipolo L, Ziogas J, Miura S, Karnik S, et al. (2002). Side‐chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol Pharmacol 61: 768–777. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Gesty‐Palmer D (2010). Beyond desensitization: physiological relevance of arrestin‐dependent signaling. Pharmacol Rev 62: 305–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymperopoulos A, Bathgate A (2013). Arrestins in the cardiovascular system. Prog Mol Biol Transl Sci 118: 297–334. [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Zincarelli C, Soltys S, Koch WJ (2008). Modulation of adrenal catecholamine secretion by in vivo gene transfer and manipulation of g protein‐coupled receptor kinase‐2 activity. Mol Ther 16: 302–307. [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Zincarelli C, Kim J, Soltys S, Koch WJ (2009). An adrenal beta‐arrestin 1‐mediated signaling pathway underlies angiotensin II‐induced aldosterone production in vitro and in vivo. Proc Natl Acad Sci USA 106: 5825–5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Zincarelli C, Kim J, Koch WJ (2011). Adrenal beta‐arrestin 1 inhibition in vivo attenuates post‐myocardial infarction progression to heart failure and adverse remodeling via reduction of circulating aldosterone levels. J Am Coll Cardiol 57: 356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymperopoulos A, Sturchler E, Bathgate‐Siryk A, Dabul S, Garcia D, Walklett K, et al. (2014). Different potencies of angiotensin receptor blockers at suppressing adrenal β ‐Arrestin1‐dependent post‐myocardial infarction hyperaldosteronism. J Am Coll Cardiol 64: 2805–2806. [DOI] [PubMed] [Google Scholar]
- McGuinness D, Malikzay A, Visconti R, Lin K, Bayne M, Monsma F, et al. (2009). Characterizing cannabinoid CB2 receptor ligands using discoverx pathhunter beta‐arrestin assay. J Biomol Screen 14: 49–58. [DOI] [PubMed] [Google Scholar]
- Miura S, Feng YH, Husain A, Karnik SS (1999). Role of aromaticity of agonist switches of angiotensin II in the activation of the AT1 receptor. J Biol Chem 274: 7103–7110. [DOI] [PubMed] [Google Scholar]
- Nikiforovich GV, Zhang M, Yang Q, Jagadeesh G, Chen H‐C, Hunyady L, et al. (2006). Interactions between conserved residues in transmembrane helices 2 and 7 during angiotensin AT1 receptor activation. Chem Biol Drug Des 68: 239–249. [DOI] [PubMed] [Google Scholar]
- Peters MF, Knappenberger KS, Wilkins D, Sygowski LA, Lazor LA, Liu J, et al. (2007). Evaluation of cellular dielectric spectroscopy, a whole‐cell, label‐free technology for drug discovery on Gi‐coupled GPCRs. J Biomol Screen 12: 312–319. [DOI] [PubMed] [Google Scholar]
- Qian H, Pipolo L, Thomas WG (2001). Association of β ‐arrestin 1 with the type 1A angiotensin II receptor involves phosphorylation of the receptor carboxyl terminus and correlates with receptor internalization. Mol Endocrinol 15: 1706–1719. [DOI] [PubMed] [Google Scholar]
- Rainey WE, Saner K, Schimmer BP (2004). Adrenocortical cell lines. Mol Cell Endocrinol 228: 23–38. [DOI] [PubMed] [Google Scholar]
- Salazar NC, Vallejos X, Siryk A, Rengo G, Cannavo A, Liccardo D, et al. (2013). GRK2 blockade with β ARKct is essential for cardiac β 2‐adrenergic receptor signaling towards increased contractility. Cell Commun Signal 11: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Violin JD, Whalen EJ, Gesty‐Palmer D, Shenoy SK, Lefkowitz RJ (2008). Distinct conformational changes in beta‐arrestin report biased agonism at seven‐transmembrane receptors. Proc Natl Acad Sci USA 105: 9988–9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, et al. (2013). Structure of active β ‐arrestin‐1 bound to a G‐protein‐coupled receptor phosphopeptide. Nature 497: 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soergel DG, Subach RA, Cowan CL, Violin JD, Lark MW (2013). First clinical experience with TRV027: pharmacokinetics and pharmacodynamics in healthy volunteers. J Clin Pharmacol 53: 892–899. [DOI] [PubMed] [Google Scholar]
- Tzakos AG, Gerothanassis IP, Troganis AN (2004). On the structural basis of the hypertensive properties of angiotensin II: a solved mystery or a controversial issue? Curr Top Med Chem 4: 431–444. [DOI] [PubMed] [Google Scholar]
- Violin JD, DeWire SM, Yamashita D, Rominger DH, Nguyen L, Schiller K, et al. (2010). Selectively engaging β ‐arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther 335: 572–579. [DOI] [PubMed] [Google Scholar]
- Violin JD, Soergel DG, Boerrigter G, Burnett JC Jr, Lark MW (2013). GPCR biased ligands as novel heart failure therapeutics. Trends Cardiovasc Med 23: 242–249. [DOI] [PubMed] [Google Scholar]
- Weber KT (2001). Aldosterone in congestive heart failure. N Engl J Med 345: 1689–1697. [DOI] [PubMed] [Google Scholar]
- Wilkes BC, Masaro L, Schiller PW, Carpenter KA (2002). Angiotensin II vs its type I antagonists: conformational requirements for receptor binding assessed from NMR spectroscopic and receptor docking experiments. J Med Chem 45: 4410–4418. [DOI] [PubMed] [Google Scholar]
- Zhang H, Unal H, Gati C, Han GW, Liu W, Zatsepin NA, et al. (2015). Structure of the angiotensin receptor revealed by serial femtosecond crystallography. Cell 161: 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman B, Beautrait A, Aguila B, Charles R, Escher E, Claing A, et al. (2012). Differential β ‐arrestin‐dependent conformational signaling and cellular responses revealed by angiotensin analogs. Sci Signal 5: ra33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Schematic illustration of the DiscoveRx PathHunterTM β‐Arrestin assay.
Figure S2. Schematic illustration of the CellKey assay protocol employed in this study.
Figure S3. (A) In vitro aldosterone secretion from H295R cells overexpressing a dominant negative mutant β‐arrestin1 and stimulated for 4 h with 100 nmol/L CORSML, CORML, or CORET. Data are presented as % of the secretion induced by 10 μmol/L SII. *P < 0.05, versus SII, ^P < 0.05, versus CORML or CORSML, n = 5 independent experiments/treatment. No significant differences were observed among treatments.