

Abstract
A modular and highly efficient protocol for the synthesis of 2-aryl- and heteroaryl-2H-chromenes is described. Under base-free conditions, readily accessible 2-ethoxy-2H-chromenes undergo Csp3–O activation and Csp3–C bond formation in the presence of an inexpensive nickel catalyst and boronic acids. This new strategy enables broad access to 2-substituted-2H-chromenes and has been applied to the late-stage incorporation of complex molecules, including the pharmaceuticals loratidine and indomethacin methyl ester.
Graphical abstract
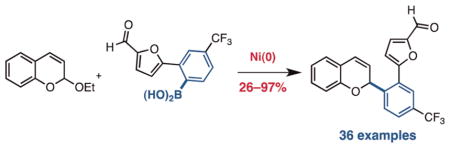
2-Substituted 2H-chromenes and their analogs are pervasive molecular structures in chemistry, biology, and medicine. Examples include fungicidal agents,1 potent antioxidants (e.g., Vitamin E),2 and pharmaceuticals that possess antitumor,3 antibacterial,4 and antiviral activity.5 The heterocycle is also used extensively as a photochromic material6 and a precursor to flavylium dyes7 (Figure 1). Accordingly, the availability of efficient and modular methods for the preparation of this privileged motif is of paramount importance.8
Figure 1.

Pharmacologically active and photochromic chromene derivatives.
Numerous synthetic approaches have been devised to access 2-substituted 2H-chromenes (Scheme 1).9 Routinely employed protocols rely on benzopyran construction from phenol or salicylaldehyde derivatives by oxa–Michael-type or olefin metathesis annulations.10 These approaches typically involve multistep sequences, incorporate the C-2 group early in the synthesis, or lack broad generality. Alternatively, nucleophilic substitution of chromene acetals, prepared by reduction of readily available coumarins, potentially offers a versatile solution to the synthesis of this scaffold where the C-2 group is introduced in the final step. Reactions with Grignard reagents are frequently used; while these obviate the need for prefunctionalized fragment synthesis,11 they often produce regioisomeric product mixtures and are intolerant of many functional groups.12 Schaus recently reported a related approach that selectively affords C-2 substituted chromenes.13,14 This organocatalytic Petasis-like reaction uses functional group tolerant boronate esters but is only effective for intrinsically nucleophilic partners such as cinnamyl and π-rich arenes. For these reasons, a concise, efficient, and general method for the synthesis of chromenes is still in demand.
Scheme 1.

Synthetic Strategies for Chromene Synthesis
- Feasibility independent of electronic/steric properties of reaction partners
- First example of Suzuki–Miyaura cross-coupling with allylic acetals
Herein, we report such a method: a nickel-catalyzed cross-coupling reaction of aryl- and heteroarylboronic acids with chromene acetals. The method represents a mechanistic alternative to nucleophilic acetal substitution that enables access to a broad range of previously inaccessible C-2 substituted chromenes by late-stage diversification of a common scaffold. To our knowledge, it also represents the first example of a Suzuki–Miyaura cross-coupling reaction with allylic acetals.
Although recent reports have described Suzuki–Miyaura reactions with allylic phenoxides, acetates, and carbonates,15 the use of allylic acetals in cross-coupling has been limited to Kumada16 and Negishi17 reactions. Slow oxidative addition of electron-rich Csp3–O bonds to a transition metal18 and the preponderance of such bonds in the substrate as well as the product of acetal cross-coupling are two likely causes for this comparative lack of success.19 We recently reported Suzuki–Miyaura reactions of quinoline-derived N,O-acetals20 and styrenyl epoxides.21 Our preliminary mechanistic data indicated that in these systems Csp3–O oxidative addition is promoted by Lewis acid complexation of the boronic acid to the Lewis basic oxygen of the electrophile. Inspired by these findings, we decided to pursue the possibility that boronic acid activation22 of an allylic acetal in the absence of a base would encourage selective oxidative addition and transmetalation. A particularly attractive aspect of this approach is that it would afford a neutral method for the synthesis of allylic ethers from widely available and stable precursors.23
In an initial demonstration of this hypothesis, we chose to evaluate chromene-based acetals for the preparation of 2-substituted 2H-chromenes (Scheme 1). Subjecting 2-ethoxy-2H-chromene 1a to 10 mol % Ni(cod)2, 10 mol% PPh3, and 2 equiv of p-fluorophenyl boronic acid at rt in dioxane/t-AmOH as solvent afforded the desired cross-coupled heterocycle 3 in 92% yield (Figure 2). In the absence of the Ni catalyst, 3 was not observed, even when the reaction was conducted at elevated temperatures (<5% yield). Furthermore, it was found that the type of organoboron nucleophile employed significantly affected the reaction outcome. The use of boronate esters or potassium aryltrifluoroborate salts also did not afford the desired product. Since these boronates are less Lewis acidic than boronic acids the results provide circumstantial support for the role of the boronic acid in activating the acetal toward oxidative addition. Notably, the chromene acetals possess two distinct ethereal leaving groups, but selective activation of the more Lewis basic exocyclic ether is observed. The reaction is also regioselective, delivering the aryl group to the C-2 rather than the C-4 position with greater than 20:1 rr (regioisomeric ratio). Overall, the method displays an expansive scope with respect to both coupling partners, is scalable, and requires short reaction times (vida infra). Moreover, the catalyst and ligand are both inexpensive and purification of the products is straightforward because the only side products arise from acetal hydrolysis and protodeboronation.24
Figure 2.

Scope of Ni-catalyzed coupling of aryl- and heteroarylboronic acids with 2-ethoxy-2H-chromene 1a.a
a General conditions (yields are the average of two runs, 0.50 mmol scale): Ni(cod)2 (10 mol %), PPh3 (10 mol %), 0.02 M, 23 or 40 °C. b10 mmol scale, Ni(cod)2 (5 mol %), PPh3 (5 mol %), 0.20 M, 40 °C, 14 h. c8:1 rr. dNi(cod)2 (10 mol %), PPh3 (30 mol %), 100 °C. eBoronic acid prepared from the chloride, see Supporting Information for details. f0.10 mmol, one run. gContains PPh3 as a minor impurity.
The reaction proved general for a broad range of aryl boronic acids, with yields ranging from 51 to 97% (Figure 2). High efficiency was observed regardless of the electronic nature of the para- or meta-substituted aryl boronic acid employed (e.g., π-neutral boronic acids 2–7, 16, 17 and π-deficient boronic acids 8–15). In this regard, the strategy holds a distinct advantage over Petasis-type reactions as its success is not governed by the intrinsic reactivity of the nucleophile. In fact, competition studies indicate that electron-deficient aryl boronic acids display higher reactivity than electron-rich ones (4-CF3 > 4-H > 4-MeO).25 Notably, the reaction proceeded with similar efficiency and high regioselectivity (8:1 rr) on a gram scale using a reduced catalyst loading and an elevated concentration, indicating that it should be a viable approach for the preparative scale synthesis of chromene derivatives (Figure 2, 2).
For ortho-substituted boronic acids, we found that the reaction is sensitive to the steric properties of the arene. While ortho-tolyl boronic acid could be used at 40 °C under the outlined conditions (Figure 2, 18, 95% yield) more sterically encumbered boronic acids were generally not reactive. This limitation was overcome by employing a higher reaction temperature (100 °C); at this temperature, a PPh3/Ni ratio of 3:1 was necessary to prevent the formation of nickel black. Under these modified conditions sterically encumbered (e.g., 2,6-(CH3)2 21, 2-Ph 22) and strongly deactivated (e.g., 2,4-(CF3)2 23) aryl boronic acids reacted smoothly.
Given the importance of heterocyclic motifs within biologically relevant molecules, our attention turned to heteroarylboronic acids as reaction partners. Dibenzofuran (19), dibenzothiophene (20), and indole (25) boronic acids smoothly underwent coupling (Figure 2). However, 3-pyridylboronic acid failed to deliver product. Given our success with electron-deficient aryl boronic acids, we assumed that catalyst deactivation by the basic nitrogen, rather than the electron-deficient nature of the heterocycle, was responsible for its relative lack of reactivity. Pleasingly, we found that substituted pyridyl boronic acids (Figure 2, 26–28) were compatible with the reaction conditions. The success of these substrates appears to be the result of steric shielding, as both trifluoromethyl (28) and methoxy (26)-substituted pyridyl boronic acids undergo coupling with similar efficiency. However, the use of related heteroaryl boronic acids bearing exposed Lewis basic nitrogens (quinoline, pyrimidine, isoxazole, imidazole) remains an important limitation of the methodology.
The scope of the chromene acetal component in this cross-coupling protocol has also been studied (Figure 3). We found that chromene acetals substituted at the 5- and 6-positions with methoxy (30), methyl (31), fluoro (32), and chloro (33) functionalities are suitable substrates. Attempts to use sterically congested chromene acetals resulted in low reactivity (3-Ph, 34), although a modest yield could be achieved at 100 °C and a PPh3/Ni ratio of 3:1. Notably, aryl chlorides are well tolerated on both reaction partners, allowing for a direct entry toward the antiviral BW683C and providing handles for further functionalization by metal-catalyzed cross-coupling (35).
Figure 3.

Scope of 2-ethoxy-2H-chromenes.a
a Conditions (yields are the average of two runs, 0.50 mmol): Ni(cod)2 (10 mol %), PPh3 (10 mol %), 0.02 M, 40 °C. bNi(cod)2 (10 mol %), PPh3 (30 mol %), 100 °C (one run).
Perhaps the most important demonstration of the capacity of the outlined method is late-stage functionalization of molecules that possess functionality incompatible with existing methods for chromene and ether syntheses. As a validation of the principle, we selected the pharmaceutical drugs loratadine and indomethacin methyl ester, in addition to a known furfuryl-benzotrifluoride, as test substrates. Collectively, these substrates possess acidic C–H bonds, Lewis basic heteroatoms, nucleophilic heterocycles, and an aldehyde. Utilizing Molander’s recently disclosed method for the direct synthesis of aryl boronic acids from aryl chlorides,26 we were able to successfully isolate the requisite boronic acids. Under the reaction conditions described for heteroaryl substrates, the desired coupling occurred in 26–79%yield (Figure 2, 29, and Scheme 2, 36 and 37). Importantly, these compounds would be impossible to construct by methods that involved strong bases at any stage of the chromene synthesis, including the addition of Grignard reagents to chromene acetals.
Scheme 2.

Late-Stage Chromene Acetal Couplinga
aConditions: (a) acetal 1a, 2 equiv of aryl boronic acid, Ni(cod)2 (10 mol %), PPh3 (30 mol %), dioxane/t-AmOH (10:1), 100 °C; see Supporting Information for further details.
In conclusion, we have reported a modular synthesis of 2-aryl and 2-heteroaryl 2H-chromene derivatives from readily available chromene acetals that is characterized by a broad scope, mild conditions, and an inexpensive metal/ligand system. The reaction also represents the first example of a transition-metal-catalyzed cross-coupling reaction between allylic acetals and boronic acids. Experiments to delineate the generality of this acid and base-free strategy for the construction of ethers will be the focus of our future efforts.
Supplementary Material
Acknowledgments
Financial support was provided by Princeton University and kind gifts from Eli Lilly and Sanofi-Aventis. Allychem and Frontier Scientific are acknowledged for generous donations of boronic acids. BASF is acknowledged for a generous donation of B2(OH)4.
Footnotes
The authors declare no competing financial interest.
Supporting Information Available. Experimental procedures and full spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lago JHG, Ramos CS, Casanova DCC, Morandim AD, Bergamo DCB, Cavalheiro AJ, Bolzani VD, Furlan M, Guimaraes EF, Young MCM, Kato MJ. J Nat Prod. 2004;67:1783–1788. doi: 10.1021/np030530j. [DOI] [PubMed] [Google Scholar]
- 2.Mukai K, Okabe K, Hosose H. J Org Chem. 1989;54:557–560. [Google Scholar]
- 3.Gauthier S, Caron B, Cloutier J, Dory YL, Favre A, Larouche D, Mailhot J, Ouellet C, Schwerdtfeger A, Leblanc G, Martel C, Simard J, Merand Y, Belanger A, Labrie C, Labrie F. J Med Chem. 1997;40:2117–2122. doi: 10.1021/jm970095o. [DOI] [PubMed] [Google Scholar]
- 4.Schneider P, Hawser S, Islam K. Biorg Med Chem Lett. 2003;13:4217–4221. doi: 10.1016/j.bmcl.2003.07.023. [DOI] [PubMed] [Google Scholar]
- 5.Bauer DJ, Selway JWT, Batchelor JF, Tisdale M, Caldwell IC, Young DAB. Nature. 1981;292:369–370. doi: 10.1038/292369a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Nigel Corns S, Partington SM, Towns AD. Color Technol. 2009;125:249–261. [Google Scholar]; (b) Bamfield P, Hutchings MG. Chromic Phenomena: The Technological Applications of Colour Chemistry. The Royal Society of Chemistry; Cambridge: 2010. [Google Scholar]
- 7.(a) Iacobucci GA, Sweeny JG. Tetrahedron. 1983;39:3005–3038. [Google Scholar]; (b) Pina F, Melo MJ, Laia CAT, Parola J, Lima JC. Chem Soc Rev. 2012;41:869–908. doi: 10.1039/c1cs15126f. [DOI] [PubMed] [Google Scholar]
- 8.(a) Welsch ME, Snyder SA, Stockwell BR. Curr Opin Chem Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nicolaou KC, Pffefferkorn JA, Roecker AJ, Cao GQ, Barluenga S, Mitchell HJ. J Am Chem Soc. 2000;122:9939–9953. [Google Scholar]
- 9.Majumdar N, Korthals KA, Wulff WD. J Am Chem Soc. 2012;134:1357–1362. doi: 10.1021/ja210655g.and references therein.
- 10.For examples in the context of target synthesis, see ref 3 and: Brimble M, Jay-Smith M, Furkert D, Sperry J. Synlett. 2011;2011:1395–1398.Wipf P, Weiner WS. J Org Chem. 1999;64:5321–5324. doi: 10.1021/jo990352s.
- 11.Li X, Reuman M, Russell RK, Adams R, Ma R, Branum S, Youells S, Roberts J, Jain N, Kanojia R, Sui Z. Org Process Res Dev. 2007;11:414–421. [Google Scholar]
- 12.Grese TA, Pennington LD. Tetrahedron Lett. 1995;36:8913–8916. [Google Scholar]
- 13.Moquist PN, Kodama T, Schaus SE. Angew Chem, Int Ed. 2010;49:7096–7100. doi: 10.1002/anie.201003469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For a related approach, see: Doodeman R, Rutjes FPJT, Hiemstra H. Tetrahedron Lett. 2000;41:5979–5983.
- 15.For a review, see: Pigge F. Synthesis. 2010;2010:1745–1762.For specific examples, see: Nishikata T, Lipshutz BH. J Am Chem Soc. 2009;131:12103–12105. doi: 10.1021/ja905082c.Chung KG, Miyake Y, Uemura S. J Chem Soc, Perkin Trans. 2000;1:15–18.Kobayashi Y, Mizojiri R, Ikeda E. J Org Chem. 1996;61:5391–5399.
- 16.(a) Wenkert E, Ferreira TW. Organometallics. 1982;1:1670–1673. [Google Scholar]; (b) Sugimura H, Takei H. Chem Lett. 1985:351–354. [Google Scholar]; (c) Moineau C, Bolitt V, Sinou D. Chem Commun. 1995:1103–1104. [Google Scholar]; (d) Gomez-Bengoa E, Heron NM, Didiuk MT, Luchaco CA, Hoveyda AH. J Am Chem Soc. 1998;120:7649–7650. [Google Scholar]; (e) Guagnano V, Lardicci L, Malanga C, Menicagli R. Tetrahedron Lett. 1998;39:2025–2026. [Google Scholar]; (f) Moineau C, Bolitt V, Sinou D. J Org Chem. 1998;63:582–591. doi: 10.1021/jo9714674. [DOI] [PubMed] [Google Scholar]
- 17.Chatterjee S, Negishi E. J Org Chem. 1985;50:3406–3408. [Google Scholar]
- 18.Yamashita Y, Gopalarathnam A, Hartwig JF. J Am Chem Soc. 2007;129:7508–7509. doi: 10.1021/ja0730718. [DOI] [PubMed] [Google Scholar]
- 19.Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita AM, Garg NK, Percec V. Chem Rev. 2011;111:1346–1416. doi: 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graham TJA, Shields JD, Doyle AG. Chem Sci. 2011;2:980–984. [Google Scholar]
- 21.Nielsen DK, Doyle AG. Angew Chem, Int Ed. 2011;50:6056–6059. doi: 10.1002/anie.201101191. [DOI] [PubMed] [Google Scholar]
- 22.(a) Hall DG. Boronic Acids. Wiley-VCH; Weinheim: 2005. [Google Scholar]; (b) Zheng H, Lejkowski M, Hall DG. Chem Sci. 2011;2:1305–1310. [Google Scholar]
- 23.(a) Vo CVT, Mitchell TA, Bode JW. J Am Chem Soc. 2011;133:14082–14089. doi: 10.1021/ja205174c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mitchell TA, Bode JW. J Am Chem Soc. 2009;131:18057–18059. doi: 10.1021/ja906514s. [DOI] [PubMed] [Google Scholar]
- 24.Determined by LC/MS.
- 25.Ratio of products for 4-CF3/4-H/4-MeO was 3:1:0.6. See Supporting Information for details.
- 26.Molander GA, Trice SLJ, Dreher SD. J Am Chem Soc. 2010;132:17701–17703. doi: 10.1021/ja1089759. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.