

Abstract
Background:
Wolf-Hirschhorn syndrome (WHS) is a contiguous gene syndrome that is typically caused by a deletion of the distal portion of the short arm of chromosome 4. However, there are few reports about the features of Chinese WHS patients. This study aimed to characterize the clinical and molecular cytogenetic features of Chinese WHS patients using the combination of multiplex ligation-dependent probe amplification (MLPA) and array comparative genomic hybridization (array CGH).
Methods:
Clinical information was collected from ten patients with WHS. Genomic DNA was extracted from the peripheral blood of the patients. The deletions were analyzed by MLPA and array CGH.
Results:
All patients exhibited the core clinical symptoms of WHS, including severe growth delay, a Greek warrior helmet facial appearance, differing degrees of intellectual disability, and epilepsy or electroencephalogram anomalies. The 4p deletions ranged from 2.62 Mb to 17.25 Mb in size and included LETM1, WHSC1, and FGFR3.
Conclusions:
The combined use of MLPA and array CGH is an effective and specific means to diagnose WHS and allows for the precise identification of the breakpoints and sizes of deletions. The deletion of genes in the WHS candidate region is closely correlated with the core WHS phenotype.
Keywords: Array Comparative Genomic Hybridization, Multiplex Ligation-dependent Probe Amplification, Wolf-Hirschhorn Syndrome
INTRODUCTION
Wolf-Hirschhorn syndrome (WHS, OMIM 194190) is caused by the deletion of contiguous genes from the short arm of chromosome 4 and characterized by the Greek warrior helmet profile facial appearance, growth and development delays, intellectual disability, and seizures or electroencephalographic (EEG) anomalies.[1,2] The WHS patients have been examined by novel technologies, such as array comparative genomic hybridization (array CGH) and multiplex ligation-dependent probe amplification (MLPA).[3,4] Array CGH enables the detection of the precise sizes of deletions in the WHS candidate region (WHSCR) with greater accuracy compared with either fluorescence in situ hybridization (FISH) or conventional G-banded chromosome analysis alone.[5] In addition, MLPA allows for a more comprehensive determination of gene dosages with increased convenience and a relatively low cost compared with subtelomeric FISH analysis.[6] Moreover, MLPA can be used to detect micro-deletions that may not be identified by FISH.[7,8,9]
Here, we reported the characteristics of ten Chinese patients with WHS, as determined by the combination of MLPA and array CGH.
METHODS
Patients
Clinical information was collected for ten patients at the Genetics Clinic of the Peking University First Hospital between July 2008 and June 2014. Examining clinical manifestations and performing genetic analyses are necessary to diagnose WHS. Informed consent was obtained from all the patients’ parents. The procedures followed in this investigation were approved by the Human Research Ethics Committee of the Peking University First Hospital.
Genetics
Conventional cytogenetic analysis
Routine G-banding techniques at the 400 bands of resolution were performed. Genomic DNA was extracted from the peripheral blood of the patients using a QuickGene-610L system (FUJIFILM, Osaka, Japan). DNA purification was performed using a QIAamp DNA Kit (Qiagen, Germany), according to the manufacturer's protocol. Quantitative DNA analysis was performed using an ultraviolet NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, Delaware, USA).
Multiplex ligation-dependent probe amplification
MLPA analysis was performed to screen subtelomeric rearrangements using SALSA P070 and P036, and micro-deletion syndromes using SALSA P245 probemix (MRC-Holland, Amsterdam, the Netherlands). Both P070 and P036 contained one specific probe for 4p subtelomeric regions. The P245 contained special 2 probes for WHS. MLPA analysis was performed according to the manufacturer's instructions. The amplification products were identified and quantified by capillary electrophoresis using a 3130 XL genetic analyzer (Applied Biosystems, Foster City, USA). The fluorescence signal intensities of the PCR products were determined with Gene Marker l. 6 software (Softgenetics, State College, PA, USA).
Array comparative genomic hybridization
The kit of Agilent SurePrint G3 Human CGH Microarray 8×60K (Agilent Technologies, CA, USA) was used for genetic analysis according to the manufacturer's instructions. This microarray included 55,077 probes with a median probe space of 41 kb for intergenic genomic sequences. DNA hybridization was performed according to the standard procedures after the labeling of 500 ng of DNA from each of the patients with cyanine-5 and the labeling of control DNA (Promega, USA) with cyanine-3. The signals were captured according to the instructions of the Agilent SurePrint G3 CGH Microarray Kit. Microarray data were analyzed using Feature Extraction software and Workbench genomics software (Agilent Technologies, CA, USA). Probe alignments were performed using National Center for Biotechnology Information 37 (http://www.ncbi.nlm.nih.gov/), University of California, Santa Cruz hg19 build (http://genome.ucsc.edu/), and DECEPHER v8.8 (https://decipher.sanger.ac.uk/).
RESULTS
Clinical features of Chinese patients with Wolf-Hirschhorn syndrome
Detailed clinical data are presented in Table 1. The patients were sorted by their distal chromosome 4p deletion sizes. Intrauterine growth restriction (IUGR) was detected in 5 patients. Hypotonia was detected in 9 patients, but was not observed in patient 1, who had smaller deletions. Postnatal growth delay occurred more frequently than IUGR. This severe growth delay was present in all patients. The Greek warrior helmet appearance was obvious in almost all the patients. Only two patients had cleft lip and palate. Patients 9 and 10 were excluded from the investigation of congenital heart defects due to insufficient information. A total of 5 patients displayed congenital heart defects, including atrial septal defects in three patients and pulmonary stenosis in two patients. The Gesell Developmental Schedule was administered to all patients to assess their levels of intellectual disability, except for patient 6 because of her age. All patients showed intellectual disability. The array CGH results revealed that among these 6 patients with disability, one patient with mild disability had small deletions (<3 Mb), 3 with moderate disability had moderate-sized deletions (3–14 Mb), and 2 with severe disability had large deletions (>14 Mb). Epilepsy or EEG anomalies were detected in 8 patients. Motor developmental delay was detected in all patients, including difficulties in controlling the head and in sitting and walking. The feeding difficulty was found in 3 patients, renal abnormalities was found in 1 patient, and skeletal abnormalities was found in 1 patient.
Table 1.
Summary of clinical data of ten Chinese WHS patients
| Characteristics | Patient number | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8[10]* | 9[10]* | 10[10]* | |
| Gender | Male | Male | Female | Male | Female | Female | Female | Male | Female | Female |
| Age | 14 months | 56 months | 39 months | 4 months | 81 months | 6 days | 37 months | 14 months | 30 months | 42 months |
| IUGR | + | + | + | − | + | + | − | − | − | − |
| Hypotonia | − | + | + | + | + | + | + | + | + | + |
| Postnatal growth delay | <P3 | <P3 | <P10 | <P3 | <P3 | <P3 | <P3 | <P10 | <P3 | <P3 |
| High or broad forehead | + | + | + | + | + | + | + | + | + | + |
| Prominent glabella | + | + | + | + | + | + | + | + | + | + |
| Hypertelorism | + | + | + | + | + | + | + | + | + | + |
| Prominent or slanting eyes | + | − | + | + | + | +/− | + | + | +/− | + |
| High arched eyebrows | + | + | + | + | + | +/− | + | + | + | + |
| Broad nasal bridge | + | + | + | + | + | + | + | + | + | + |
| Short philtrum | + | + | + | + | + | + | + | − | + | + |
| Distinct mouth | + | +/− | + | − | +/− | +/− | + | − | + | + |
| Micrognathia | + | − | − | − | − | + | + | − | − | + |
| Dysplastic ears | − | − | − | − | − | − | − | + | − | + |
| Cleft lip and/palate | − | − | + | − | + | − | − | − | − | − |
| Congenital heart defect | PS | − | ASD | ASD | − | − | PS | ASD | NI | NI |
| Intellectual disability | Mild | Moderate | Moderate | Moderate | Severe | − | Severe | Moderate | Moderate | Moderate |
| Epilepsy/EEG anomalies | + | + | + | − | + | − | + | + | + | + |
| Motor developmental delay | + | + | + | + | + | + | + | + | + | + |
| Feeding difficulties | − | + | − | − | − | − | − | + | + | − |
| Renal abnormalities | − | − | − | − | − | + | − | − | − | − |
| Skeletal anomalies | − | + | − | − | − | − | − | − | − | − |
*Array CGH test results were not available for patient 8, 9, or 10. The deletion sizes were determined mainly by MLPA by calculating the distances between probes. P3 and P10 stand for the 3rd and 10th percentile length/height for age. IUGR: Intrauterine growth restriction; PS: Pulmonary stenosis; ASD: Atrial septal defect; NI: Not investigated; +: Positive; −: Negative; WHS: Wolf-Hirschhorn syndrome; EEG: Electroencephalogram; CGH: Comparative genomic hybridization; MLPA: Multiplex ligation-dependent probe amplification.
Molecular and cytogenetic analyses
The genetic results, including MLPA and array CGH, are displayed in detail [Figures 1–3 and Table 2]. Deletions of the 4p terminus that were smaller than 5 Mb could not be recognized by karyotypic analysis. Deletions of 4p were detected in all patients with MLPA. The 4p16.3 deletions detected by array CGH ranged in size from 2.62 Mb to 17.25 Mb, with breakpoints at 0.07 Mb from the distal 4p telomere and at 17.32 Mb from the 4p telomere. LETM1 (OMIM 604407), WHSC1 (OMIM 602952), and FGFR3 (OMIM 134934) were deleted in all patients evaluated by array CGH.
Figure 1.

The molecular diagnostic findings of patients. The array comparative genomic hybridization results showed a 2.62 Mb 4p deletion in patient 1 (a), a 5.44 Mb 4p deletion in patient 3 (b), and a 17.25 Mb 4p deletion in patient 7 (c).
Figure 3.
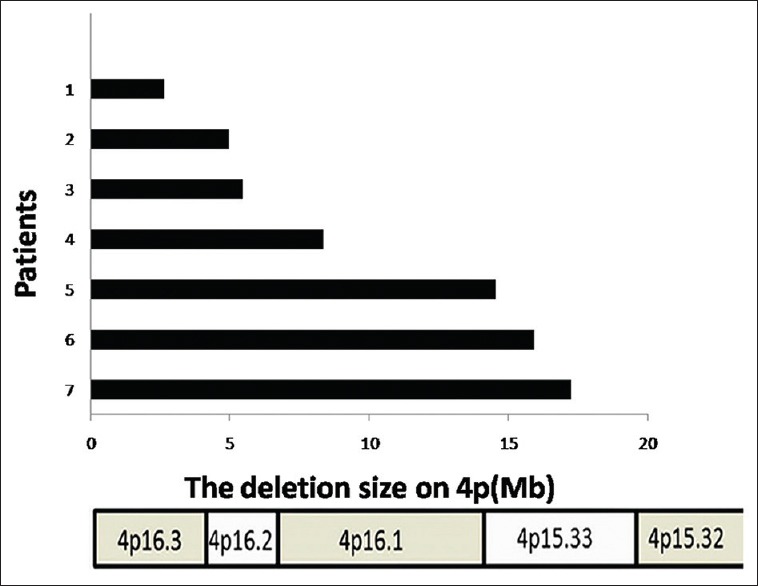
The size of 4p deletion in seven patients detected by array comparative genomic hybridization. Ordinate represents the corresponding patient. Abscissa represents the deletion size on chromosome 4. Black bars mean the deleted length of 4p.
Table 2.
Results of molecular and cytogenetic analyses of ten Chinese patients with WHS
| Patient number | G-band karyotype ≥400 bands | Array CGH analysis | MLPA analysis | |
|---|---|---|---|---|
| Boundaries of deletion (kb) | Size of deletion (Mb) | |||
| 1 | 46, XY | 71,552–2,691,306 | 2.62 | 4pdel |
| 2 | 46, XY | 71,552–5,036,718 | 4.97 | 4pdel |
| 3 | 46, XX | 71,552–5,506,588 | 5.44 | 4pdel |
| 4 | 46, XY, del(4)(p16) | 71,552–8,416,608 | 8.35 | 4pdel |
| 5 | 46, XX, del(4)(p16.1) | 71,552–14,631,433 | 14.56 | 4pdel |
| 6 | 46, XX, del(4)(p15.3) | 71,552–15,992,938 | 15.92 | 4pdel |
| 7 | 46, XX, del(4)(p15.3) | 71,552–17,320,084 | 17.25 | 4pdel |
| 8* | 46, XY | Not applicable | >2.00 | 4pdel |
| 9* | 46, XX | Not applicable | >2.00 | 4pdel |
| 10* | 46, XX | Not applicable | >2.00 | 4pdel |
*Array CGH test results were not available for patient 8, 9 or 10. Their deletion sizes were identified mainly by MLPA by calculating the range between probes. WHS: Wolf-Hirschhorn syndrome; MLPA: Multiplex ligation-dependent probe amplification; CGH: Comparative genomic hybridization.
Figure 2.

The results of MLPA P036, P070, and P245 with terminal 4p deletion in patient 1. Ordinate represents the peak ratio. Abscissa represents the size of probes. Each dot stands for the signal of corresponding probe. Each probe has a probe space of 6–8 bases pairs. Green dots, between the two green lines, stand for the normal signal of probes, the peak ratio of which range from 0.7 to 1.3. Red dots, below the green lines, stand for deletion of gene dosage. The results of MLPA P036 (a), MLPA P070 (b), and MLPA P245 (c) show that the 4p subtelomere is deleted. MLPA: Multiplex ligation-dependent probe amplification.
DISCUSSION
WHS is one of the most common deletion syndromes, and it is caused by a deletion of the short arm of chromosome 4. Here, we discussed the genotype–phenotype correlation of WHS in Chinese patients, as assessed by genetic analyses and comprehensive clinical evaluation in ten patients. The frequency of WHS was 60.0% (6/10) in the females in our study, consistent with previous reports.[10,11] The frequency of the core phenotype of WHS, including severe growth delay (IUGR and postnatal growth delay), distinctive facial features, intellectual disability, and epilepsy or EEG anomalies, ranged from 50.0% (5/10) to 80.0% (8/10), which was consistent with previous reports conducted in Asia.[12,13] The corresponding deletion breakpoints ranged from the 4p terminus to 17.32 Mb from the distal 4p telomere, including the critical 1.9 Mb region of 4p16.3, which was described as WHSCR-2. WHSC1 and FGFR3 were deleted in the patients detected by array CGH, and the absence of expression of these two gene has been associated with the typical craniofacial features of WHS, obvious growth delay, and skeletal disorders.[14,15] Several studies have reported a delicate relationship between the WHS phenotype and deletion size. The clinical manifestations of this syndrome have been classified into three groups according to the size of the distal chromosome 4p deletion as follows: small (<3.5 Mb), intermediate (5–18 Mb), and large (22–25 Mb).[11,16] Our data showed that midline fusion defects, such as congenital heart defects and cleft lip/palate, were present at higher frequencies when the deletion size was larger than 5 Mb. A similar trend was observed in the proportion of patients with severe intellectual disability, which was higher in those with a larger deletion size. Patients detected by array CGH analysis have the deletion of LETM1. LETM1 has been reported to regulate ATP levels by acting on a protein involved in Ca2+/H+ exchange in the mitochondria, affecting seizure activity in the brain.[17] However, only 8 of 10 patients had epilepsy or EEG abnormalities. LETM1 is currently considered the major but not the unique gene for seizures.[16,17] Seizures were observed in some patients with 4p deletion without the LETM1 deletion, for example, smaller than 1.4 Mb terminal.[18] Comparing the patients with terminal 4p deletions preserving LETM1 or with interstitial 4p deletions encompassing LETM1, Zollino et al.[12] found that the breakpoints nearby the LETM1 locus may lead to the subtle difference in phenotypes between patients with LETM1 deletion and without LETM1 deletion. Epilepsy was not found in 2 of 10 patients in this study, both of whom were within their first 6 months of life. These results were also consistent with those of a previous study, reporting that seizures generally begin in WHS patients during the first 3 years of life, especially at around 6–12 months of age.[19] It is a pity that we do not collect the spectrum of epilepsy. The spectrum of epilepsy in WHS was various, including generalized tonic-clonic seizures, tonic spasms, complex partial seizures, and clonic seizures.[19] However, there is no proven direct link between the type of seizures and the deletion of LETM1.
Combined use of MLPA and array CGH was applied to evaluate WHS in our research. Routine G-banding techniques at the 400 bands of resolution were performed for karyotype. It was available and observable only when the deletion size was larger than 5 Mb. MLPA is more accurate to describe the karyotype due to its delicate probe spacing. A screening strategy is required for MLPA analysis of WHS, that is, using SALSA P070 and P036 for subtelomeric rearrangements first, and SALSA P245 or other further investigated kits second.[20] Array CGH is advanced in detecting the deletion genes and corresponding breakpoints in WHSCR, which is not only a complementary method for MLPA, but also helpful for precise medical therapy in the future. Therefore, the combined use of MLPA and array CGH facilitates the evaluation of WHS with greater accuracy than conventional cytogenetic methods.
The series reports of WHS are rare, especially for the Asian populations. Compared with the domestic reports of four patients with WHS,[13] the patients in the current study have a wide range of deletions, although ranging from less than 5 Mb to more than 15 Mb, but similar phenotypes. The main reason is that haploinsufficiency of the core candidate genes, WHSC1, LETM1, and FGFR3, is responsible for most of the WHS characteristics.[21,22] All genes deleted in patients detected by array CGH were supplied [Table 3]. This article aimed to help clinicians to recognize the features of Chinese WHS and diagnose it with appropriate molecular genetic methods through summing the corresponding phenotypes of different size of deletions. The results emphasized the relationship between genotype and phenotype which was consistent with the previous reports in Europe.[11] It also highlighted the importance of accurate diagnosis of WHS although the recurrence of WHS is low.[20]
Table 3.
The genes deleted in ten Chinese patients with WHS
| Patient number | Deletion genes |
|---|---|
| 1 | ZNF595, ZNF718, ZNF876P, ZNF732, ZNF141, ABCA11P, ZNF721, PIGG, PDE6B, ATP5I, MYL5, MFSD7, PCGF3, LOC100129917, CPLX1, GAK, TMEM175, DGKQ, SLC26A1, IDUA, FGFRL1, RNF212, TMED11P, SPON2, LOC100130872, CTBP1, C4orf42, MAEA, KIAA1530, CRIPAK, FAM53A, SLBP, TMEM129, TACC3, FGFR3, LETM1, WHSC1, SCARNA22, WHSC2, MIR943, C4orf48, NAT8L, POLN, HAUS3, MXD4, ZFYVE28, LOC402160, RNF4, FAM193A |
| 2 | ZNF595, ZNF718, ZNF876P, ZNF732, ZNF141, ABCA11P, ZNF721, PIGG, PDE6B, ATP5I, MYL5, MFSD7, PCGF3, LOC100129917, CPLX1, GAK, TMEM175, DGKQ, SLC26A1, IDUA, FGFRL1, RNF212, TMED11P, SPON2, LOC100130872, CTBP1, C4orf42, MAEA, KIAA1530, CRIPAK, FAM53A, SLBP, TMEM129, TACC3, FGFR3, LETM1, WHSC1, SCARNA22, WHSC2, MIR943, C4orf48, NAT8L, POLN, HAUS3, MXD4, ZFYVE28, LOC402160, RNF4, FAM193A, TNIP2, SH3BP2, ADD1, MFSD10, C4orf10, NOP14, GRK4, HTT, C4orf44, RGS12, HGFAC, DOK7, LRPAP1, LOC100133461, ADRA2C, LOC348926, OTOP1, TMEM128, LYAR, ZBTB49, D4S234E, STX18, LOC100507266, MSX1, CYTL1 |
| 3 | ZNF595, ZNF718, ZNF876P, ZNF732, ZNF141, ABCA11P, ZNF721, PIGG, PDE6B, ATP5I, MYL5, MFSD7, PCGF3, LOC100129917, CPLX1, GAK, TMEM175, DGKQ, SLC26A1, IDUA, FGFRL1, RNF212, TMED11P, SPON2, LOC100130872, CTBP1, C4orf42, MAEA, KIAA1530, CRIPAK, FAM53A, SLBP, TMEM129, TACC3, FGFR3, LETM1, WHSC1, SCARNA22, WHSC2, MIR943, C4orf48, NAT8L, POLN, HAUS3, MXD4, ZFYVE28, LOC402160, RNF4, FAM193A, TNIP2, SH3BP2, ADD1, MFSD10, C4orf10, NOP14, GRK4, HTT, C4orf44, RGS12, HGFAC, DOK7, LRPAP1, LOC100133461, ADRA2C, LOC348926, OTOP1, TMEM128, LYAR, ZBTB49, D4S234E, STX18, LOC100507266, MSX1, CYTL1, STK32B |
| 4 | ZNF595, ZNF718, ZNF876P, ZNF732, ZNF141, ABCA11P, ZNF721, PIGG, PDE6B, ATP5I, MYL5, MFSD7, PCGF3, LOC100129917, CPLX1, GAK, TMEM175, DGKQ, SLC26A1, IDUA, FGFRL1, RNF212, TMED11P, SPON2, LOC100130872, CTBP1, C4orf42, MAEA, KIAA1530, CRIPAK, FAM53A, SLBP, TMEM129, TACC3, FGFR3, LETM1, WHSC1, SCARNA22, WHSC2, MIR943, C4orf48, NAT8L, POLN, HAUS3, MXD4, ZFYVE28, LOC402160, RNF4, FAM193A, TNIP2, SH3BP2, ADD1, MFSD10, C4orf10, NOP14, GRK4, HTT, C4orf44, RGS12, HGFAC, DOK7, LRPAP1, LOC100133461, ADRA2C, LOC348926, OTOP1, TMEM128, LYAR, ZBTB49, D4S234E, STX18, LOC100507266, MSX1, CYTL1, STK32B, C4orf6, EVC2, EVC, CRMP1, JAKMIP1, LOC285484, WFS1, PPP2R2C, MAN2B2, MRFAP1, LOC93622, S100P, MRFAP1L1, CNO, KIAA0232, TBC1D14, LOC100129931, CCDC96, TADA2B, GRPEL1, FLJ36777, SORCS2, PSAPL1, MIR4274, AFAP1-AS1, AFAP1, ABLIM2 |
| 5 | ZNF595, ZNF718, ZNF876P, ZNF732, ZNF141, ABCA11P, ZNF721, PIGG, PDE6B, ATP5I, MYL5, MFSD7, PCGF3, LOC100129917, CPLX1, GAK, TMEM175, DGKQ, SLC26A1, IDUA, FGFRL1, RNF212, TMED11P, SPON2, LOC100130872, CTBP1, C4orf42, MAEA, KIAA1530, CRIPAK, FAM53A, SLBP, TMEM129, TACC3, FGFR3, LETM1, WHSC1, SCARNA22, WHSC2, MIR943, C4orf48, NAT8L, POLN, HAUS3, MXD4, ZFYVE28, LOC402160, RNF4, FAM193A, TNIP2, SH3BP2, ADD1, MFSD10, C4orf10, NOP14, GRK4, HTT, C4orf44, RGS12, HGFAC, DOK7, LRPAP1, LOC100133461, ADRA2C, LOC348926, OTOP1, TMEM128, LYAR, ZBTB49, D4S234E, STX18, LOC100507266, MSX1, CYTL1, STK32B, C4orf6, EVC2, EVC, CRMP1, JAKMIP1, LOC285484, WFS1, PPP2R2C, MAN2B2, MRFAP1, LOC93622, S100P, MRFAP1L1, CNO, KIAA0232, TBC1D14, LOC100129931, CCDC96, TADA2B, GRPEL1, FLJ36777, SORCS2, PSAPL1, MIR4274, AFAP1-AS1, AFAP1, ABLIM2, SH3TC1, HTRA3, ACOX3, METTL19, GPR78, CPZ, HMX1, LOC650293, USP17, USP17L6P, DEFB131, MIR548I2, DRD5, SLC2A9, WDR1, MIR3138, ZNF518B, CLNK, MIR572, HS3ST1, HSP90AB2P, RAB28, LOC285547, NKX3-2, LOC285548, BOD1L, LOC152742 |
| 6 | ZNF595, ZNF718, ZNF876P, ZNF732, ZNF141, ABCA11P, ZNF721, PIGG, PDE6B, ATP5I, MYL5, MFSD7, PCGF3, LOC100129917, CPLX1, GAK, TMEM175, DGKQ, SLC26A1, IDUA, FGFRL1, RNF212, TMED11P, SPON2, LOC100130872, CTBP1, C4orf42, MAEA, KIAA1530, CRIPAK, FAM53A, SLBP, TMEM129, TACC3, FGFR3, LETM1, WHSC1, SCARNA22, WHSC2, MIR943, C4orf48, NAT8L, POLN, HAUS3, MXD4, ZFYVE28, LOC402160, RNF4, FAM193A, TNIP2, SH3BP2, ADD1, MFSD10, C4orf10, NOP14, GRK4, HTT, C4orf44, RGS12, HGFAC, DOK7, LRPAP1, LOC100133461, ADRA2C, LOC348926, OTOP1, TMEM128, LYAR, ZBTB49, D4S234E, STX18, LOC100507266, MSX1, CYTL1, STK32B, C4orf6, EVC2, EVC, CRMP1, JAKMIP1, LOC285484, WFS1, PPP2R2C, MAN2B2, MRFAP1, LOC93622, S100P, MRFAP1L1, CNO, KIAA0232, TBC1D14, LOC100129931, CCDC96, TADA2B, GRPEL1, FLJ36777, SORCS2, PSAPL1, MIR4274, AFAP1-AS1, AFAP1, ABLIM2, SH3TC1, HTRA3, ACOX3, METTL19, GPR78, CPZ, HMX1, LOC650293, USP17, USP17L6P, DEFB131, MIR548I2, DRD5, SLC2A9, WDR1, MIR3138, ZNF518B, CLNK, MIR572, HS3ST1, HSP90AB2P, RAB28, LOC285547, NKX3-2, LOC285548, BOD1L, LOC152742, CPEB2, C1QTNF7, CC2D2A, FBXL5, FAM200B, BST1, CD38, FGFBP1, FGFBP2, PROM1 |
| 7 | ZNF595, ZNF718, ZNF876P, ZNF732, ZNF141, ABCA11P, ZNF721, PIGG, PDE6B, ATP5I, MYL5, MFSD7, PCGF3, LOC100129917, CPLX1, GAK, TMEM175, DGKQ, SLC26A1, IDUA, FGFRL1, RNF212, TMED11P, SPON2, LOC100130872, CTBP1, C4orf42, MAEA, KIAA1530, CRIPAK, FAM53A, SLBP, TMEM129, TACC3, FGFR3, LETM1, WHSC1, SCARNA22, WHSC2, MIR943, C4orf48, NAT8L, POLN, HAUS3, MXD4, ZFYVE28, LOC402160, RNF4, FAM193A, TNIP2, SH3BP2, ADD1, MFSD10, C4orf10, NOP14, GRK4, HTT, C4orf44, RGS12, HGFAC, DOK7, LRPAP1, LOC100133461, ADRA2C, LOC348926, OTOP1, TMEM128, LYAR, ZBTB49, D4S234E, STX18, LOC100507266, MSX1, CYTL1, STK32B, C4orf6, EVC2, EVC, CRMP1, JAKMIP1, LOC285484, WFS1, PPP2R2C, MAN2B2, MRFAP1, LOC93622, S100P, MRFAP1L1, CNO, KIAA0232, TBC1D14, LOC100129931, CCDC96, TADA2B, GRPEL1, FLJ36777, SORCS2, PSAPL1, MIR4274, AFAP1-AS1, AFAP1, ABLIM2, SH3TC1, HTRA3, ACOX3, METTL19, GPR78, CPZ, HMX1, LOC650293, USP17, USP17L6P, DEFB131, MIR548I2, DRD5, SLC2A9, WDR1, MIR3138, ZNF518B, CLNK, MIR572, HS3ST1, HSP90AB2P, RAB28, LOC285547, NKX3-2, LOC285548, BOD1L, LOC152742, CPEB2, C1QTNF7, CC2D2A, FBXL5, FAM200B, BST1, CD38, FGFBP1, FGFBP2, PROM1, TAPT1, FLJ39653, LDB2 |
WHS: Wolf-Hirschhorn syndrome.
In conclusion, the combined use of array CGH and MLPA have increased the detection rate of submicroscopic chromosomal aberrations, and is an effective and specific means to diagnose WHS and allows for the precise identification of the breakpoints and sizes of deletions. This study presented detailed genotype–phenotype correlations in ten Chinese patients with WHS. More clinical and molecular cytogenetic data need to be collected for the study of genotype–phenotype correlations in Chinese patients.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
We thank the patients and their families for participating in this study.
Footnotes
Edited by: Xiu-Yuan Hao and Xin Chen
REFERENCES
- 1.Battaglia A, Filippi T, Carey JC. Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome: Experience with 87 patients and recommendations for routine health supervision. Am J Med Genet C Semin Med Genet. 2008;148C:246–51. doi: 10.1002/ajmg.c.30187. doi:10.1002/ajmg.c.30187. [DOI] [PubMed] [Google Scholar]
- 2.Hirschhorn K, Cooper HL, Firschein IL. Deletion of short arms of chromosome 4-5 in a child with defects of midline fusion. Humangenetik. 1965;1:479–82. doi: 10.1007/BF00279124. [DOI] [PubMed] [Google Scholar]
- 3.Kjaergaard S, Sundberg K, Jørgensen FS, Rohde MD, Lind AM, Gerdes T, et al. Diagnostic yield by supplementing prenatal metaphase karyotyping with MLPA for microdeletion syndromes and subtelomere imbalances. Prenat Diagn. 2010;30:995–9. doi: 10.1002/pd.2604. doi:10.1002/pd.2604. [DOI] [PubMed] [Google Scholar]
- 4.Hemmat M, Hemmat O, Anguiano A, Boyar FZ, El Naggar M, Wang JC, et al. Genotype-phenotype analysis of recombinant chromosome 4 syndrome: An array-CGH study and literature review. Mol Cytogenet. 2013;6:17. doi: 10.1186/1755-8166-6-17. doi:10.1186/1755-8166-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.South ST, Whitby H, Battaglia A, Carey JC, Brothman AR. Comprehensive analysis of Wolf-Hirschhorn syndrome using array CGH indicates a high prevalence of translocations. Eur J Hum Genet. 2008;16:45–52. doi: 10.1038/sj.ejhg.5201915. [DOI] [PubMed] [Google Scholar]
- 6.Boggula VR, Shukla A, Danda S, Hariharan SV, Nampoothiri S, Kumar R, et al. Clinical utility of multiplex ligation-dependent probe amplification technique in identification of aetiology of unexplained mental retardation: A study in 203 Indian patients. Indian J Med Res. 2014;139:66–75. [PMC free article] [PubMed] [Google Scholar]
- 7.Ahn JW, Ogilvie CM, Welch A, Thomas H, Madula R, Hills A, et al. Detection of subtelomere imbalance using MLPA: Validation, development of an analysis protocol, and application in a diagnostic centre. BMC Med Genet. 2007;8:9. doi: 10.1186/1471-2350-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monfort S, Orellana C, Oltra S, Roselló M, Guitart M, Martínez F. Evaluation of MLPA for the detection of cryptic subtelomeric rearrangements. J Lab Clin Med. 2006;147:295–300. doi: 10.1016/j.lab.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Pohovski LM, Dumic KK, Odak L, Barisic I. Multiplex ligation-dependent probe amplification workflow for the detection of submicroscopic chromosomal abnormalities in patients with developmental delay/intellectual disability. Mol Cytogenet. 2013;6:1755–8166. doi: 10.1186/1755-8166-6-7. doi:10.1186/1755-8166-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li MR, Wang XZ, Yang YL, Zhang YH, Xiong H, Bao XH, et al. Multiplex ligation-dependent probe amplification analysis of subtelomeric chromosome rearrangements in children with idiopathic mental retardation (in Chinese) Natl Med J China. 2009;89:2839–42. [PubMed] [Google Scholar]
- 11.Zollino M, Murdolo M, Marangi G, Pecile V, Galasso C, Mazzanti L, et al. On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: Genotype-phenotype correlation analysis of 80 patients and literature review. Am J Med Genet C Semin Med Genet. 2008;148C:257–69. doi: 10.1002/ajmg.c.30190. doi:10.1002/ajmg.c.30190. [DOI] [PubMed] [Google Scholar]
- 12.Zollino M, Orteschi D, Ruiter M, Pfundt R, Steindl K, Cafiero C, et al. Unusual 4p16.3 deletions suggest an additional chromosome region for the Wolf-Hirschhorn syndrome-associated seizures disorder. Epilepsia. 2014;55:849–57. doi: 10.1111/epi.12617. doi:10.1111/epi.12617. [DOI] [PubMed] [Google Scholar]
- 13.Ji TY, Chia D, Wang JM, Wu Y, Li J, Xiao J, et al. Diagnosis and fine localization of deletion region in Wolf-Hirschhorn syndrome patients. Chin Med J. 2010;123:1663–7. [PubMed] [Google Scholar]
- 14.Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, et al. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature. 2009;460:287–91. doi: 10.1038/nature08086. doi:10.1038/nature08086. [DOI] [PubMed] [Google Scholar]
- 15.Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390–7. doi: 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- 16.Shimizu K, Wakui K, Kosho T, Okamoto N, Mizuno S, Itomi K, et al. Microarray and FISH-based genotype-phenotype analysis of 22 Japanese patients with Wolf-Hirschhorn syndrome. Am J Med Genet A. 2014;164A:597–609. doi: 10.1002/ajmg.a.36308. doi:10.1002/ajmg.a.36308. [DOI] [PubMed] [Google Scholar]
- 17.Jiang D, Zhao L, Clish CB, Clapham DE. Letm1, the mitochondrial Ca2+/H+antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc Natl Acad Sci U S A. 2013;110:28. doi: 10.1073/pnas.1308558110. doi:10.1073/pnas.1308558110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maas NM, Van Buggenhout G, Hannes F, Thienpont B, Sanlaville D, Kok K, et al. Genotype-phenotype correlation in 21 patients with Wolf-Hirschhorn syndrome using high resolution array comparative genome hybridisation (CGH) J Med Genet. 2008;45:71–80. doi: 10.1136/jmg.2007.052910. [DOI] [PubMed] [Google Scholar]
- 19.Battaglia A, Filippi T, South ST, Carey JC. Spectrum of epilepsy and electroencephalogram patterns in Wolf-Hirschhorn syndrome: Experience with 87 patients. Dev Med Child Neurol. 2009;51:373–80. doi: 10.1111/j.1469-8749.2008.03233.x. doi:10.1111/j.1469-8749.2008.03233.x. [DOI] [PubMed] [Google Scholar]
- 20.Yang W, Pan H, Wang S, Li L, Wu H, Qi Y. Detection of recurrent 4p16.3 microdeletion with 2p25.3 microduplication by MLPA and aCGH in a fetus from a family with Wolf-Hirschhorn syndrome. Taiwan J Obstet Gynecol. 2016;55 doi: 10.1016/j.tjog.2015.12.006. In press. [DOI] [PubMed] [Google Scholar]
- 21.Zollino M, Lecce R, Fischetto R, Murdolo M, Faravelli F, Selicorni A, et al. Mapping the Wolf-Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region, WHSCR-2. Am J Hum Genet. 2003;72:590–7. doi: 10.1086/367925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okamoto N, Ohmachi K, Shimada S, Shimojima K, Yamamoto T. 109 kb deletion of chromosome 4p16.3 in a patient with mild phenotype of Wolf-Hirschhorn syndrome. Am J Med Genet A. 2013;161A:1465–9. doi: 10.1002/ajmg.a.35910. [DOI] [PubMed] [Google Scholar]