

Abstract
Background:
Stellate ganglion (SG) plays an important role in cardiovascular diseases. The electrical activity of SG neurons is involved in the regulation of the autonomic nervous system. The aim of this research was to evaluate the effects of fluvastatin on the electrophysiological characteristics of SG neurons in a rabbit model of myocardial ischemia (MI).
Methods:
The MI model was induced by abdominal subcutaneous injections of isoproterenol in rabbits. Using whole-cell patch clamp technique, we studied the characteristic changes of ion channels and action potentials (APs) in isolated SG neurons in control group (n = 20), MI group (n = 20) and fluvastatin pretreated group (fluvastatin group, n = 20), respectively. The protein expression of sodium channel in SG was determined by immunohistochemical analysis.
Results:
MI and the intervention of fluvastatin did not have significantly influence on the characteristics of delayed rectifier potassium channel currents. The maximal peak current density of sodium channel currents in SG neurons along with the characteristics of activation curves, inactivation curves, and recovery curves after inactivation were changed in the MI group. The peak current densities of control group, MI group, and fluvastatin group (n = 10 in each group) were −71.77 ± 23.22 pA/pF, −126.75 ± 18.90 pA/pF, and −86.42 ± 28.30 pA/pF, respectively (F = 4.862, P = 0.008). Fluvastatin can decrease the current amplitude which has been increased by MI. Moreover, fluvastatin induced the inactivation curves and post-inactive recovery curves moving to the position of the control group. But the expression of sodium channel-associated protein (Nav1.7) had no significantly statistical difference among the three groups. The percentages of Nav1.7 protein in control group, MI group, and fluvastatin group (n = 5 in each group) were 21.49 ± 7.33%, 28.53 ± 8.26%, and 21.64 ± 2.78%, respectively (F = 1.495, P = 0.275). Moreover, MI reduced the electrical activity of AP and increased amplitude of AP, fluvastatin pretreatment could recover amplitude and electrical activity of AP. The probability of neurons induced continuous APs were 44.44%, 14.29%, and 28.57% in control group, MI group, and fluvastatin group, respectively.
Conclusions:
Fluvastatin pretreatment can recover electrophysiology characteristics of ion channel and AP in SG neurons in a rabbit model of MI. It could be considered as potential method for treating coronary heart diseases.
Keywords: Action Potential, Delayed Rectifier Potassium Channel, Fluvastatin, Myocardial Ischemia, Sodium Channel, Stellate Ganglion
INTRODUCTION
Autonomic nervous system activity exerts potent and diverse effects on cardiac rhythm through elaborate neurocircuitry that is integrated at multiple levels.[1] Among them, parts of the nerve fibers from stellate ganglion (SG) can dominate the heart and be in closely associated with the cardiovascular system. The SG blockade has been used for the treatment of heart disease and protection of the heart,[2,3,4,5,6] which indicated that SG is involved in the signaling transmission of myocardial ischemia (MI) injury. Ion channels of SG neurons are the basis of nerve impulses. Among them, the main physiological functions of sodium channels are propagated action potential (AP).[7] Delayed rectifier potassium channel is one of the major components of AP repolarization course.[8,9] The ion channel directly affects the generation of AP in neurons, also directly affects the release of neurotransmitter in the heart. Therefore, research on characteristics of ion channels and AP of SG neurons in heart diseases is important for the regulation of sympathetic nerve system in the heart.
MI is one of the most common heart diseases in clinical practice. No doubt, sympathetic nerve, including the SG, could regulate the blood pressure and heart rate. A various receptor and protein in sympathetic ganglion participate in the transmission of nociceptive information in MI.[10] Besides prevention of cardiovascular diseases by lipid-regulating effects,[11,12] statins could reduce the nervous injury due to ischemia-reperfusion. They also may prevent postoperative atrial fibrillation through autonomic modulation.[13] Therefore, the aim of this study was to investigate the effects of statins on sympathetic ganglion, and proposed statins would be a novel strategy for the treatment of MI. In this study, we will investigate the electrophysiological characteristics of the sodium channel and delayed rectifier potassium channel, and AP of neurons in a rabbit model of MI pretreated with fluvastatin. Our study will provide a new experimental evidence for the potential role of statins in the treatment of the coronary heart disease.
METHODS
Animal and treatments
Young rabbits (aging 20–40 days, weighing 200–450 g, and no limit to the gender) were provided by China Academy of Military Medical Sciences (Beijing, China). All experimental protocols were approved by the Institutional Animal Care and Use Committee at Tianjin Medical University.
The rabbits were randomly assigned to the following groups: (a) control group (n = 20); (b) isoproterenol (ISO)-induced MI group (MI group, n = 20); and (c) fluvastatin pretreatment group (fluvastatin group, n = 20).
MI was induced by abdominal subcutaneous injections of ISO (85 mg·kg−1·d−1, twice at an interval of 24 h)[14] for 2 consecutive days. The rabbits in fluvastatin group were administered with fluvastatin (10 mg·kg−1·d−1) for consecutive 7 days and received ISO (85 mg·kg−1·d−1, twice at an interval of 24 h) on the 6th and 7th days. The rabbits in the control group were injected with saline. Twenty-four hours after the last treatment, the rabbits were sacrificed.
Stellate ganglion neurons preparation
The method used here was referred to the result described by Schofield and Ikeda[15] and our previous study.[8,16] First, we injected 3% pentobarbital sodium (1 ml/kg) through the auricle vein for anesthesia. The rabbits were executed, neck sympathetic trunk was exposed, then the the SG was removed and placed in the incubation solution. Incubation solution (including NaCl 130 mmol/L, KCl 5 mmol/L, glucose 10 mmol/L, HEPES 10 mmol/L, NaH2PO4 1.5 mmol/L, NaHCO3 25 mmol/L, MgCl2 1 mmol/L, and CaCl2 2 mmol/L) were prepared and continuously bubbled with 95% O2 + 5% CO2 for 15 min, then adjusted the pH to 7.3. Then, SG slices were cut with the thickness of 400–500 μm on the ice pillow, and then incubated for 30 min in above incubation solution at room temperature, at the same time, 95% O2 + 5% CO2 were continuously bubbled. After that, SG slices were taken out and digested in another incubation solution (4 ml, adding 1.9–2.0 g/L collagenase type II [Sigma, USA], 0.600–0.751 g/L pronase E [Sigma] and 7.4–8.0 g/L bovine serum albumin) at 37°C, 95% O2 + 5% CO2 environment for 50–60 min. After digestion, tissue pieces were washed 3 times with incubation solution and SG neurons were dispersed gently by the fine Pasteur glass tubes with different calibers.
The neurons suspension was transferred into a 35-mm culture dish attaching for 30 min and then washed with an external solution and ready for the following experiments. The external solution was composed of NaCl 130 mmol/L, KCl 5 mmol/L, MgCl2 1 mmol/L, glucose 10 mmol/L, HEPES 10 mmol/L, CaCl2 2 mmol/L, which continuously bubbled with 95% O2 + 5% CO2 for 30 min, and then adjusted the pH to 7.3.
Patch-clamp recordings
At 20–25°C room temperature, the data of whole-cell patch clamp experiment was recorded with Axopach 200B patch clamp amplifier (Axon Inc., USA), and stored in a computer with an interface Clampex 10.2 (Axon Inc., USA) data acquisition software. Channel current was recorded by using voltage-clamp mode, the AP was recorded by using current-clamp mode. The glass microelectrodes were pulled by a microelectrode puller (Sutter Instrument Co., USA) and electrodes had resistances of 2–5 MΩ. The tips of glass electrodes were filled with a filtered pipette solution, which composed of CsCl 134 mmol/L, NaCl 5.8 mmol/L, MgCl2 1 mmol/L, HEPES 10 mmol/L, EGTA 3 mmol/L, pH 7.2 (records of sodium channel currents [IK]) or KCl 134 mmol/L, CaCl2 1 mmol/L, MgCl2 2 mmol/L, HEPES 10 mmol/L, EGTA 10 mmol/L, Na2 ATP 2 mmol/L, pH 7.2 (records of delayed rectifier potassium currents [INa] and AP). After forming a conventional “gigaseal,” the membrane was ruptured with a gentle suction to obtain the whole-cell voltage clamp configuration. Capacitances were rapidly canceled, and the series resistance was compensated. Then, stimulus voltages were given, and the activations of channel currents were observed. The leakage currents were subtracted by the P/N procedure. In the records of INa, 2 mmol/L 4-AP, 30 mmol/L TEA-Cl and 0.1 mmol/L CdCl2 were included in the external solution to block transient outward potassium currents, IK, and calcium currents. In the records of IK, 0.1 mmol/L CdCl2 were included in the external solution and neurons were held at a holding potential of −40 mV to block calcium currents, transient outward potassium currents and INa. The blocker was not added in the records of AP. Under an inverted microscope, neurons with a smooth appearance and intense stereoscopic sence were selected for recording. The neurons activity in tissue pieces could maintain within 4–6 h.
Immunohistochemical staining
In this part of the experiment, SGs of three groups were immediately placed in 4% paraformaldehyde. These parts were then embedded in paraffin, sliced perpendicular to the vertical axis of SGs, and then subjected to immunohistochemical staining. Antibodies were anti-Nav1.7 antibody (the dilution ratio: 1:200). Details of the staining techniques were referred to the reference.[17] Nav1.7 densities were determined by a computer-assisted image analysis system (Image Pro Plus 7.0, Media Cybernetics Inc., USA). Each SG tissue was chosen from three slices (between two slices at least 30 μm interval). Because the cross-section of SG was small, each slide was examined under a microscope to select only one field. The computer then automatically calculated the area occupied by the Nav1.7 in the field. The Nav1.7 density was the area occupied by the Nav1.7 area divided by the actual tissue area examined. The mean density of Nav1.7 in these slides was used to represent the Nav1.7 density of that slide.
Statistical analysis
All data were analyzed with Clampfit 10.2 (Axon Inc., USA) for data processing, Origin 6.0 (OriginLab Inc., USA) for image processing, and SPSS 17.0 (SPSS Inc., USA) for statistical analysis. The variables (including peak current densities, potential at half-activation, slope factor and resting potential, etc.) were expressed as mean ± standard deviation (SD), and were compared using two-sample Student's t-test between two groups or one-way analysis of variance (ANOVA) among three groups. P < 0.05 was considered to be statistically significant.
RESULTS
Effects of myocardial ischemia and fluvastatin on current curve of IK
Effects of myocardial ischemia and fluvastatin on I–V curves of IK
The baseline information of all studied rabbits is shown in Table 1. IK was recorded using above pipette solution and an external solution containing blocker. Neurons were held at a holding potential of −40 mV, and stimulated by a series of 160 ms depolarizing steps from −40 mV to + 50 mV (10 mV increment at each step, frequency 0.2 Hz). A series of slowly activated and scarcely inactivated outward currents were obtained. The outward currents appeared slowly and stabilized after 20–40 ms [Figure 1a]. Because the using of 30 mmol TEA-Cl in external solution can completely and reversibly block the outward currents, these outward currents were conformed to be IK.
Table 1.
The baseline information of rabbits in the three groups
| Items | Control group | MI group | Fluvastatin group |
|---|---|---|---|
| Age (days), range | 20–40 | 20–40 | 20–40 |
| Weight (g), range | 200–450 | 200–450 | 200–450 |
| SBP (mmHg), mean ± SD | 105.4 ± 3.3 | 106.6 ± 3.4 | 108.4 ± 3.3 |
| DBP (mmHg), mean ± SD | 87.4 ± 4.0 | 86.8 ± 3.0 | 84.2 ± 2.2 |
| Heart rate (beats/min), mean ± SD | 272.8 ± 30.5 | 283.0 ± 12.4 | 271.4 ± 22.5 |
MI: Myocardial ischemia; SBP: Systolic blood pressure; DBP: Diastolic blood pressure; SD: Standard deviation.
Figure 1.

Effects of myocardial ischemia and fluvastatin on IK of stellate ganglion neurons. (a) Typical examples of IK curves. (b) Effects of myocardial ischemia and fluvastatin on I–V curves of stellate ganglion neurons IK. The peak current densities of control group, myocardial ischemia group, and fluvastatin group were 90.99 ± 31.38 pA/pF, 104.33 ± 22.45 pA/pF, and 91.39 ± 7.73 pA/pF, respectively (n = 10 in each group, F = 0.902, P = 0.407). (c) Effects of myocardial ischemia and fluvastatin on activation curves of IK. The half-activation voltages of the activation curves in control group, myocardial ischemia group, and fluvastatin group were −6.29 ± 0.57 mV, 1.79 ± 0.44 mV, and 0.76 ± 0.44 mV (n = 10 in each group, F = 0.289, P = 0.751), and the slope factors were 19.35 ± 0.60 mV, 19.28 ± 0.59 mV, and 18.95 ± 0.57 mV, respectively (n = 10 in each group, F = 1.141, P = 0.335). Each point represents mean ± standard deviation.
Under the same stimulation mentioned above, IK of control group, MI group, and fluvastatin group were recorded separately. Current-voltage (I–V) curves are shown in Figure 1b, which were plotted by peak current density (peak current amplitude/membrane capacitance) versus different test potentials. The results indicated that peak current densities of control group, MI group, and fluvastatin group (n = 10 in each group) were 90.99 ± 31.38 pA/pF, 104.33 ± 22.45 pA/pF, and 91.39 ± 7.73 pA/pF, respectively (F = 0.902, P = 0.407). In term of I–Vcurve, peak current density of MI group was slightly increased compared with the control group (t = −1.228, P = 0.221). Peak current density of fluvastatin group was reduced to control group. By one-way ANOVA, peak current density between three groups was found no statistically significant difference (P > 0.05).
Effects of myocardial ischemia and fluvastatin on activation kinetics of IK
At a holding potential of −40 mV, IK were obtained with a series of 20 ms step pulses from −40 mV to + 50 mV (10 mV increment for each step, frequency 0.2 Hz), following a hyperpolarizing prepulse of −110 mV for 200 ms. The control group, MI group, and fluvastatin group were given the same stimulation, and then the currents of three groups were recorded. The activation curves for IK of three groups are shown in Figure 1c. Peak amplitude for IK evoked by the step pulses from −40 mV to + 50 mV were converted into conductance by use of the equation:, where G is conductance, V is membrane potential, and Vrev is reversal potential. The activation curves were well fitted to Boltzmann equation: G = I / (V–Vrev), where Gmax is maximum conductance, V1/2 is membrane potential at half-activation, and kis slope factor. Consequently, the values of V1/2 in control group, MI group, and fluvastatin group (n = 10 each group) were −6.29 ± 0.57 mV, 1.79 ± 0.44 mV, and 0.76 ± 0.44 mV, respectively (F = 1.141, P = 0.335). In addition, the values of k in control group, MI group, and fluvastatin group (n = 10 each group) were 19.35 ± 0.60 mV, 19.28 ± 0.59 mV, and 18.95 ± 0.57 mV, respectively (F = 0.289, P = 0.751). Byone-way ANOVA, V1/2 and kamong three groups were found no significantly statistical difference (P > 0.05).
Effects of myocardial ischemia and fluvastatin on current curve of INa
Effects of myocardial ischemia and fluvastatin on I–V curves of INa
INa was recorded using above pipette solution and an external solution containing blocker. Neurons were held at a holding potential of –100 mV, and stimulated by a series of 20 ms depolarizing steps from −100 mV to + 60 mV (10 mV increment at each step, frequency of 0.2 Hz). A series of fast activated and inactivated inward currents were obtained [Figure 2a–2c], so these inward currents were conformed to be INa.
Figure 2.

Effects of myocardial ischemia and fluvastatin on current curves and I–V curves of INa in stellate ganglion neurons. (a-c) Typical examples of INa in control group, myocardial ischemia group, and fluvastatin group. Membrane capacitances are same in three cells. (d) Effects of myocardial ischemia and fluvastatin on I–V curves of INa in stellate ganglion neurons. Currents were elicited with a series of 20 ms depolarizing steps from holding potential of −100 mV to + 60 mV (10 mV increment each step). The amplitude of INa increased after myocardial ischemia and recovered to control level in a certain extent by fluvastatin. The peak current densities of control group, myocardial ischemia group, and fluvastatin group were −71.77 ± 23.22 pA/pF, −126.75 ± 18.90 pA/pF, and −86.42 ± 28.30 pA/pF, respectively (n = 10 in each group, F = 4.862, P = 0.008). Each point represents mean ± standard deviation.
Under the same stimulation mentioned above, sodium currents of control group, MI group, and fluvastatin group were recorded separately. INa traces of three groups are shown in Figure 2a–2c, their membrane capacitance of three curves correspond to the cells were same. I–V curves are shown in Figure 2d, which were plotted by the method mentioned above. The results indicated that peak current densities of control group, MI group, and fluvastatin group (n = 10 in each group) were −71.77 ± 23.22 pA/pF, −126.75 ± 18.90 pA/pF, and −86.42 ± 28.30 pA/pF, respectively (F = 4.862, P = 0.008). There was a statistically significant difference in peak current densities between MI group and control group (n = 10 in each group, t = 2.529, P = 0.012), but no significant difference was found between fluvastatin group and control group (n = 10 in each group, t = −0.066, P = 0.947). In term of I–Vcurve, peak current density of MI group was significantly increased compared with the control group. The amplitudes of INa were increased differently at different membrane potential in a voltage-dependent manner. Peak current density of fluvastatin group was reduced to the level of control group in a voltage-dependent manner.
Effects of myocardial ischemia and fluvastatin on activation and inactivation kinetics of INa
At a holding potential of −100 mV, INa was obtained with a series of 20 ms step pulses from −80 mV to −20 mV (10 mV increment for each step, frequency of 0.2 Hz), following a hyperpolarizing prepulse of −110 mV for 200 ms. Control group, MI group, and fluvastatin group were given the same stimulation, and then the currents of three groups were recorded. The activation curves for INa of three groups are shown in Figure 3a. Consequently, the values of V1/2 in control group, MI group, and fluvastatin group (n = 10 each group) were −50.99 ± 0.18 mV, −40.39 ± 0.12 mV, and –38.45 ± 0.26 mV, respectively (F = 16.087, P < 0.001). In addition, the values of k in control group, MI group, and fluvastatin group (n = 10 in each group) were 2.13 ± 0.34 mV, 1.99 ± 0.48 mV, and 2.13 ± 0.31 mV (F = 1.910, P = 0.168). There was a statistically significant difference in potential at half-activation between MI group and control group (t = −4.686, P < 0.001), but no significant difference was found between fluvastatin group and MI group (t = −1.163, P = 0.260). The activation curve of MI group was shifted toward positive potential, and the activation process was inhibited, and activation curve of fluvastatin group close to MI level. Thus, fluvastatin did not recover the change of activation characteristics of INa by MI.
Figure 3.

Effects of myocardial ischemia and fluvastatin on activation and inactivation process of INa. (a) Effects of myocardial ischemia and fluvastatin on activation process of INa. The half-activation voltages of the activation curves of control group, myocardial ischemia group, and fluvastatin group were −50.99 ± 0.18 mV, −40.39 ± 0.12 mV, and −38.45 ± 0.26 mV (n = 10 in each group, F = 16.087, P < 0.001), and the slope factors were 2.13 ± 0.34 mV, 1.99 ± 0.48 mV, and 2.13 ± 0.31 mV (n = 10 in each group, F = 1.910, P = 0.168). (b) Effects of myocardial ischemia and fluvastatin on inactivation process of INa. The half-inactivation voltages of the inactivation curves of control group, myocardial ischemia group, and fluvastatin group were −77.16 ± 0.47mV, −60.00 ± 0.34 mV, and −72.50 ± 0.45 mV (n = 10 in each group, F = 0.848, P = 0.440), and the slope factors were 10.67 ± 0.44 mV, 7.59 ± 0.31 mV, and 10.74 ± 0.44 mV (n = 10 in each group, F = 14.115, P = 0.036). (c) Effects of myocardial ischemia and fluvastatin on recovery curves after inactivation of INa. The time constants of the recovery curves after inactivation in control group, myocardial ischemia group, and fluvastatin group were 3.96 ± 0.35 ms, 2.55 ± 0.31 ms, and 4.02 ± 0.42 ms (n = 10 in each group, F = 2.704, P = 0.045). Each point represents mean ± standard deviation.
At a holding potential of −100 mV, currents were obtained with a 12 ms test pulse of −30 mV, following prepulse from −120 mV to −20 mV for 500 ms. The inactivation curves for INa of three groups are shown in Figure 3b. Peak amplitudes of INa were normalized. The curves were well-fitted with Boltzmann equation: I/Imax=1/{1+exp[(v-v1/2)/k]}, where V is prepulse potential, V1/2 is membrane potential at half-inactivation, and k is slope factor. The results indicated that the values of V1/2 in control group, MI group, and fluvastatin group (n = 10 in each group) were −77.16 ± 0.47 mV, −60.00 ± 0.34 mV, and −72.50 ± 0.45 mV, respectively (F = 0.848, P = 0.440). In addition, the values of k in control group, MI group, and fluvastatin group (n = 10 in each group) were 10.67 ± 0.44 mV, 7.59 ± 0.31 mV, and 10.74 ± 0.44 mV, respectively (F = 8.115, P = 0.036). There was a statistically significant difference in k between MI group and control group (t = −3.638, P = 0.023), but no significant difference was found between fluvastatin group and MI group (t = −0.893, P = 0.384). The inactivation curve of MI group was shifted toward positive potential, and the inactivation process was inhibited, and the inactivation curve of fluvastatin group recovered to control level. Thus, fluvastatin could recover the change of inactivation characteristics of INa by MI.
At a holding potential of −100 mV, currents were obtained by a series of double-pulses (breadth: 20 ms, amplitude: −20 mV). The intervals of double-pulses were 1, 2, 3, 5, 8, 12, 20, 50, and 100 ms. The recovery curves after inactivation of INa are shown in Figure 3c. Peak amplitudes of INa were normalized. The curves were well-fitted with the monoexponential equation: I/Imax = 1 − exp (−t/τ), where t is recovery time, and τis time constant of channel recovery. The results indicated that the τof the recovery curves after inactivation in control group, MI group, and fluvastatin group (n = 10 in each group) were 3.96 ± 0.35 ms, 2.55 ± 0.31 ms, and 4.02 ± 0.42 ms (F = 6.704, P = 0.045), respectively. For the τ of the recovery curves after inactivation, there was a statistically significant difference between MI group and control group (t = 2.638, P = 0.020), but no significant difference was found between fluvastatin group and control group (t = 0.161, P = 0.874). The recovery curve after inactivation of MI group was shifted toward negative potential, and the recovery process after inactivation was promoted. Recovery curve after inactivation of fluvastatin group was shifted to the level of control group.
Effects of myocardial ischemia and fluvastatin on expression of sodium channel protein
The protein expressions of Nav1.7 in SGs of control group, MI group, and fluvastatin group are shown in Figure 4. The percentages of Nav1.7 protein in control group, MI group, and fluvastatin group (n = 5 in each group) were 21.49 ± 7.33%, 28.53 ± 8.26%, and 21.64 ± 2.78%, respectively [F = 1.495, P = 0.275, Figure 4d], without significant difference. Therefore, the changes of protein distribution of Nav1.7 and sodium channel current in three groups should be relative, fluvastatin also has a certain effect on the distribution of Nav1.7 protein.
Figure 4.
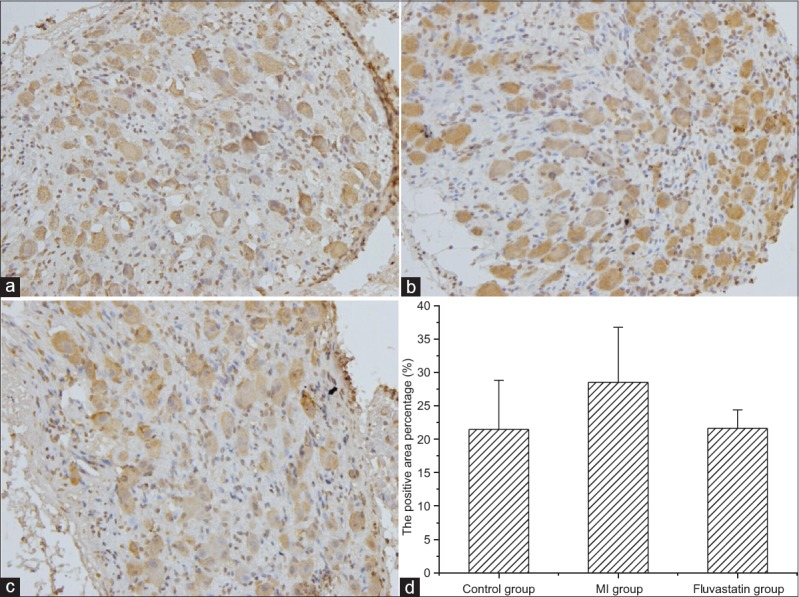
Immunohistochemical staining of Nav1.7 protein in stellate ganglion (original magnification, ×400). Typical examples of Nav1.7 protein in control group (a), myocardial ischemia group (b), and fluvastatin group (c). (d) Histogram of the percentages of Nav1.7 protein in three groups. The percentages of Nav1.7 protein in control group, myocardial ischemia group, and fluvastatin group were 21.49 ± 7.33%, 28.53 ± 8.26%, and 21.64 ± 2.78%, respectively (n = 5 in each group, F = 1.495, P = 0.275).
Effects of myocardial ischemia and fluvastatin on action potential
In current-clamp mode, APs of SG neurons were elicited by positive current 200 pA for 500 ms. Resting potential of control group, MI group, and fluvastatin group (n = 10 in each group) were −59.08 ± 7.60 mV, −64.64 ± 9.39 mV, and −61.57 ± 8.40 mV, respectively, without significant difference (F = 1.368, P = 0.270). The amplitude of AP in control group, MI group, and fluvastatin group (n = 10 in each group) were 87.14 ± 10.00 mV, 105.10 ± 12.57 mV, and 85.13 ± 9.70 mV, respectively (F = 6.668, P = 0.045). For the amplitude of AP, there was a statistically significant difference between MI group and control group (t = −2.515, P = 0.029), but no significant difference was found between fluvastatin group and control group (t = 0.307, P = 0.765). The probability of neurons induced continuous AP (the number of neurons induced continuous AP/the total number of neurons recorded correct AP) in control, MI and fluvastatin groups were 44.44%, 14.29%, and 28.57%, respectively. Thus, electrical activities of SG neurons were reduced, and the amplitude of AP was increased in MI group. Fluvastatin plays a protective effect on the amplitude of AP and electrical activities of SGs.
DISCUSSION
Our study indicated that the characteristics of IK in control group, MI group, and fluvastatin group were not changed significantly. The characteristics (including change of activation and inactivation characteristics in sodium channel) of INa in control group, MI group, and fluvastatin group were changed, but expression of sodium channel-associated protein had no significantly statistical difference among three groups. At the same time, the characteristics of AP in control group, MI group, and fluvastatin group were changed. In MI, the amplitude of AP was increased, but the electrical activity was decreased. Moreover, fluvastatin may recover the changes of AP to a certain degree.
MI is a common heart disease, it will lead to various diseases including cardiac sympathetic nerve remodeling.[17] These sympathetic nerves not only include the sympathetic nerve in the heart, but also include the distal sympathetic nerve innervating the heart such as SGs. According to the latest reports, clinical application of SG blockade has been expanded to patients with heart disease.[2,3,4,5,6] Sympathetic ganglion, especially, the SG, plays the main role in controlling the cardiac sympathetic nerve remodeling. Researches on the mechanism of SG are much less than those in cardiac sympathetic nerves. The objective of this research was to study the distant sympathetic nerves that can dominate heart through the ion channel and AP of SG sympathetic neurons. The distribution of sympathetic neurons in the heart is small, and the single sympathetic neuron is difficult to be isolated. In opposite, the SG tissue block is fit for single neuron isolation and patch-clamp experiments. Therefore, we used SG neurons in neck sympathetic trunk but not in heart sympathetic nerves, and we investigated the effect of fluvastatin on ion channel and AP of SG in MI rabbits. In this study, MI led to significant changes in sodium channel and AP of SG neurons. Therefore, sympathetic ganglions innervating the heart are tightly linked to heart functions under certain conditions. It may serve as a novel target for drug intervention in heart diseases.
The sympathetic ganglions play an important role in the process of dominating the heart, whose efferent impulses would control heart beat frequency and contraction strength. SG neurons could integrate partial information coming from efferent and afferent impulse in heart diseases. In SG, ion channel of neurons influence the issuance of nerve impulses, and it is the basis of electrical activity innervating heart. Nerve impulses, i.e., AP conduction, are an important way to transfer information in the nervous system. Several ion channels participate in this process. Based on previous reports, many diseases are closely associated with abnormal ion channels. There are various ion channels on the membrane of neurons, and different ion channel plays different physiological function. For example, the physiological function of the sodium ion channels is to produce the spread of AP and participate in the zero phase of AP. Delayed rectifier potassium channel is one of the main component of repolarization in AP, and it influenced rate adaptive of AP.[8,9] Our results showed that the electrophysiological characteristics and protein expression of sodium channel were altered in MI. Delayed rectifier potassium channel did not change. Finally, the change of ion channel led to a change of AP, which also affected the transfer of information.
The increase of AP amplitude is associated with an increase of peak current density of INa. The decrease of the probability of neurons induced continuous AP was related not only to a change of diversiform ion channels, but also to the regulation of the central nervous system. For example, the hypothalamus can control function of the autonomic nervous system. Microglial P2X7 receptor in the hypothalamic paraventricular nuclei contributes to sympathoexcitatory responses in acute myocardial infarction rat.[18] The nociceptive information in MI will transfer to the central nervous system. The feedback effect of the central nervous system further affected the electrical activity of SG neurons. In addition, a variety of protein or receptor in SG had a direct impact on release of AP in SG.[19,20] Therefore, the decreasing rate of inducing continuous AP in our results was associated with a variety of mechanisms.
The AP of SG neurons can directly affect the nerve terminal neurotransmitter release in myocardial tissue. MI will lead to serious accumulation of norepinephrine (NE), which has a toxic effect on myocardium and nerve, causing increased morbidity of arrhythmia. The release of NE is closely related to the excitatory of the cell membrane.[17] As our results, MI may induce the change of AP characteristics of SG neurons, which may also cause the regulation of sympathetic nerve on the heart and aggravate the injury of heart diseases.
Statins may favorably possess cardiovascular protective effects in those patients with cardiovascular disease, which is independent of their cholesterol-lowering effects. Statins not only has lipid-lowering efficacy but also protect cardiovascular system directly or indirectly through anti-inflammatory effects, anti-oxidation, anti-thrombus, improving endothelial dysfunction, etc.[21] Fluvastatin is a typical statin possessing protective effect on the heart.[22,23] For instance, fluvastatin ameliorates cardiac sympathetic neural dysfunction in diabetic rats in association with attenuation of increased myocardial oxidative stress.[24] Statins may prevent postoperative atrial fibrillation through autonomic modulation.[13] But its mechanism of cardiovascular protection has not been fully revealed. Our study showed that fluvastatin could partially repair the characteristics of the sodium channel. Moreover, fluvastatin could improve the electrical activity of the sympathetic nerve in MI. The protection of sympathetic nerve electricity activities of SG neurons may be a new target for cardiovascular protection of statins.
In order to ensure that SG neurons for patch-clamp experiments, we need to adopt a young rabbit, which could obtain high quality of neurons compared with the adult rabbits. MI model can be established by ligation operation or drug injection. The ligation operation needs thoracotomy, this can cause a higher risk of death during thoracotomy of young rabbits. MI model induced by ISO injection can significantly reduce the mortality rate of young rabbits. In this method, MI was induced by ISO through shrinking coronary artery, increasing myocardial contraction force and increasing myocardial oxygen consumption, etc. The vasoconstriction process of MI can be simulated by this method, which has some similarities on the pathological features and pathogenesis compared with the human ischemia anoxic MI.[24] Therefore, we chose the ISO injection to induce MI model in young rabbits.
In conclusion, fluvastatin pretreatment before acute MI can recover the characteristics of ion channel and AP in SG neurons of rabbits. The protective mechanism of statins could be achieved through its influence on sympathetic ganglion neurons.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Xin Chen
REFERENCES
- 1.Tan AY, Verrier RL. The role of the autonomic nervous system in cardiac arrhythmias. Handb Clin Neurol. 2013;117:135–45. doi: 10.1016/B978-0-444-53491-0.00012-2. doi: 10.1016/B978-0-444-53491-0.00012-2. [DOI] [PubMed] [Google Scholar]
- 2.Gu Y, Wang L, Wang X, Tang Y, Cao F, Fang Y. Assessment of ventricular electrophysiological characteristics at periinfarct zone of postmyocardial infarction in rabbits following stellate ganglion block. J Cardiovasc Electrophysiol. 2012;23(Suppl 1):S29–35. doi: 10.1111/j.1540-8167.2012.02437.x. doi: 10.1111/j.1540-8167.2012.02437.x. [DOI] [PubMed] [Google Scholar]
- 3.Patel RA, Priore DL, Szeto WY, Slevin KA. Left stellate ganglion blockade for the management of drug-resistant electrical storm. Pain Med. 2011;12:1196–8. doi: 10.1111/j.1526-4637.2011.01167.x. doi: 10.1111/j.1526-4637.2011.01167.x. [DOI] [PubMed] [Google Scholar]
- 4.Chen YQ, Jin XJ, Liu ZF, Zhu MF. Effects of stellate ganglion block on cardiovascular reaction and heart rate variability in elderly patients during anesthesia induction and endotracheal intubation. J Clin Anesth. 2015;27:140–5. doi: 10.1016/j.jclinane.2014.06.012. doi: 10.1016/j.jclinane.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 5.Boe BA, Webster G, Asher Y, Tsao S, Suresh S, Steinhorn DM. Percutaneous, ultrasound-guided stellate ganglion nerve block suppresses recurrent ventricular fibrillation in an infant awaiting heart transplant. Circ Arrhythm Electrophysiol. 2012;5:e93–4. doi: 10.1161/CIRCEP.112.974329. doi: 10.1161/CIRCEP. 112.974329. [DOI] [PubMed] [Google Scholar]
- 6.Na S, Kim OS, Ryoo S, Kweon TD, Choi YS, Shim HS, et al. Cervical ganglion block attenuates the progression of pulmonary hypertension via nitric oxide and arginase pathways. Hypertension. 2014;63:309–15. doi: 10.1161/HYPERTENSIONAHA.113.01979. doi: 10.1161/HYPERTENSIONAHA.113.01979. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Tu H, Zhang D, Zheng H, Li YL. Voltage-gated sodium channel expression and action potential generation in differentiated NG108-15 cells. BMC Neurosci. 2012;13:129. doi: 10.1186/1471-2202-13-129. doi: 10.1186/1471-2202-13-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li G, Cheng L, Qiao X, Ling L. Characteristics of delayed rectifier potassium channels exposed to 3 mT static magnetic field. IEEE Trans Magn. 2010;46:2635–8. doi: 10.1109/TMAG.2010.2045389. [Google Scholar]
- 9.Tian YT, Liu ZW, Yao Y, Yang Z, Zhang T. Effect of alpha-cypermethrin and theta-cypermethrin on delayed rectifier potassium currents in rat hippocampal neurons. Neurotoxicology. 2009;30:269–73. doi: 10.1016/j.neuro.2009.01.001. doi: 10.1016/j.neuro.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Liu S, Xu B, Li G, Li G, Huang A, et al. Study of baicalin on sympathoexcitation induced by myocardial ischemia via P2X3 receptor in superior cervical ganglia. Auton Neurosci. 2015;189:8–15. doi: 10.1016/j.autneu.2014.12.001. doi: 10.1016/j.autneu.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Laufs U, Custodis F, Böhm M. HMG-CoA reductase inhibitors in chronic heart failure: Potential mechanisms of benefit and risk. Drugs. 2006;66:145–54. doi: 10.2165/00003495-200666020-00002. doi: 10.2165/00003495-200666020-00002. [DOI] [PubMed] [Google Scholar]
- 12.Ambrosi P. Short-term clinical effects of periprocedural statin therapy. Presse Med. 2013;42:261–8. doi: 10.1016/j.lpm.2012.05.016. doi: 10.1016/j.lpm.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 13.Liu T, Li GP. Statins may prevent postoperative atrial fibrillation through autonomic modulation. Am J Cardiol. 2006;97:1266. doi: 10.1016/j.amjcard.2005.11.016. doi: 10.1016/j.amjcard.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 14.Senthil S, Sridevi M, Pugalendi KV. Cardioprotective effect of oleanolic acid on isoproterenol-induced myocardial ischemia in rats. Toxicol Pathol. 2007;35:418–23. doi: 10.1080/01926230701230312. doi: 10.1080/01926230701230312. [DOI] [PubMed] [Google Scholar]
- 15.Schofield GG, Ikeda SR. Sodium and calcium currents of acutely isolated adult rat superior cervical ganglion neurons. Eur J Physiol. 1988;411:481–90. doi: 10.1007/BF00582368. doi: 10.1007/BF00582368. [DOI] [PubMed] [Google Scholar]
- 16.Li G, Cheng LJ, Qiao XY, Lin L. Characteristics of Delayed Rectifier Potassium Channels Exposed to 3 mT Static Magnetic Field. IEEE Transactions on Magnetics. 2010;46:2635–8. doi: 10.1109/TMAG.2010.2045389. [Google Scholar]
- 17.Wu X, Jiang H, Yu L, Hu X, Liu W. Desipramine pretreatment improves sympathetic remodeling and ventricular fibrillation threshold after myocardial ischemia. J Biomed Biotechnol 2012. 2012 doi: 10.1155/2012/732909. 732909. doi: 10.1155/2012/732909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du D, Jiang M, Liu M, Wang J, Xia C, Guan R, et al. Microglial P2X7 receptor in the hypothalamic paraventricular nuclei contributes to sympathoexcitatory responses in acute myocardial infarction rat. Neurosci Lett. 2015;587:22–8. doi: 10.1016/j.neulet.2014.12.026. doi: 10.1016/j.neulet.2014.12.026. [DOI] [PubMed] [Google Scholar]
- 19.Hernández-Ochoa EO, Prosser BL, Wright NT, Contreras M, Weber DJ, Schneider MF. Augmentation of Cav1 channel current and action potential duration after uptake of S100A1 in sympathetic ganglion neurons. Am J Physiol Cell Physiol. 2009;297:C955–70. doi: 10.1152/ajpcell.00140.2009. doi: 10.1152/ajpcell.00140.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luther JA, Birren SJ. Nerve growth factor decreases potassium currents and alters repetitive firing in rat sympathetic neurons. J Neurophysiol. 2006;96:946–58. doi: 10.1152/jn.01078.2005. doi: 10.1016/j.compositesa.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 21.Hu YF, Chen YC, Cheng CC, Higa S, Chen YJ, Chen SA. Fluvastatin reduces pulmonary vein spontaneous activity through nitric oxide pathway. J Cardiovasc Electrophysiol. 2009;20:200–6. doi: 10.1111/j.1540-8167.2008.01281.x. doi: 10.1111/j.1540-8167.2008.01281.x. [DOI] [PubMed] [Google Scholar]
- 22.Schouten O, Boersma E, Hoeks SE, Benner R, van Urk H, van Sambeek MR, et al. Fluvastatin and perioperative events in patients undergoing vascular surgery. N Engl J Med. 2009;3(361):980–9. doi: 10.1056/NEJMoa0808207. doi: 10.1056/NEJMoa0808207. [DOI] [PubMed] [Google Scholar]
- 23.Zhou R, Xu Q, Zheng P, Yan L, Zheng J, Dai G. Cardioprotective effect of fluvastatin on isoproterenol-induced myocardial infarction in rat. Eur J Pharmacol. 2008;586:244–50. doi: 10.1016/j.ejphar.2008.02.057. doi: 10.1016/j.ejphar.2008.02.057. [DOI] [PubMed] [Google Scholar]
- 24.Matsuki A, Nozawa T, Igarashi N, Sobajima M, Ohori T, Suzuki T, et al. Fluvastatin attenuates diabetes-induced cardiac sympathetic neuropathy in association with a decrease in oxidative stress. Circ J. 2010;74:468–75. doi: 10.1253/circj.cj-09-0402. doi: 10.1253/circj.CJ-09-0402. [DOI] [PubMed] [Google Scholar]