

Abstract
Mitral valve disease is a frequent cause of heart failure and death. Emerging evidence indicates that the mitral valve is not a passive structure, but—even in adult life—remains dynamic and accessible for treatment. This concept motivates efforts to reduce the clinical progression of mitral valve disease through early detection and modification of underlying mechanisms. Discoveries of genetic mutations causing mitral valve elongation and prolapse have revealed that growth factor signalling and cell migration pathways are regulated by structural molecules in ways that can be modified to limit progression from developmental defects to valve degeneration with clinical complications. Mitral valve enlargement can determine left ventricular outflow tract obstruction in hypertrophic cardiomyopathy, and might be stimulated by potentially modifiable biological valvular–ventricular interactions. Mitral valve plasticity also allows adaptive growth in response to ventricular remodelling. However, adverse cellular and mechanobiological processes create relative leaflet deficiency in the ischaemic setting, leading to mitral regurgitation with increased heart failure and mortality. Our approach, which bridges clinicians and basic scientists, enables the correlation of observed disease with cellular and molecular mechanisms, leading to the discovery of new opportunities for improving the natural history of mitral valve disease.
Introduction
Mitral valve insufficiency is a major source of morbidity and death worldwide and a frequent cause of heart failure, with complications that include arrhythmia, endocarditis, and sudden cardiac death.1,2 Structural deficiencies in the mitral valve and secondary changes induced by abnormal ventricular size and deformation are implicated in the development of these valvular lesions.3 However, the effect of mitral insufficiency on cardiac function is more than purely mechanical, whereby pump function is maintained at the expense of elevated filling pressures, but extends to impaired contractility and electrical instability.
Mitral valve diseases (MVDs) that lead to valve insufficiency have long been conceived as inexorable processes (Figure 1a) for which therapeutic options are limited to surgical valve repair or replacement, with the underlying mechanisms inaccessible. The under lying genetic mutations and mechanisms of mitral valve dysfunction associated with myxomatous degeneration have remained elusive.4 In mitral valve lesions associated with ventricular disease, the leaflets are considered biologically passive and fixed in size relative to the enlarged or narrowed ventricle and not an available target for therapy.5 Valve plasticity—defined as the potential for change in cellular phenotype and behaviour—and altered leaflet matrix and micromechanics, have been considered in studies of valve development. However, MVD had not been associated with modified adult valve biology until the 2004 discovery in a mouse model of Marfan syndrome, in which a mutation in an extra cellular matrix (ECM) protein alters the regulation of aorta and valve cell biology and thereby creates opportunities for modifying the course of disease.6 We believe that the mitral valve is not a passive structure, but—even in adult life—a ‘living valve’ that undergoes active changes.3 The interface between valve imaging, genetics, biology, and biomechanics can help us to understand the mechanisms of mitral valve function and those that might be applied to novel therapies for mitral regurgitation (Figure 1b). Our mission is to reduce the clinical progression of MVD by early detection and modification of underlying mechanisms.
Figure 1.
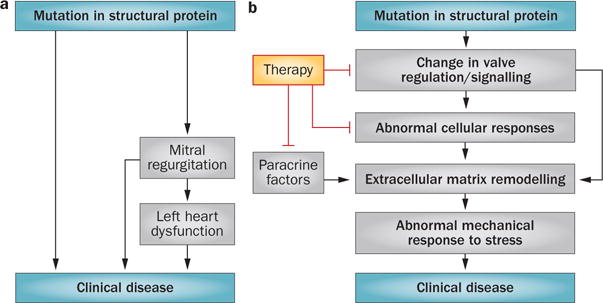
Models of mitral valve disease. a | Previous conceptual model of mitral valve disease caused by mutations in structural proteins. In this model, the mutations impair valve biomechanical integrity and thereby cause mitral regurgitation and clinical disease. Therapeutic options are limited to surgical valve repair or replacement, as the underlying mechanisms are inaccessible. b | Our conceptual model of mitral valve disease caused by mutations in structural proteins. This model is amenable to molecular therapy. In addition to impairing valve biomechanical integrity, mutations in structural proteins can also alter valve regulatory and signalling pathways, leading to abnormal cellular responses and extracellular matrix remodelling. Paracrine factors provide further therapeutic targets.
This approach can also be applied to conditions that lead to mitral regurgitation, in which the mitral leaflets are either too long or small in relation to the remodelling left ventricle, or where they are too stiff or compliant. For example, in patients with mitral valve prolapse (MVP), the leaflets are excessively long and typically show myxomatous degeneration with increased leaflet compliance.7–9 In ischaemic and functional mitral regurgitation, which often occurs in patients surviving a myocardial infarction (MI) or with dilated cardiomyo‐pathy,10 the leaflet area is deficient relative to the dilated left ventricle,11,12 and the leaflets are stiffer and undergo fibrosis,13 so that any compensatory leaflet remodelling is insufficient to prevent regurgitation. In individuals with hypertrophic cardiomyopathy (HCM), although the underlying genetic mutation is associated with a sarcomeric protein, elongated leaflets contribute to both mitral regurgitation and left ventricular outflow tract (LVOT) obstruction, and are disproportionate to the reduced cavity size and abnormal LVOT.14–16
Imaging, most commonly cardiac ultrasonography, has a major role in the understanding of these structural mechanisms,17 and reveals mitral valve biology by allowing correlations of valve structure, biomechanics, and haemodynamics with basic cellular, molecular, and genetic findings and, consequently, the development of new physiological therapies. In this Review, we provide an understanding of the mitral valve as a functional and interactive organ, beginning with consideration of how normal valve structure and biomechanical function are ideally matched as a result of choreographed development. We then review how inherited and acquired perturbations alter valve function, and consider how we can access controlling pathways to maintain physiological heart function and prevent clinically apparent disease.
Mitral valve structure
The mitral valve has two leaflets: the anterior leaflet, contiguous with the aorto‐mitral curtain; and the posterior leaflet composed of three scallops, P1–P3 (Figure 2a). Both leaflets attach to the dynamic mitral annulus at their basal ends, whereas multiple chordae tendineae emerge from the ventricular surfaces and attach distally to the papillary muscles. This complex shape enables a delicate balance of force to sustain unidirectional blood flow through the large and dynamic orifice of the mitral valve. Both leaflets are divided into three radially located zones from annulus to free edge, with locally unique ultrastructural composition and biomechanics. The annulus‐to‐leaflet transition zone, which spans ~1 cm from the mitral annulus radially toward the leaflet edge, contains atrial myocytes18,19 and cells that express smooth muscle αactin.20 Nerve fibres and terminals extend into this region from the mitral annulus, maintaining electro‐physiological continuity with the rest of the heart,21,22 and small blood vessels span this leaflet region.19,23–25 The translucent smooth zone that forms the central belly of the leaflet consists of thin, but dense, collagenous networks and vimentin‐positive fibroblasts that actively syn‐thesize the ECM in response to mechanical loading.26 The distal rough zone at the leaflet edge is thick and rich in glycosaminoglycans and retains water to sustain the compressive stresses from coaptation.27 The cross‐sectional structure of the normal mitral leaflets is analogous to the aortic valve, with three well‐defined tissue layers—the atrialis, spongiosa, fibrosa/ventricularis (Figure 2b and Figure 3)—each with a different thickness, characteristic cells, matrix composition, and biomechanical features that contribute to its proper function. The atrialis layer is covered with endothelium and contains a subendothelial connective tissue layer of lamellar collagen and elastin sheets, which extends from the left atrial endocardium into the leaflet. This layer sustains high‐frequency, large tensile deformations in systole, with the interspersed elastin enabling elastic recoil in diastole. The presence of left atrial myocytes and smooth muscle cells adjacent to nerve endings can transiently modulate leaflet stiffness and deformation during closure.28 Just beneath the atrialis is the spongiosa, which contains loosely arranged collagen and is rich in glycosaminoglycans; this layer gets thinner towards the mitral annulus, and becomes prominent towards the leaflet free edge. Retention of water in the spongiosa aids in resisting compressive loading imposed by coaptation of the leaflets.27 The fibrosa is connected to the mitral annulus and faces the high‐pressure left ventricle; this layer is composed predominantly of collagen fibres that are arranged parallel to the leaflet free edge, are densely packed, and flexibly crimped or wavy.29 At the sites of chordal insertion, the fibrosa transitions from a planar collagenous structure to a cylindrical collagenous chord that enables the gradual transition of forces between the chordae and leaflets without regional kinking.24,30 Conversely, the chordae tendineae are composed of a cylindrical collagen core within an elastin sheath, and have demonstrable viscoelastic properties—their extensibility decreases with increasing strain rates and varies with chordal size and location.31,32
Figure 2.
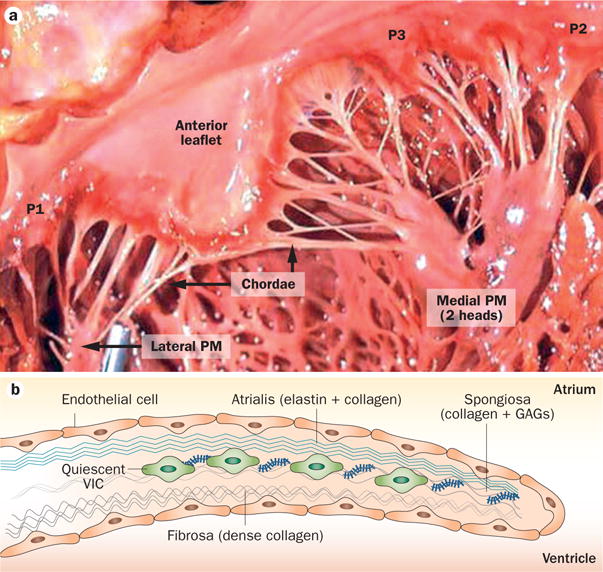
Mitral valve structure. a | The mitral valve has two leaflets—the anterior leaflet below the aortic valve, and the posterior leaflet composed of three scallops: P1, P2, and P3. At the leaflet edges, the chordae tendineae link the leaflets to the PM anchors on the left ventricular wall. b | The leaflet structure is well organized, with a dense fibrous layer of collagen, known as the fibrosa, which arises from the mitral annulus and faces the left ventricle. The fibrosa is thicker close to the annulus and thinner at the edge of the leaflet. The fibrosa is covered by the spongiosa, a loose connective tissue layer rich in GAGs. The spongiosa is thicker at the leaflet edge and thinner at the annulus. The atrialis is made up of lamellar collagen and elastin sheets, which extend from the left atrial endocardium into the leaflet. Endothelial cells cover the valve. The deep subendothelial layers contain quiescent VICs—noncontractile, fibroblast-like cells that originate from endocardial endothelial cells. Abbreviations: GAG, glycosaminoglycan; PM, papillary muscle; VIC, valvular interstitial cell.
Figure 3.
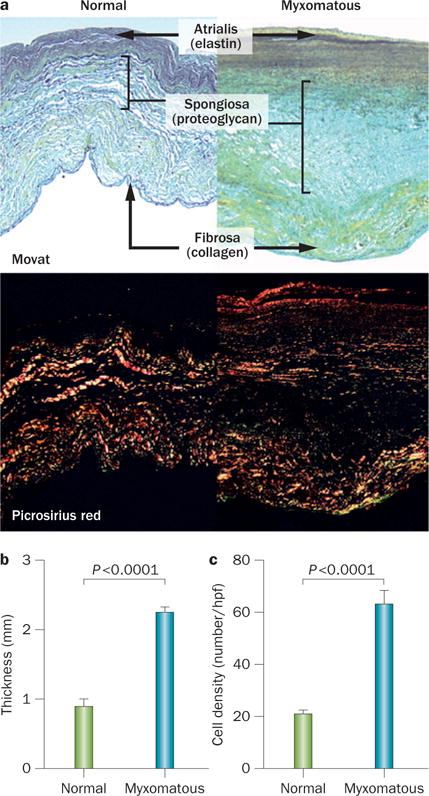
Morphological features of normal and myxomatous mitral valves. a | Normal mitral valves (left) and valves with myxomatous degeneration (right). Myxomatous valves have an abnormal layered architecture: loose collagen in fibrosa, expanded spongiosa strongly positive for proteoglycans, and disrupted elastin in atrialis. Top: Movat pentachrome stain (collagen stains yellow; proteoglycans, blue-green; and elastin, black). Bottom: Picrosirius red staining viewed under polarized light detected disruption and lower birefringence of collagen fibres in myxomatous leaflets. Magnification ×100. b | Quantitative analysis of valve thickness, demonstrating thickening of myxomatous valves. c | Increased density of interstitial cells in myxomatous spongiosa. Bars indicate SEM. Abbreviation: hpf, high-power field. Reprinted from Rabkin, E. et al. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation 104 (21), 2525–2532 (2001).
The leaflets are populated with cells that have unique features and sustain valve homeostasis throughout life.33 The leaflets are covered by a continuous endothelial cell layer on both atrial and ventricular aspects. The deep sub‐endothelial layers contain mainly valvular interstitial cells (VICs), a population of cells unique to cardiac valves34 that originate from endocardial endothelial cells.35–38 Quiescent VICs are noncontractile fibroblastlike cells, do not express smooth muscle αactin, but are positive for vimentin and chondromodulin1, and contribute to homeostatic remodelling of matrix constituents with relatively low turnover.39 During tissue remodelling in embryonic development and conditions such as myxomatous degeneration, epicardial‐derived cells migrate into the posterior leaflet,40,41 and VICs transition to an activated myofibroblast‐like phenotype, expressing smooth muscle‐associated contractile proteins, matrix metallo‐proteinases, and inflammatory cytokines that rapidly remodel the extracellular milieu.42–44 Many of the same factors that modulate postnatal VIC phenotype in MVD also have important functions during embryonic development, suggesting that the VIC phenotype is a common link between primitive embryonic mesenchyme and postnatal plasticity to adapt to the dynamic valve environment. Mitral valve endothelial and interstitial cells have reciprocal interactions that favour normal homeostasis.45
Mitral valve function during the cardiac cycle is remarkably dynamic in response to myocardial contraction and varying transvalvular pressure. The mitral annulus undergoes a reduction in presystolic area contributed by left atrial contraction, which facilitates early leaflet motion and adequate leaflet coaptation.46 When such atriogenic annular reduction is ablated, equivalent reduction in the mitral annulus is possible via ventriculogenic contraction, but this contraction is delayed and results in increased mitral regurgitation.46 The dynamic interaction between the surrounding myocardial structures and the valve leaflets governs the leaflet coaptation47 and, ultimately, the mechanical strain and stress on the valve extracellular matrix and cells.48–50 Under normal conditions in healthy individuals, the anterior leaflet undergoes large deformations with peak circumferential strains (parallel to the mitral annulus) ranging from 2.5% to 5.0%, and peak radial strains (perpendicular to the mitral annulus) ranging from 7.8% to 22.0%.29,51–53 In the circumferential direction, peak strains are concentrated in the free edge of the leaflet, as the leaflets are stretched between the marginal chordae. In the radial direction, peak strains are concentrated in the soft belly region of the anterior leaflet, because that leaflet has the largest systolic curvature.51,54 Principal leaflet strains rotate from the circumferential direction during diastole to the radial direction in systole; this rotation corresponds with the diastolic tension in the secondary chordae that controls leaflet motion into the LVOT, and the large systolic leaflet curvature and chordal tethering that increase radial leaflet strain.51,54 These dynamic shifts in leaflet strains from diastole to systole align with orientation of the collagen and elastin fibril networks in the leaflet. Although tempting to postulate that optimal valve biomechanics are supported by such favourable fibre orientation, this orientation might actually be the result of deformations during fetal and neonatal valve development. Anecdotal evidence of this hypothesis is seen in patients with stunted left ventricular growth, in whom loss of transmitral blood flow or strain results in an immature valve with chordal fusion and immobile leaflets.55 An abundance of glycosaminoglycans in the spongiosa indicates that an inherent viscoelastic and strain rate‐dependent behaviour exists in the valve leaflets.
The adaptive matching of leaflet biomechanics to the needs for normal leaflet excursion and closure indicates why altered biomechanics in disease might impair coaptation. Transfer of the mechanical strains imposed on the ECM to the resident valve cells might also govern valve homeostasis and behaviour. While the matrix might dampen the magnitude of cellular strains, VICs cultured at cyclic matrixlevel strains respond with altered expression of collagens, proteoglycans, and growth factors such as those of the transforming growth factor β (TGFβ) family.56–60 Furthermore, VIC‐mediated tissue stiffness can be modulated by the endothelium‐dependent mediators nitric oxide and endothelin acting on contractile VIC phenotypes.61 In addition to this interplay between mechanics and biology, nonphysiological oscillatory flows induce inflammatory activation of the valvular endothelium that can induce VIC activation, but the precise mechanisms are still unknown.62 Different shear stress patterns on the inflow and outflow valve surfaces might induce heterogeneous mechanical responses.33 The inherent dynamism of valve action, involving both tissue and fluid stresses, strongly suggests that these diverse signals are integrated during normal valvular cell function. Disease states perturb the mechanical strains and ECM composition and can shift these dynamic equilibria towards pathological remodelling. Understanding this interplay of biomechanics and valve biology can substantially inform long‐term repair strategies, to promote beneficial as opposed to detrimental changes.
Normal mitral valve dynamics and competence also determine the fluid dynamics of left ventricular filling and ejection. During early‐to‐mid diastolic filling, vortical flow structures that form beneath the anterior leaflet direct the inflow towards the aortic valve and enable systolic ejection without substantial kinetic energy losses.63 Elongation of the anterior leaflet, which occurs in patients with hypertrophic obstructive cardiomyopathy, or loss of annular dynamics and restricted leaflet motion, as seen in individuals with functional mitral regurgitation, might alter these vortical structures and impair efficient ventricular pumping.64 Loss of normal vortical structures might also increase residual blood in the left ventricle between consecutive cardiac beats, and therefore increase diastolic filling pressures, cause adverse ventricular dilatation, and biological remodelling.65 As flow‐based imaging to detect cardiac dysfunction emerges, the role of the mitral valve and its lesions in governing efficient ventricular pumping might be further revealed.66
Mitral valve development
Early mitral valve development is complex and errors in early morphogenesis can contribute to MVD.67–72 The first cell‐fate decision in the embryo that contributes to mitral valve formation is the segregation of the endocardial and myocardial lineages.73,74 This process occurs before, or at the onset of, gastrulation within the cranial and cardiac lateral plate mesoderm, marked by the transcription factor mesoderm posterior protein 1, or earlier in the mesoderm when haematopoietic and cardiac lineages are determined by brachyury protein‐expressing cells.74 These prospective valve cells emerge as vascular endothelial growth factor receptor 2 (VEGFR2)‐positive cells.74 Some VEGFR2‐positive cells arising when cells of the second heart field migrate from the pharyngeal mesoderm are also likely to contribute to the endocardium (Figure 4).74
Figure 4.
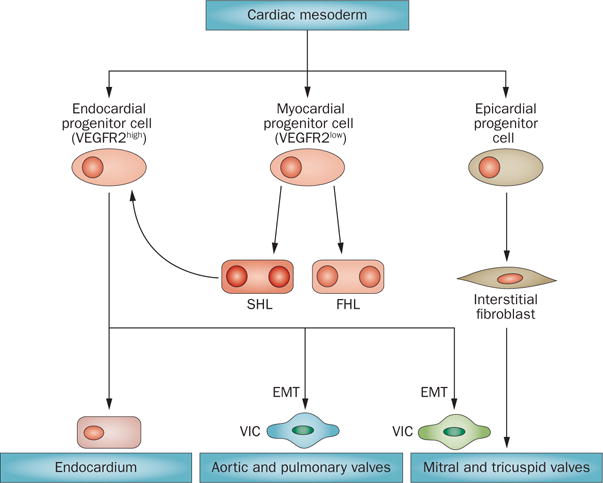
Cell lineages that contribute to valve formation. The myocardial and endocardial cells are segregated at the onset or just after gastrulation in the E7.5 mouse embryo. This process occurs before, or at the onset of, gastrulation within the cardiac mesoderm. Endocardial progenitor cells emerge highly expressing VEGFR2. These cells undergo EMT to give rise to different cell types that participate in the valve leaflets. Some of these prospective valve cells are also likely to contribute to the endocardium. Myocardial progenitor cells, expressing only low levels of VEGFR2, give rise to both the FHL and SHL. SHL cells increase their expression of VEGFR2 and become endocardial progenitor cells. Epicardial progenitors also give rise to cells found within the valves. Abbreviations: EMT, endothelial-to-mesenchymal transition; FHL, first heart lineage; SHL, second heart lineage; VEGFR2, vascular endothelial growth factor receptor 2; VIC, valve interstitial cell.
Mitral valve development begins soon after looping of the early embryonic heart tube. In response to growth factors including several proteins of the TGFβ super family (TGFβ1–3, BMP2–4), a subset of the endocardial cells of the early embryonic heart become activated and transform into a mesenchymal phenotype, known as endothelial‐to‐mesenchymal transition (EMT; Figure 5).71,75 The endothelial cells that do not undergo EMT express NFATc1, which suppresses important transcriptional factors needed for the phenotypic transition of the cells to proliferate and elongate to form the maturing leaflets.76 NFATc1 limits EMT by suppression of its positive regulators Snail and Slug.69 The molecular and cellular events that occur after this initial EMT are complex and poorly understood. Post‐EMT leaflet maturation involves migration and proliferation of mesenchymal cells to distend the forming cushions into the lumen, under the control of growth factors,72,75,77 followed by their gradual thinning and elongation. One hypothesis for this leaflet maturation is that cell proliferation results in polarized, aligned cell arrays that collectively migrate and extrude leaflet tissue into the lumen, referred to as a convergent extension process.69 Indeed, microscopic analysis of developing valves shows alignment of cells during post‐EMT and neonatal development.69 Concomitant with the early remodelling phase is differentiation of the cushion mesenchyme into interstitial cells.74 Based on this fundamental change in cell phenotype, the matrix surrounding these fibroblasts matures into a highly organized lamellar and fibrous tissue, making the valve more rigid and thereby able to handle the increasing haemodynamic load of the beating heart.74 Slight defects in any of these fundamental processes might lead to structural and functional valve disease that presents in postnatal life and can progress into degenerative changes in adult life.
Figure 5.
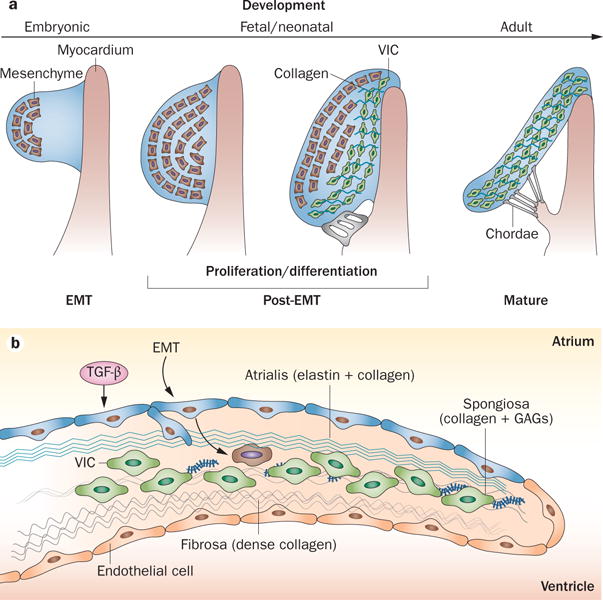
Mitral valve growth and development. a | Mitral valve development from an endocardial cushion. The cells undergo a process of EMT to generate mesenchyme (brown cells) that invades the underlying matrix. These cells undergo proliferative expansion and differentiate into collagen-secreting VICs (green) that become progressively aligned along the proximal–distal tissue axis. Finally, the valve elongates and thins resulting in a mature valve comprised of stratified and aligned cells and matrix. b | Valve growth and response to stress. In a growing or stressed valve, or a valve stimulated by TGF-β, endothelial cells undergo EMT, increasing the number of matrix-producing interstitial cells. Abbreviations: EMT, endothelial-to-mesenchymal transition; GAG, glycosaminoglycan; TGF-β, transforming growth factor β; VIC, valve interstitial cell.
Mitral valve ageing
While mitral valve structure remains largely preserved with age, ultrastructural and cellular changes that indicate chronic mechanical loading have been reported. For example, some investigators have reported high cellularity, closely packed, parallel, and thick collagen fibres, very few elastic fibres, and dense muco‐polysaccharides in the valves of children aged >10 years.78 Conversely, less cellular, thick, dense, nonparallel collagen bundles, with slightly increased number of elastic fibres, and reduced density of muco‐polysaccharides were observed in mitral valves of individuals in their second to fifth decades of life.78 Finally, marked reduction in cellularity, disoriented collagen fibres, and increased elastin fibres with severely reduced muco‐polysaccharides were seen in valves of those aged ≥60 years.78 Other investigators have identified age‐related changes that differ between the anterior and posterior leaflets.79 A study from patients aged 72–91 years without cardiac disease confirmed the well‐known calcification concentrated along the annulus or chordal insertion regions, but remarkably minimal changes in leaflet mechanical properties.80 Unlike the semilunar cardiac valves, age‐dependent biological changes in the mitral valve are not well understood.
Mitral valve prolapse
Echocardiographic diagnosis
MVP affects nearly one in 40 individuals,81 and is the leading surgical indication for mitral regurgitation.82 Although screening of the general population indicates that many individuals with MVP have few clinical symptoms,81,83 in large clinical populations the disease is strongly associated with heart failure, need for valve surgery, endocarditis, and death in the ~20% of those with moderate‐to‐severe mitral regurgitation and left ventricular ejection fraction <50%.84
Echocardiography has been central to the diagnosis of MVP as leaflet displacement beyond the normal range relative to the mitral annulus.17 Diagnostic specificity is required to study the genetic mutations associated with this MVP. Older studies lacked this specificity and suggested that MVP prevalence was as high as 10–15% or more,85 often on the basis of 2D views that seemed to show prolapse because of the normal saddle shape of the valve, without actual pathological leaflet displacement.81,85–89 3D echocardiographic demonstration of this saddle shape led to recognition that prolapse could be defined most specifically by leaflet displacement relative to the anterior and posterior high points of the annular saddle when imaged in long‐axis views (Figure 6a).87–89 Additional assessment focuses on leaflet displacement relative to each other, leading to malcoaptation.90,91 New diagnostic criteria have revised the prevalence of MVP to 2.4% without loss of sensitivity.81 Improved diagnostic specificity has also provided a firm foundation for genetic studies of this frequently inherited disease.92
Figure 6.
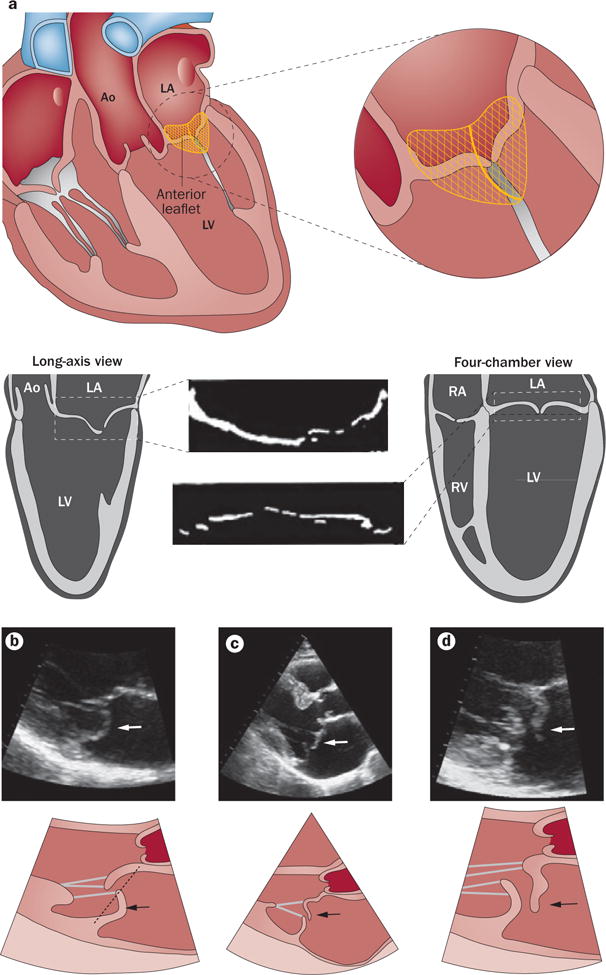
Echocardiographic diagnosis of mitral valve prolapse. a | Diagnosis of mitral valve prolapse must take into account the normal saddle shape of the valve and annulus, which produces opposite leaflet–annular relationships in perpendicular views. Mitral valve prolapse is most specifically diagnosed by leaflet displacement above the annular high points, imaged in long-axis views; and by leaflet misalignment at their point of coaptation. b | Parasternal long-axis echocardiographic view of posterior leaflet prolapse (arrows) beyond the annular hinge points (dashed line). c | Anterior leaflet prolapse and partial flail (partial eversion of the leaflet tip into the dilated LA; arrows) relative to the posterior leaflet, which is restricted, tethered by the dilated LV.90 These opposite leaflet displacements increase the regurgitant gap between the leaflets. d | Patient with extensive leaflet thickening and anterior leaflet flail (arrows). Abbreviations: Ao, aorta; LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
For cardiologists or sonographers, mitral prolapse is defined by an abnormal systolic bulging or ‘billowing’ of one or both leaflets towards the left atrium (≥2 mm beyond a line connecting the annular hinge points in a long‐axis view; Figures 6b,c).88,93 For cardiac surgeons, ‘billowing’ designates bulging of the leaflet bodies into the atrium while the leaflet tips and coaptation point remain on the ventricular side. ‘Prolapse’ indicates an overriding of the annulus by a leaflet edge with displacement of the coaptation point into the atrium, and is therefore restricted to MVP complicated by moderate‐to‐severe mitral regurgitation, which does not include the overall spectrum of MVP disease, particularly in familial studies.93 Leaflet ‘flail’ implies coaptation failure with eversion of the free edge of a leaflet into the left atrium, usually consequent to chordal rupture (Figure 6d). Flail most frequently affects the posterior leaflet and is generally associated with severe mitral regurgitation, leading to ventricular remodelling and conveying a poor outcome with medical management.94–99
Other valve characteristics evaluated by echocardio‐graphy in MVP are leaflet length and thickness, mitral annulus diameter, and mitral regurgitation severity. Normal leaflet length averages are 22–23 mm for the anterior and 12–13 mm for the posterior leaflet; normal annulus diameter is 28–30 mm.100,101 In patients with MVP, leaflets are generally elongated and thickened, and the annulus is enlarged.102 Mid‐leaflet thickness in the parasternal long‐axis view in diastole (which is normally 2–3 mm) is increased in 14% of patients with moderate MVP (to ~3–8 mm) and up to 40% of those with severe forms of the disease.88 Notably, leaflet thickness measured by echocardiography depends on the acquisition mode (harmonics) and acoustic wavelength,103 typically overestimating the dimensions measured during surgery. Thickness also decreases as the leaflet elongates in systole by Poisson’s effect in soft materials.104 ‘Classic’ MVP refers to superior leaflet overshoot >2 mm and echocardiographic leaflet thickness ≥5 mm, associated with more severe dysfunction.81,105 Mitral regurgitation varies from trivial to severe according to the failure of coaptation driven by chordal elongation or rupture and annular enlargement.106 Regurgitation in patients with MVP is typically greatest in mid‐to‐late systole, whereas functional mitral regurgitation is often seen during early‐to‐mid systole.107,108 Severe mitral regurgitation seems to occur in ~10% of patients with MVP and most with flail. Mitral valve lesions also seem to worsen with patient age, with leaflet thickening and increasing mitral regurgitation, in about one‐quarter to one‐third of patients, although an asymptomatic phase limits accurate knowledge of natural history.109,110 Interactions among mitral apparatus components augment mitral regurgitation. For example, annular flattening, observed with severe mitral regurgitation, can increase chordal stresses and promote chordal rupture;111,112 left ventricular remodelling can exacerbate malcoaptation through apical tethering of nonprolapsed leaflet segments (Figure 6c);90 and mitral regurgitation can beget mitral regurgitation through leaflet remodelling induced by nonphysiological strains and shear stress.
Genetic studies of MVP
Familial genetic studies have improved our understanding of MVP and its diagnosis.113 First, some patients with MVP do not have diagnostic leaflet displacement into the left atrium, but have features that suggest early, potentially prodromal forms.114,115 These prodromal forms are based on anterior displacement of the coaptation point as a manifestation of excessive posterior leaflet length; in the familial context, such individuals share genetic variation with relatives manifesting fully developed MVP.114,115 The prodromal morphology of MVP has allowed identification of a new locus for autosomal dominant MVP on chromosome 13.115 Although the natural history of prodromal forms of MVP is unknown, they might indicate genetically susceptible individuals in whom early therapy can limit clinical progression. Recognizing that leaflet thickness, displacement, and mitral regurgitation vary widely within the same family allows us to include all family members who will be informative for genetic linkage.114–116
Histopathology and biomechanics
Degenerative MVD leads to mitral valve incompetence with annular dilatation, excess valvular tissue resulting in hooding, doming, or billowing of a leaflet portion or entire leaflet, especially the posterior, and chordal rupture resulting in a flail leaflet with prominent mitral regurgitation (Figures 6 and 7). Two forms of degenerative MVD have been reported. So-called Barlow disease is associated with excess connective tissue, with redundant, thickened, yellow, and gelatinous leaflets, marked annular dilatation, elongated and thin (sometimes ruptured) or thick (frequently calcified) chordae, disrupted collagen and elastic layers, and excess acidic mucopolysaccharides. Fibroelastic deficiency is characterized by thin, translucent leaflets deficient in collagen, elastin, and proteoglycans with only moderate annular dilatation and with focal chordal elongation or rupture.44,117–120 Pathologically thickened lesions in these otherwise thin leaflets appear in leaflet segments exposed to mitral regurgitation, leading to the hypothesis that these are secondary leaflet changes.44
Figure 7.
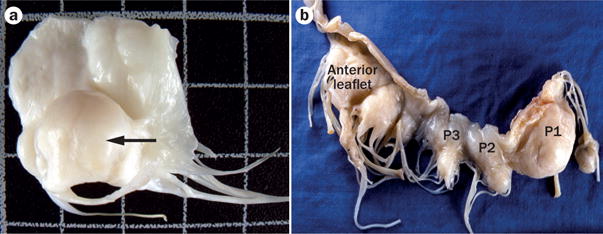
Anatomy of mitral valve prolapse. a | A resected mitral valve with prominent leaflet thickening and opacity and a prolapsing and domed central scallop (P2) of the posterior leaflet (arrow). b | Mitral valve removed from a patient with mitral valve prolapse. Note that the lesions, reflected by leaflet bulging towards the observer, are present throughout and most prominent in the medial scallop (P1) of the posterior leaflet.
In myxomatous leaflet degeneration, leaflet thickness varies with the degree of fibrosis in the atrialis layer and the accumulation of myxoid ECM rich in glycosaminoglycans.121–127 The most consistent marker of degenerative MVD is disruption of the fibrosa by myxomatous ECM (Figure 8).44,121 Movat’s stain, which delineates ECM components, shows expansion of the spongiosa by loose, amorphous ECM strongly positive for proteoglycans, with diminished staining for collagen and fragmented elastin.122 Similarly, myxoid replacement of the collagen core of chordae tendineae is frequent, leading to their rupture (Figure 8a).122 Loss or severe disruption of the elastic lamina beneath the atrialis and the spongiosa is prominent (Figure 8a).122 Expansion of the spongiosa and plaque formation lead myxomatous leaflets to thicken (2.3 mm versus 0.9 mm in the normal valve; P <0.0001), with increased cell density.122 VICs in diseased valves are mostly in an activated myofibroblastlike phenotype characterized by expression of vimentin and smooth muscle αactin, but not smooth muscle myosin heavy chain isoforms 1 or 2, markers of differentiated smooth muscle cells.123 Cells that stain positive for the haematopoietic marker CD45+ are also detected and might be fibrocytes, which are engrafted blood‐borne cells responsible for fibrosis in several diseases (such as fibrotic lung disease and nephropathy) and can differentiate into myofibroblasts that can both secrete matrix and degrade collagen and elastin.122,123 In myxomatous degeneration, expression of proteoglycans and catabolic enzymes is increased, including matrix metalloprotein‐ases (MMP‐1, MMP‐2, MMP‐9, and MMP‐13) and cathepsin K, which contributes to fragmentation of collagen and elastin fibres (Figure 8b).121–124 A substantial increase in hyaluronic acid content with reduced sulphated glycosaminoglycan content has been reported in degenerated posterior mitral leaflets in comparison with normal valve leaflets in humans.125,126 These biological changes alter the mechanical properties of the diseased leaflets, with an increase in circumferential extensibility from 15.5% in healthy valves to 37.2% in myxomatous valves, increase in radial extensibility from 24.1% to 39.7%, and an important reduction in leaflet stiffness and failure strain.7 However, echocardiographic classification of flail versus nonflail valves did not demonstrate differences in the leaflet thickness, length or thicknesses, volume or weight, but measurable differences in leaflet extensibility and basal chordal stiffness.127 In that study, flail leaflets had highly extensible leaflets and less stiff basal chordae than nonflail valves.127
Figure 8.
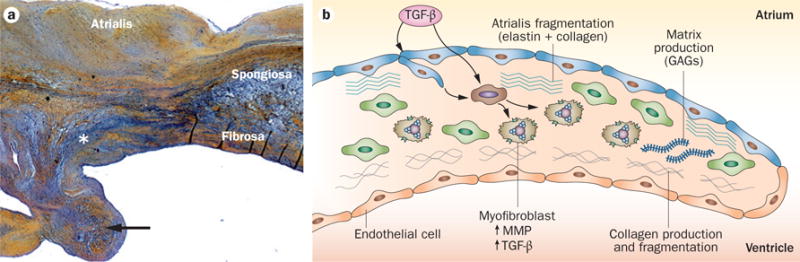
Mechanisms of mitral valve prolapse. a | A mitral valve stained with haemotoxylin and eosin to define the lesion of mitral valve prolapse as disruption of the fibrosa by myxoid extracellular matrix (*), which also infiltrates the collagen core of the chordae tendineae, one of which was ruptured (arrow). The elastin lamina beneath the atrialis is also disrupted. b | A schematic showing the mechanism of myxomatous degeneration, with activation of valve interstitial cells to myofibroblasts that increase matrix production and turnover, secrete MMPs that drive collagen and elastin fragmentation, and release TGF-β that in turn promotes further cell proliferation and myofibroblast differentiation. Abbreviations: GAG, glycosaminoglycan; MMP, matrix metalloprotease; TGF-β, transforming growth factor β.
Surgical presentation
Surgical mitral valve repair involves removal, restraint, or reconstruction of a part or whole valve scallop that is most redundant (Figure 7a). Degenerative processes resulting in mitral regurgitation have two main phenotypes: diffuse myxomatous degeneration (so‐called Barlow disease), which can present as a genetic disorder, and fibroelastic deficiency (FED),44,117–119,128,129 which might be caused by an accelerated ageing process. Patients with Barlow disease usually present in their fourth or fifth decade for surgery after a long history of murmur and regurgitation; those with FED are typically older with more‐abrupt onset of mitral regurgitation and associated symptoms owing to chordal rupture and flail.44,117,119,120 Although most of the leaflet is thin in FED, localized myxomatous thickening occurs within the typically posterior flail scallop; whether this change is primary or secondary to the regurgitant lesion is unknown.44 Other differences between these two expressions of mitral valve degeneration exist (Table 1). Of note, however, the echocardiographic definition of these entities has not been standardized or validated, nor are there known genetic differences or recognized variations in basic mechanisms.
Table 1.
Echocardiographic differences between so-called Barlow disease and fibroelastic deficiency44
| Lesion | Barlow disease | Fibroelastic deficiency |
|---|---|---|
| Primary effects | Excess valve tissue | No excess valve tissue |
| Posterior leaflet height >annulus septal–lateral diameter* | Excess tissue and thickness if present limited to the prolapsing segment | |
| Anterior leaflet height >38 mm* | Thin leaflets in nonprolapsing segments | |
| Leaflet thickness >3 mm | No leaflet billowing | |
| Leaflet billowing | Prolapse often limited to one scallop (usually P2)* | |
| Prolapse of multiple scallops* | Elongated chordae | |
| Atrialization of the base of the posterior leaflet* Chordae thickened, elongated, and calcified Chordal rupture infrequent |
Chordal rupture* | |
|
| ||
| Secondary effects | Leaflet or annular calcifications* Constant annular dilatation involving preferentially the posterior region |
No calcifications* Inconsistent or only moderate annular dilatation |
Inconsistent, but frequent lesions.
Genetic presentation
Prolapse and mitral valve leaflet thickening occur in several inherited connective tissue disorders, including Marfan syndrome, Ehlers–Danlos syndrome, osteogenesis imperfecta, and pseudo‐xanthoma elasticum.130 Familial occurrence of MVD without syndromic features was first described >40 years ago.130 In addition to the X‐linked form of the disease, to date, only three autosomal loci have been reported to be associated with familial MVD: chromosome 16p12.1‐p11.2, chromosome 11p15.4, and chromosome 13q31.3‐q32.1.115,131,132 Identifying genes with mutations in these chromosomal locations will eventually lead to improved understanding of the complex pathogenesis of MVD.92 A promoter polymorphism in the matrix metalloproteinase 3 gene (MMP3) is a modifying factor that affects the severity MVD.133
The frequency of familial MVD inheritance is unknown, but familial clustering has been proved.134 While myxomatous and prolapsing mitral valves often seem to be isolated, familial studies reveal that many affected individuals are not clinically recognized, so heritability might be more frequent than is evident without screening.135 The pattern of inheritance is typically autosomal dominant with reduced, age‐dependent, penetrance.136 The genetic susceptibility of heterogeneous MVD might be complex, with several common susceptibility variants explaining its frequency of 2.4% in the general population and rare, penetrant variants present in families.81,137 A genome‐wide case–control study is underway, but several loci have been identified in families with Mendelian transmission of MVD, which is consistent with heterogeneous genetic pathways that lead to the myxomatous phenotype. Genome‐wide association studies might reveal the interrelated pathways responsible for normal and disordered valve development that can be targeted to improve disease natural history.138
Marfan syndrome, fibrillin-1, and TGF-β
Investigations into the pathogenesis of Marfan syndrome, in particular, have led to improved understanding of genetic contributions to MVD. Marfan syndrome results from heterozygous mutation in FBN1, the gene that encodes fibrillin‐1, which is the principal component of microfibrils in the ECM.139 The occurrence of myxomatous mitral valve thickening and MVP in this disorder has been variably reported as >50%140 versus 28% with more‐recent diagnostic criteria.141 MVD has also been reported to be associated with subdiagnostic variants of Marfan syndrome.142,143 Affected individuals often have mitral valve elongation and thickening similar to Barlow disease morphology.44,143 Some phenotypic manifestations such as ocular lens dislocation are readily explained by a deficiency of fibrillin‐1, a structural protein. Although such a structural deficiency might explain leaflet prolapse itself, the myxomatous leaflet thickening suggests additional changes have occurred in nonstructural pathways.144
Several lines of evidence indicate that excessive signalling by members of the TGFβ superfamily leads to many features of Marfan syndrome.6,145,146 Fibrillin1 is similar to the latent TGFβbinding proteins (LTBP), and binds to several LTBP isoforms (Figure 9).147 Mice with a cysteine‐substitution mutation in Fbn1 have longer and thicker mitral valves than wild‐type control mice.6 Antagonism of TGFβ signalling prevents the pathological prolongation and thickening of mitral valves.6 TGFβ family members can also be inhibited by blocking type1 angiotensin II receptor,148,149 a pathway that is also implicated in nonsyndromic myxomatous MVP.150
Figure 9.
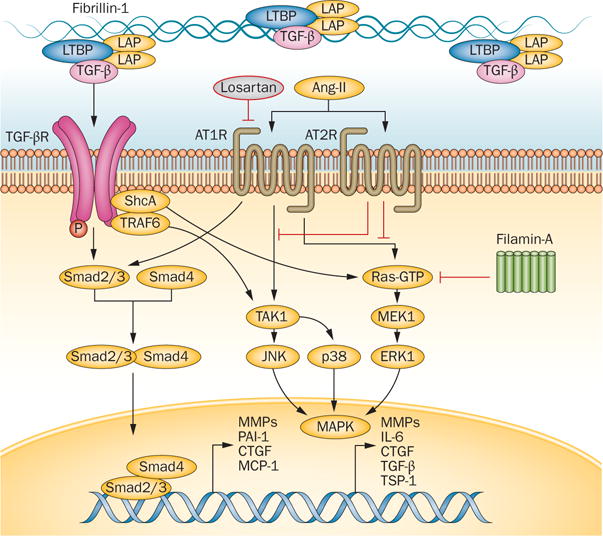
Proposed mechanism of mitral valve disease in Marfan syndrome. A mutation in fibrillin-1 decreases local binding of the TGF-β LLC, consisting of LAP, LTBP, and TGF-β ligands. TGF-β binds to its receptor (TGFBR1/2), activating canonical (Smad2/3, Smad4) and noncanonical (ERK1, JNK1, MAPK, MEK1, p38, Ras-GTP, ShcA, TAK1, TRAF6) signalling. Activation of these pathways leads to increased nuclear transcription of several downstream products, including CTGF, IL6, MCP1, MMPs, PAI-1, TSP1, and TGF-β itself. Filamin A inhibits activation of ERK1 and MEK1, and its deficiency increases these pathways and might contribute to mitral valve proliferation through this mechanism. Cross-talk between the Ang II receptors (AT1R and AT2R) and TGF-β receptors provides a therapeutic benefit of selective AT1R antagonism for aortic aneurysms in mice with fibrillin deficiency, and this approach is under investigation for treatment of myxomatous mitral valve disease. Abbreviations: Ang-II, angiotensin II; AT1R, angiotensin II type 1 receptor; AT2R, angiotensin II type 2 receptor; CTGF, connective tissue growth factor; ERK1, mitogen-activated protein kinase 3; JNK, mitogen-activated protein kinase 8; LAP, latency-associated peptide; LLC, large latent complex; LTBP, latent TGF-β-binding protein; MAPK, mitogen-activated protein kinase; MCP-1, C-C motif chemokine 2 (also known as monocyte chemoattractant protein 1); MEK1, dual specificity mitogen-activated protein kinase kinase 1; MMP, matrix metalloproteinase; PAI-1, plasminogen activator inhibitor 1; ShcA, SHC-transforming protein 1; TGF-β, transforming growth factor β; TGF-βR, transforming growth factor β receptor; TAK1, TGF-β-activated kinase 1; TRAF6, TNF receptor-associated factor 6; TSP-1, thrombospondin-1.
Many Marfan syndrome manifestations can be treated by antagonism of TGFβ signalling in mouse models, which has led to the investigation of this pathway in human patients with this disease. Loeys–Dietz syndrome, which has phenotypic overlap with Marfan syndrome, leads to aortic aneurysms, dissection, and arterial tortuosity in addition to midline facial abnormalities such as widely spaced eyes (hypertelorism), often with long bone overgrowth.151 This disease is associated with heterozygous mutations in genes encoding the components of the TGFβ receptors, TGFBR1 and TGFBR2, or the TGFβ2 ligand, TGFB2.151,152 Four of 14 (29%) affected individuals with Loeys–Dietz syndrome had substantial mitral valve prolapse.151 In another investigation in which the prevalence of MVP among individuals with FBN1 mutations was compared with that in individuals with TGFBR2 mutations, more people with FBN1 mutation had MVP: 105/232 (45%) in those with FBN1 mutations versus 15/66 (21%) with TGFBR2 mutations.153 Furthermore, in a report that identified mutations in MADH3, which encodes mothers against decapentaplegic homologue 3 (commonly known as Smad3), this gene was associated with a very similar condition to Loeys–Dietz syndrome that was termed aneurysms–osteoarthritis syndrome,154 in which MVP has been reported in 18 of 36 (50%) patients.154,155
Examination of affected tissues in Marfan, Loeys–Dietz, and aneurysms–osteoarthritis syndromes consistently shows increased TGFβ activation and signalling.153–156 The loss of TGFβ receptor function or its Smad3 signalling factor should have the opposite effect, but paradoxically does not. An attempt to address and understand this paradox arose from investigation of mice with a fibrillin‐1 mutation and haploinsufficiency for Smad4.157 This combination of mutated genes was expected to improve the phenotype, but the mice actually had more aortic dissection and shorter lifespan than mice with the fibrillin‐1 mutation alone.157 Further investigation led to the discovery that this combination of mutant alleles leads to a unique increase in mitogen‐activated protein kinase signalling, most notably the extracellular signal‐regulated kinase (ERK), and that selective antagonism of this pathway reverses this effect.156,157 Several potential mechanisms have been outlined in another review.158 One possibility is that mutations in TGFβ receptors selectively decrease canonical signalling that leads to a loss of feedback inhibition.158 Consequently, increased production and activation of the TGFβ ligand might lead to excessive noncanonical signalling. Another hypothesis posits that impaired TGFβ receptor function leads to upregulation of ligand production and activation.156 This situation might have disparate effects on different cell types depending on their embryological origin and their relative potential to maintain TGFβ signalling. The interaction between different cell types seems to correlate with specific regions of the vasculature and specific tissues that are typically affected in these conditions of increased TGFβ signalling, namely Marfan, Loeys–Dietz, and aneurysms–osteoarthritis syndromes.158
Filamin A mutation syndrome
An X‐linked form of myxomatous valvular dystrophy which affects all four cardiac valves was first reported in 1969,159 and has led to exploration of its molecular pathogenesis. Affected men typically develop dysfunction of the mitral or aortic valves or both, which requires replacement with a prosthetic valve.160 A large extended pedigree was identified in western France, and the trait mapped along with mild haemophilia A using techniques of genetic genealogy.160 In this family, the identification of two cousins surgically treated for severe valvulopathy, both also affected by mild haemophilia A, led to familial screening that identified 10 out of the 44 tested men as having progressive MVP, and four with moderate‐to‐severe aortic regurgitation.161 Among 47 women in the same family, 10 had asymptomatic mitral or aortic valve abnormalities. All individuals with valvular disease had mild haemophilia A (factor VIII activity 15–50%).161 To increase the size of this family for genetic studies, databases were screened to identify patients with mild haemophilia A, surgically treated for valvulopathy, with the same geographical origin as the initial family. This genealogical approach linked two kindreds to a common ancestor born in the 18th century. All family members with MVP had mild haemophilia A, except one individual whose factor VIII activity was normal (>50%), demonstrating that the myxomatous valvulopathy was a separate entity.160 Finally, an 8 cM interval on chromosome Xq28 was investigated and the association refined to the FLNA gene encoding filamin A.161,162 Three distinct missense mutations and a 1,944 bp deletion were identified in this gene, each of which is suspected to affect the same antiparallel βstrand organization of filamin A.161,162 Notably, individuals with these mutations do not have nodular periventricular heterotopia, a neurological phenotype previously associated with mutations in other regions of the filamin A molecule.161–163
Filamin proteins have an Nterminal actinbinding domain, followed by 24 immunoglobinlike filamin repeats that fold into antiparallel βsheets that function as interfaces for protein interactions.163 Filamin A is an actin‐binding protein that stabilizes cortical F‐actin networks and links them to cellular membranes, thereby conferring membrane integrity and protecting cells against mechanical stress.163 Filamin A binds to >70 additional cellular proteins, including trans‐membrane receptors and signalling molecules. FLNA, therefore, has essential scaffolding functions and integrates multiple cellular behaviours during embryonic development, cellular migration, and mechanical stress responses.164–166 N‐terminal filamin A mutations causing valvular dystrophy also regulate small Rho‐GTPases, so that cells with these mutations are less able to spread and migrate on culture surfaces. Mutated filamin A alters the balance between RhoA and Rac1 GTPases in favour of RhoA, potentially altering the role of Rac1‐specific Rho GTPaseactivating protein 24 (also known as FilGAP) and downstream trafficking of β1integrins to the cellular membrane, and thus mechanobiology.167
Complete loss of FLNA leads to embryonic lethality with a pleomorphic array of cardiac malformations involving the cardiac ventricles, atria, and outflow tracts as well as widespread aberrant vascular patterning.168,169 Abnormal epithelial and endothelial organization and aberrant adherens junctions in developing blood vessels in heart and other tissues support the essential roles for FLNA in intercellular junctions and provide a possible mechanism for the valvular defects.168,169 The human disease mutations in FLNA also influence many signalling pathways that modulate valve growth, causing excessive TGFβ and 5‐hydroxytryptamine activation that provides a developmental basis and potential therapeutic opportunities for valve disease.170
Progressive mitral valve degeneration
While genetic mutations are increasingly shown to underlie the pathogenesis of MVP,92,171,172 severe MVP that requires surgical correction often manifests after the fifth decade of life, frequently with a decades‐long progressive increase in severity.173 Mechanical and biological factors can contribute to disease. Mitral annular flattening has been reported in patients with MVP who develop greater mitral regurgitation compared with those with less regurgitation and normal individuals.111 Such annular flattening increases stress on the leaflets and chordae,112,174 which can accelerate the degenerative processes initiated by genetic mutations.
In studies of surgically excised human myxomatous tissues, members of the TGFβ superfamily are overexpressed, and VICs are transformed into myofibroblasts that increase expression of matrix metalloproteases that can propagate degeneration of collagen and elastin structures.122,150,175–177 Changes in these severely affected valves might be a secondary effect, and amendable only to surgical therapy. However, genetic associations between particular genes and MVP have the potential to reveal pathways and modifying factors that can be modulated to limit valve degeneration, even before it is triggered by developmental and signalling defects.173 Clues might also be gathered from correlative studies with heritable canine MVP, a cause of severe heart failure and mortality.178,179
Mitral valve in HCM
Phenotype and LVOT obstruction
Echocardiography has enabled the understanding that the mitral valve contributes to subaortic obstruction in patients with HCM via systolic anterior motion (SAM) of the mitral leaflets towards the hypertrophied upper septum. SAM has two main consequences: firstly, flow is accelerated within the narrowed LVOT, creating a pressure gradient measurable by Doppler echocardiography; and secondly, disruption of normal leaflet coaptation, causing mitral regurgitation of variable severity.91,180 Subaortic obstruction is present in two‐thirds of patients at rest or with exercise, and leads to symptoms of, and predicts progression to, heart failure and sudden cardiac death.181,182
SAM, first described as a diagnostic feature of obstructive HCM by echocardiography, was long thought to be caused simply by mitral valve suction towards the hypertrophied septum owing to accelerated LVOT velocities (known as the Venturi effect).183 The importance of structural abnormalities in the mitral valve in generating SAM was recognized after observations that SAM can be present without HCM;9 and that SAM begins before LVOT flow acceleration.64,184 Furthermore, isolated anterior and inward displacement of the papillary muscles combined with leaflet elongation, especially of the posterior leaflet, can lead to SAM, the degree of which is related to leaflet length, even in the absence of septal hypertrophy.15,17,185–192
SAM results from the combination of septal hypertrophy, creating accelerated, posteriorly‐directed left ventricular outflow velocities, with a mitral valve susceptible to SAM because of elongated leaflets that are positioned anteriorly by displaced papillary muscles (Figure 10). The use of 3D echocardiography has confirmed that the mitral valve area is increased in vivo, and that LVOT obstruction relates to a combination of upper septal hypertrophy, leaflet elongation, and papillary muscle displacement anteriorly and towards each other.15 This situation creates a long, slack distal residual leaflet portion interposed into the outflow stream at the beginning of ejection, so that ejection pushes this leaflet portion towards the septum (Figure 11).64,184,185 Secondary systolic acceleration as SAM narrows the LVOT can further propagate SAM via the Venturi effect.185,188 These findings explain why obstructive SAM can accompany modest septal hypertrophy when excessive or segmental leaflet elongation and papillary muscle malposition is present.187,192 They also explain why SAM, subaortic obstruction, and mitral regurgitation persist after septal ablation owing to anterior papillary muscle displacement and leaflet malcoaptation;193 and why, for similar degrees of SAM, mitral regurgitation is greater if the posterior leaflet is limited in its capacity to move anteriorly to coapt effectively.14 These mechanistic insights translate into new therapeutic approaches.194–200
Figure 10.
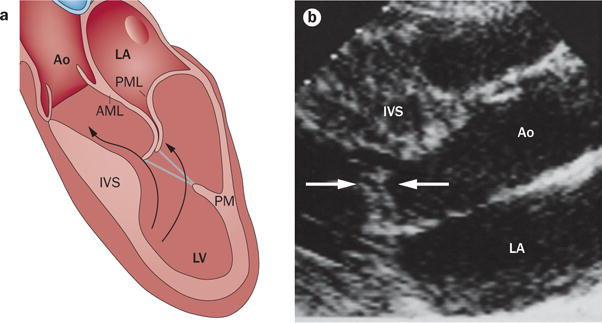
Mitral valve enlargement in SAM. a | SAM relates to a combination of a susceptible mitral valve, which is anteriorly positioned and elongated, encountering a posteriorly shifted left ventricular outflow stream, diverted by hypertrophy of the upper IVS. Anterior PM displacement decreases the posterior restraining force on the leaflets. Leaflet elongation allows greater leaflet excursion to create obstructive SAM. b | SAM of elongated leaflets (arrows) approaching the IVS and obstructing left ventricular outflow. Abbreviations: AML, anterior mitral leaflet; Ao, aorta; IVS, interventricular septum; LA, left atrium; LV, left ventricle; PM, papillary muscle; PML, posterior mitral leaflet; SAM, systolic anterior motion.
Figure 11.
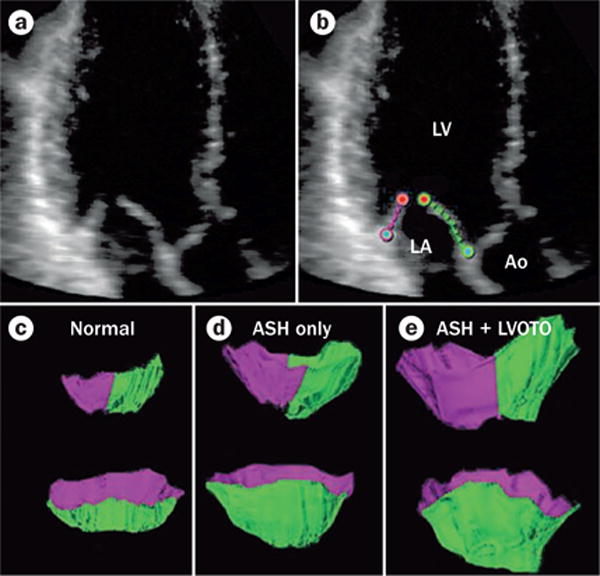
3D echocardiography showing increase in systolic anterior motion and LVOTO with increased mitral leaflet area. a,b | Component view from a healthy control individual with diastolic leaflet traces for 3D reconstruction. c–e | Representative open mitral leaflet area measurements in green and purple for anterior and posterior leaflets viewed from the side. Top row: lateral commissure in foreground. Bottom row: left ventricular outflow tract aspect. Smallest values are seen in healthy controls (c), intermediate values with ASH and no resting LVOTO (d), and greatest leaflet areas in patients with ASH and resting LVOTO (e). Abbreviations: Ao, aorta; ASH, asymmetrical septal hypertrophy; LA, left atrium; LV, left ventricle; LVOTO, left ventricular outflow tract obstruction. Reprinted from Kim, D. H. et al. In vivo measurement of mitral leaflet surface area and subvalvular geometry in patients with asymmetrical septal hypertrophy: insights into the mechanism of outflow tract obstruction. Circulation 122 (13), 1298–1307 (2010).
The use of echocardiography has clearly demonstrated that mitral valve abnormalities have an important role in determining LVOT obstruction. In patients with HCM, the mitral apparatus can have a number of diverse structural differences. For example, the mitral valve and papillary muscles can be mispositioned;185 the leaflets and chordae can be elongated, and the papillary muscles hypertrophied;201 papillary muscles can be directly inserted onto the anterior leaflet;202 or there can be accessory mitral valve tissue.203 Furthermore, the leaflets might be degenerated with increased leaflet or chordal thickness,194 ruptured chordae,204 and mitral valve prolapse.205 Investigators using MRI have confirmed leaflet elongation that is independent of left ventricular wall thickness, and which correlates with resting and inducible LVOT gradient.206 The pattern of obstruction can vary with mitral valve structure. For example, elongated leaflets sharply bend at their coaptation point and their distal portions move excessively to contact the septum, whereas shorter, thickened leaflets contact the septum primarily through posterior septal motion.207 Mitral valve anomalies must, therefore, be addressed to achieve comprehensive physiological repair in patients with HCM, and limit SAM after mitral valve repair for MVP when elongated leaflets are interposed into the LVOT.9,14,188,208
Pathology
In anatomical examinations of the mitral valve in 94 highly symptomatic patients with HCM undergoing surgery, 66% of patients had intrinsic valve abnormalities, 36% had increased posterior leaflet scallop number, leaflet thickness, and mass (twice that of healthy patients), and 10% had direct papillary muscle insertion into the leaflet that was not directly linked to hypertrophy or degree of obstruction.209 In a histopathological investigation by Klues and colleagues, no myxomatous changes in the spongiosa of excised mitral valves were seen.209 Conversely, myxomatous mitral valve degeneration was found in case reports of patients with HCM who had massive mitral regurgitation owing to chordal rupture,204 and echocardiographic findings in a surgical cohort showed abnormalities that often resembled degenerative changes.194
Leaflet elongation
Given the central role of LVOT obstruction via systolic anterior motion of the mitral leaflet, how might mutations that lead to HCM cause mitral valve leaflet elongation? Several theories have been proposed. Firstly, leaflet elongation might be governed by the mutated gene that results in HCM. For example, posterior leaflet length is often increased despite the absence of obstruction in healthy patients who carry a morbid HCM mutation15,206,210 and in Mybpc3 knock‐out mice.211 However, sarcomeric proteins are not present within the mitral valve apparatus, except the papillary muscles. Secondly, leaflet elongation might be stimulated by increased endothelial shear rate in the narrowed LVOT, or is an adaptive process due to repeated episodes of SAM, as mechanical stretch can reactivate embryonic growth pathways.212 However, many patients with leaflet enlargement do not have SAM (presumably owing to a lack of anterior malposition into the LVOT flow).209 Thirdly, leaflet elongation might be an independent disorder with tightly linked genetic contributions. However, the chromosomal loci reported to date in MVP do not overlap with genes that are associated with HCM.115 Finally, elongation might be the result of paracrine effects from the abnormal left ventricular wall, which is consistent with evidence that valvulogenesis is influenced by molecular signals generated by the surrounding myocardium as well as the valve.213
Another potential mechanism of leaflet elongation might involve pluripotent epicardial‐derived cells (EPDC), a subset of extracardiac mesenchymal cells derived from coelomic mesothelium.214,215 The cells might abnormally differentiate into fibroblast‐like cells rather than cardiomyocytes, with consequent increased synthesis of periostin that contributes to the increased myocardial fibrosis observed in patients with HCM.214,215 Markedly elevated levels of periostin are indeed expressed in the left ventricular walls of HCM mice,216–218 secreted by nonmyocyte elements, and periostin promotes VIC proliferation, differentiation, and matrix production, which might drive leaflet elongation.68,72,218 Periostin might also be part of a molecular switch that determines whether a mesodermal progenitor cell becomes a cardiomyocyte or fibroblast,72 while blocking expression of the serum response factor transcription factor required for initiating contractile protein synthesis.219 Patterns of periostin expression in mouse models of HCM exactly correlate with the hypothesis that EPDC‐derived cardiomyocytes become fibroblasts when a gene encoding a contractile protein is mutated.220 Elevated periostin in close proximity to the mitral valve might increase collagen production and elongate the leaflets, perhaps also driven by reawakened EMT.212 These correlated molecular studies are consistent with the theme of a living valve with a developmental abnormality beginning in early life and continuing into adulthood, relating to its interactions with an abnormal ventricle. Improved understanding of the responsible adaptive mechanisms might enable studies targeting prevention and treatment of MVD in HCM.
Ischaemic mitral regurgitation
Definition and mechanisms
2D echocardiography has been used to demonstrate that apically restricted leaflet closure in the ventricle, owing to papillary muscle displacement causing tethering on the mitral leaflets, is the mechanism of ischaemic mitral regurgitation (IMR) in patients with either localized inferior wall ischaemia or global dilatation and failure.10,17,221,222 The use of 3D echocardiography in experimental studies has proved that global left ventricular dysfunction alone is insufficient to cause IMR.223–225 Displacement and abnormal contraction of the left ventricular wall underlying the papillary muscles, along with decreased shortening of the distance between the papillary muscles, causes mitral leaflet tethering and restricted closure that leads to IMR (Figures 12a,b),223–226 which is referred to as a Carpentier type IIIb lesion. Isolated annular dilation, such as occurs in patients with lone atrial fibrillation, also impairs coaptation (Carpentier type I), but relatively mildly unless the leaflets are tethered at both annular and ventricular ends.227 Tethering geometry also predicts progression of IMR.228 The posterior location of the papillary muscles explains why IMR is seen predominantly with inferior, as opposed to anterior, MIs unless the anterior infarctions are sufficiently large to cause global dilatation or are associated with anterior dyskinesia causing papillary muscle traction.229–233 Investigators using 3D imaging have mapped leaflet tenting relative to papillary muscle displacement by echocardiography and CT, and have shown that asymmetric papillary muscle displacement in the apical direction causes asymmetric leaflet tenting, and is most likely to increase the severity of mitral regurgitation.234–237 The substantially increased mortality in patients with IMR is independent of left ventricle size and function, and occurs even in those with mild IMR.238–243 IMR also reduces exercise capacity.244 IMR can exacerbate left ventricular remodelling, with adverse changes in the noninfarcted myocardium in correlated cellular and molecular studies;245–248 the use of cardiac MRI has also quantified this remodelling.249 IMR is a dynamic disease that varies with multiple factors, such as with progressive LV remodelling and dysfunction,250–253 with varied haemodynamic loading under exercise conditions,254,255 reduced tethering under anaesthesia,256,257 and vasodilating thermal therapy.258 Dynamic increases in IMR can explain reduced survival in patients with apparently mild resting mitral regurgitation,259 and suggest a need for exercise testing in patients with exertional dyspnoea out of proportion to their resting mitral regurigitation.260
Figure 12.
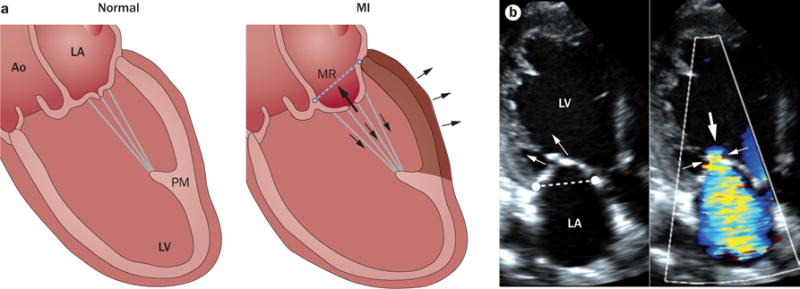
Mechanism of ischaemic mitral regurgitation. a | Mechanism of ischaemic mitral regurgitation caused by increased mitral leaflet tethering owing to left ventricular remodelling after MI (shaded wall) with outward bulging (three arrows on the outer surface of the heart). Leaflet closure is restricted by increased tethering forces on the leaflets exerted via the chordae (arrows within the heart, exceeding normal on the left). b | Echocardiogram from a patient with inferior wall infarction and tethered mitral leaflets (arrows in left panel) with characteristic anterior leaflet bend and concavity towards the LA indicating chordal tethering, mitral regurgitation orifice (small arrows in right panel), and mitral regurgitant flow (large arrow in right panel). Abbreviations: Ao, aorta; LA, left atrium; LV, left ventricle; MI, myocardial infarction; MR, mitral regurgitation; PM, papillary muscle.
The frequent failure of annular ring reduction relates to persistent leaflet tethering by displaced papillary muscles,250,261,262 with continued left ventricular remodelling associated with recurrent mitral regurgitation.250,263 The NHLBI‐supported trial by the CardioThoracic Surgical Network indicated that 32.6% of the 126 patients who received mitral annuloplasty for severe ischaemic mitral regurgitation had recurrent regurgitation within 1 year, with recurrence predicted by the preoperative degree of tethering.264,265 Recurrence of mitral regurgitation predicts absence of the favourable reverse ventricular remodelling that occurs in patients without recurrence.264 Some investigators have shown that stenosis after annuloplasty is caused by subannular tethering and funnel formation by displaced papillary muscles,266,267 causing important exercise‐induced pressure gradients and reduced survival in more severely affected patients.267–269 Subannular repair, therefore, provides the most comprehensive and lasting reduction in mitral regurgitation,270 and the best haemodynamics for forward flow as well, guided by computational analysis of this dynamic, interactive system.271–273 Importantly, reducing IMR might also slow progression of left ventricular remodelling, ultimately leading to a decrease in volume‐ overloaded heart failure.274–278 Echocardiographic insights into the tethering mechan ism and papillary muscle–ventricular unit have led to potential new interventions to resolve postoperative mitral regurgitation by repositioning the papillary muscles. Such interventions include direct surgical left ventricular reconstruction or using an adjustable external pericardial patch or polymer injected into the subpapillary muscle of the ventricular wall, or surgical papillary muscle approximation.10,11,226,237,275,277,279–283 Chordal modi‐fication can relieve tethering by cutting the secondary chordae that most restrict leaflet motion or by implanting chords to reduce the papillary muscle‐to‐annulus tethering distance.276,278,284–289 Saddle‐shaped annuloplasty can also reduce leaflet stress48,112 and improve coaptation.47,50 Papillary muscle resynchronization and leaflet augmentation are additional options for the reduction of mitral regurgitation.290,291 Patients whose IMR will improve with revascularization of ischaemic, but viable, subpapillary muscle myocardium alone292 are being predicted using advanced cardiac perfusion imaging.293
New evidence for valve adaptation
Ischaemic mitral regurgitation reflects the insufficiency of valve area relative to the remodelling left ventricle (Figure 12b). This raises the question of whether the valve can adapt to tethering by increasing its area under the influence of the increased stress imposed by tethering,294 particularly at the chordal attachment zone.272,295–298 Early mitral valve growth is well described: the annular area increases 20‐fold from fetal life to adulthood and the mitral valve grows proportionately.299 EMT is central to this adaptation, and is finely regulated by TGFβ, BMP, vascular endothelial growth factor, and Notch signalling (Figure 4), to yield leaflets large enough to maintain effective closure, with an excess of leaflet relative to annular area to adapt to increased volume loads without regurgitation.71
Testing whether the mitral leaflet area increases in response to tethering requires 3D imaging. Initial clinical studies have confirmed that mitral valve area is 35% larger in patients with left ventricular dysfunction than in healthy individuals, and that larger leaflet area in relation to the demands imposed by leaflet tethering corresponds to less mitral regurgitation.12 The mitral valve area also increases in an animal model involving MI of the inferior myocardial wall.11
Is this mitral valve enlargement caused by passive stretch or active cell proliferation? Kunzelman and colleagues compared mitral valve leaflets from sheep with an infarct and IMR, sheep with an infarct only, and healthy sheep.297,298 The investigators reported a dynamic adaptation to increased leaflet stresses by changes in leaflet thickness and stiffness.297,298 To study the mechanisms regulating these adaptive leaflet processes in vivo, an animal model was developed to separate the effects of tethering forces, the ischaemic environment, and turbulent mitral regurgitation flow.212 The effect of isolated tethering was studied in sheep mitral valves in situ over 2 months of papillary muscle retraction. Such tethering leads to increases in both valve area and thickness, with reactivation of EMT in which endothelial cells—normally quiescent—express smooth muscle αactin and enter the valve interstitium to deposit ECM (Figures 13 and 14).212
Figure 13.
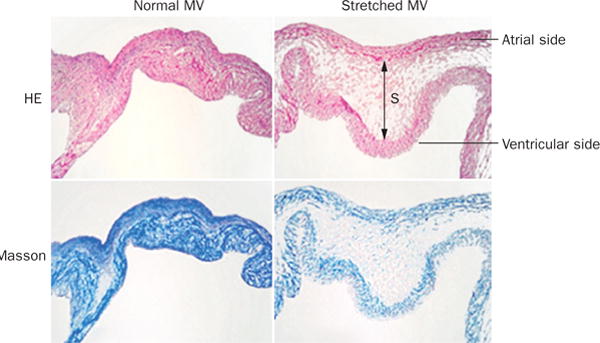
HE and Masson staining in the normal (left) and stretched (right) MV demonstrating increased leaflet thickness and spongiosa matrix after 2 months of stretching in a sheep model. Blue indicates collagen. Abbreviations: HE, hematoxylin and eosin; MV, mitral valve; S, spongiosa. Reprinted from Dal-Bianco, J. P. et al. Active adaptation of the tethered mitral valve: insights into a compensatory mechanism for functional mitral regurgitation. Circulation 120 (4), 334–342 (2009).
Figure 14.
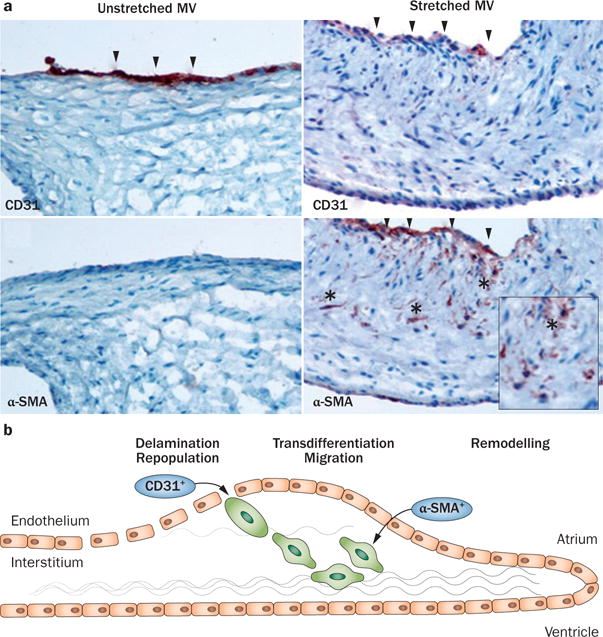
Evidence for reactivated EMT owing to leaflet stretch. a | Staining for the endothelial marker CD31 is reduced in the atrialis, and staining for smooth muscle α-actin is detected, which is normally absent, with positive cells (*) migrating into the interstitium, all features of EMT. Left panels: unstretched MV showing no staining for α-SMA along the CD31+ endothelium. Right panels: staining for α-SMA in the atrial endothelium (also CD31+) of a stretched MV, with nests of cells positive for α-SMA seeming to penetrate the interstitium (*). Upper panels: arrowheads indicate CD31+ endothelial cells. Lower panels: arrowheads indicate α-SMA+ endothelial cells, indicative of EMT. b | Schematic of active MV adaptation by EMT. Abbreviations: α-SMA, smooth muscle α-actin; EMT, endothelial-to-mesenchymal transition; MV, mitral valve. Reprinted from Dal-Bianco, J. P. et al. Active adaptation of the tethered mitral valve: insights into a compensatory mechanism for functional mitral regurgitation. Circulation 120 (4), 334–342 (2009). Bottom panel modified from Armstrong, E. J. & Bischoff, J. Heart valve development: endothelial cell signaling and differentiation. Circ. Res. 95 (5), 459–470 (2004).
These findings indicate that the mitral valve is a dynamic cellular environment that actively adapts to superimposed stresses and is influenced by ventricular pathology.43,60,71,300 Adaptive leaflet growth has been reported in other studies,301–303 as further evidence of the cellular plasticity of mitral valves.304 The compensatory adaptation might restore cellular and biomechanical leaflet homeostasis and ultimately valve function, which leads to the question of why leaflet adaptation is often insufficient to alleviate IMR in many patients. Notably, excised leaflets in patients with end-stage heart failure are thicker and stiffer than healthy valves, potentially impairing leaflet coaptation, which requires systolic leaflet stretch and bending.13,296,305 Similarly, in the experimental tethering model described above, preliminary results from adding an apical MI distal to the papillary muscles indicate that the area and thickness of the valve increase in the presence of ischaemia, with exaggerated EMT, interstitial cell activation and matrix turnover. By contrast, in patients with compensated aortic insufficiency, the mitral valve enlarges to compensate for left ventricular remodelling and seems to remain supple, making functional mitral regurgitation infrequent.306 Remodelling of the leaflet, which is beneficial at the early stage after MI, might therefore become counterproductive in the chronic setting after MI. Understanding how the valve adapts over time after MI could create opportunities for physiological interventions, for example, by modulating TGFβ, which seems to have a major regulatory role in leaflet homeostasis in IMR as well as Marfan syndrome.59,148
Therapeutics and future directions
The most effective treatment for patients with MVD is surgery, and clinicians recognize that the benefits of mitral valve repair exceed those of valve replacement, at least in the case of organic mitral regurgitation.307 The remaining clinical debate is focused on timing of surgery to preserve left ventricular contractility.94–96,308 Although current surgical repairs for organic mitral regurgitation achieve excellent haemodynamic outcomes and long‐term durability, their efficacy on the progressive degeneration or remodelling of the mitral valve leaflets remains unknown. If mechanistic interactions and their role in leaflet degeneration are understood, tailoring repair to achieve optimal haemodynamics and also restore physiological biomechanics should be possible. Such a strategy should also benefit the clinically challenging lesion of IMR, in which progressive leaflet fibrosis and restricted leaflet motion, even after mitral annuloplasty, can reduce the durability of the repair.13,250,283,305,309 Success has been achieved in this area to some extent with papillary muscle repositioning or banding, by cutting chordal structures to relieve tethering‐induced tension, or by using a saddle‐shaped annuloplasty ring to reduce tension in the leaflets and chordae as well as leaflet lengthening.226,265,278–280,284–289 However, their effect on chronic valve biomechanics at the ECM and cellular levels remains to be investigated. A ventricular approach to reducing mitral regurgitation can be achieved with vasodilators, which alter the ventricular–arterial impedance relationship, and increase the forward flow. Although this approach follows intuitive reasoning, and medications such as angiotensin‐converting‐enzyme inhibitors and angiotensin‐receptor blockers are beneficial for dilated cardiomyopathy, results are conflicting for the treatment of MVD.310,311 Medical therapies that limit left ventricular remodelling and damage to the myocardial substrate from increased adrenergic drive, such as βblockers, might be beneficial in patients with functional mitral regurgitation secondary to left ventricular dilatation, but do not address primary mitral valve dysfunction.312,313 Similarly, biventricular pacing in patients with dilated cardiomyopathy and dyssynchrony can improve left ventricular morphology and reduce mitral regurgitation.291,314,315
The exploration of novel therapies might be enabled using valve progenitor cells to recapitulate developmental abnormalities caused by genetic mutations or altered early stresses, complemented with in vivo studies using explanted endocardial cushions. Although challenging, the derivation of human embryonic stem cells from endothelial cells that are specified to become valve tissue, and which are competent to perform EMT, might be possible74,316–318 One approach to this challenge is first to derive a multipotent cardiovascular progenitor population of human embryonic stem cells.317 Using specific substrates and growth factors, these progenitors can then be differentiated into cells expressing markers of endocardial cushion endothelium and capable of undergoing EMT. Deriving such valve‐specified progenitors from induced pluripotent stem cells generated from somatic cells of patients harbouring genetic valve disease might provide invaluable early disease models that predict how mutations can lead to MVD.
Conclusions
Cardiac imaging presents valve biologists with the challenge of explaining observed valve abnormalities on the basis of cellular and molecular signalling processes. The mitral valve remains dynamic into adult life and retains the capacity to reactivate early growth processes. Inherited mutations provide clues to the mechanisms of valve growth and, as more genetic contributions are recognized, novel insights into the milieu of abnormal growth factors, cytoskeletal proteins, and mechanical strain will inevitably lead to improved therapies. We are learning that valve growth in different conditions is influenced by common factors: signals intrinsic to the valve, paracrine signals from the left ventricular wall, activation of the endothelium to populate the interstitium, and potentially engraftment of blood‐borne cells. Reciprocal interactions between the endothelial and interstitial cells of the valve can reveal signals that can be used to promote valve homeostasis.45 Current areas of investigation are focused on genetic analysis of the disease, small animal models of MVD, the role of growth factor dysregulation, abnormal EMT, and the influence of biomechanical strain in the pathogenesis of these common disorders. Novel molecular therapies have the potential to target the underlying mechanisms and limit disease progression to reduce the burden of heart failure imposed by the adverse effect of mitral regurgitation on myocardial function.
Key points.
-
▪
Mitral valve disease is a major cause of heart failure and mortality
-
▪
Even in adult life, the mitral valve is a dynamic structure and therapeutically accessible
-
▪
Genetic analysis has revealed that regulation of growth signalling and cell migration pathways could potentially be modified to limit progression from developmental defects to clinically evident valve degeneration, such as mitral valve prolapse
-
▪
Mitral valve enlargement is an important determinant of left ventricular outflow tract obstruction in hypertrophic cardiomyopathy and might be stimulated by valvular–ventricular interactions
-
▪
Mitral valve plasticity allows adaptation to ventricular remodelling, but adverse processes create relative leaflet deficiency in the ischaemic setting, leading to mitral regurgitation with worse prognosis
-
▪
Understanding these concepts can create new opportunities to reduce the clinical progression of mitral valve disease through early detection and biological modification
Acknowledgments
All the authors are part of the Leducq Transatlantic Network, which brings together clinical and basic scientists from multiple centres to identify mechanisms of both inherited and acquired mitral valve disease, with the aim of discovering therapies to modify the natural history favourably. The authors acknowledge support of grant 07CVD04 of the Leducq Foundation, Paris, France for the Leducq Mitral Transatlantic Network of Excellence, and Leducq Foundation Career Development Awards 10CDA01 to D.P.J. and 11CDA04 to M. Padala. Additional support came from the French Society of Cardiology, Paris, France (MVP France and REMY Registry: A.A.H., X.P.J.), the French National Research Agency, Paris, France (A.A.H., E.M.), the French Ministry of Health, Paris, France (A.A.H.; Clinical Research Hospital Program, J.-J.S., T.L.T., H.L.M., V.P.), the National Institutes of Health, Bethesda, MD, R01 HL72265 (R.A.L.), R01 HL109506 (R.A.L., E.A., J.B.), R01 HL114805 (E.A.), K24 HL67434 (R.A.L.), K23 HL116652 (F.N.D.), R01 HL127692 (S.A.S., D.P.M., R.A.L.), R01 HL033756 (R.R.M., R.A.N.), NIGMS P30 GM103342 (R.R.M., R.A.N.), P20 RR21949 (R.R.M., R.A.N.), AHA 11SDG5270006 (R.A.N.), the Doris Duke Charitable Trust, New York, NY (S.A.S., R.A.L.), the Ellison Foundation, Boston, MA (R.A.L.), the American Society of Echocardiography, Raleigh, NC, (J.P.D.-B.), the Austrian Health Ministry Erwin-Schrödinger Fund, Vienna (J.P.D.-B.), the Spanish Society of Cardiology, Barcelona (PROMESA: L.F.-F., J.S.), the Eurostars CARDIOMARK Project E!6490, European Union, Brussels, (J.-J.S.), the INSERM-DGOS and Nantes University Translational Research Programs (J.-J.S., C.D., T.L.T.), Fondation GenaVie, Nantes, France (T.L.T.), the French Foundation of Cardiology, Paris, France (T.L.T.), the Danish Heart Foundation, Copenhagen (M.O.J.), the Fondation de la Recherche Medicale, Paris, France (M. Pucéat), the Fonds de Recherche du Québec-Santé, Montreal, Quebec, Canada (J. Beaudoin), and the Quebec Office of the Heart and Stroke Foundation, Montreal, Quebec, Canada (J. Beaudoin).
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
R.A.L., A.A.H., and D.P.J. are joint first authors. M.P. and J.P.D.-B. are joint second authors. All authors researched data for the article, made substantial contribution to discussion of the content, and wrote, reviewed, and edited the manuscript before submission.
References
- 1.Nkomo VT, et al. Burden of valvular heart diseases: a population-based study. Lancet. 2006;368:1005–1011. doi: 10.1016/S0140-6736(06)69208-8. [DOI] [PubMed] [Google Scholar]
- 2.Enriquez-Sarano M, et al. Quantitative determinants of the outcome of asymptomatic mitral regurgitation. N Engl J Med. 2005;352:875–883. doi: 10.1056/NEJMoa041451. [DOI] [PubMed] [Google Scholar]
- 3.Judge DP, Markwald RR, Hagege AA, Levine RA. Translational research on the mitral valve: from developmental mechanisms to new therapies. J Cardiovasc Transl Res. 2011;4:699–701. doi: 10.1007/s12265-011-9320-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts R. Another chromosomal locus for mitral valve prolapse: close but no cigar. Circulation. 2005;112:1924–1926. doi: 10.1161/CIRCULATIONAHA.105.569517. [DOI] [PubMed] [Google Scholar]
- 5.Williams TH, Jew JY. Is the mitral valve passive flap theory overstated? An active valve is hypothesized. Med Hypotheses. 2004;62:605–611. doi: 10.1016/j.mehy.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Ng CM, et al. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–1592. doi: 10.1172/JCI22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barber JE, et al. Mechanical properties of myxomatous mitral valves. J Thorac Cardiovasc Surg. 2001;122:955–962. doi: 10.1067/mtc.2001.117621. [DOI] [PubMed] [Google Scholar]
- 8.Barber JE, Ratliff NB, Cosgrove DM, 3rd, Griffin BP, Vesely I. Myxomatous mitral valve chordae I: Mechanical properties. J Heart Valve Dis. 2001;10:320–324. [PubMed] [Google Scholar]
- 9.Maslow AD, Regan MM, Haering JM, Johnson RG, Levine RA. Echocardiographic predictors of left ventricular outflow tract obstruction and systolic anterior motion of the mitral valve after mitral valve reconstruction for myxomatous valve disease. J Am Coll Cardiol. 1999;34:2096–2104. doi: 10.1016/s0735-1097(99)00464-7. [DOI] [PubMed] [Google Scholar]
- 10.Levine RA, Schwammenthal E. Ischemic mitral regurgitation on the threshold of a solution: from paradoxes to unifying concepts. Circulation. 2005;112:745–758. doi: 10.1161/CIRCULATIONAHA.104.486720. [DOI] [PubMed] [Google Scholar]
- 11.Chaput M, et al. Mitral leaflet adaptation to ventricular remodeling: prospective changes in a model of ischemic mitral regurgitation. Circulation. 2009;120:S99–S103. doi: 10.1161/CIRCULATIONAHA.109.844019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaput M, et al. Mitral leaflet adaptation to ventricular remodeling: occurrence and adequacy in patients with functional mitral regurgitation. Circulation. 2008;118:845–852. doi: 10.1161/CIRCULATIONAHA.107.749440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grande-Allen KJ, et al. Mitral valve stiffening in end-stage heart failure: evidence of an organic contribution to functional mitral regurgitation. J Thorac Cardiovasc Surg. 2005;130:783–790. doi: 10.1016/j.jtcvs.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 14.Schwammenthal E, et al. Mechanism of mitral regurgitation in hypertrophic cardiomyopathy: mismatch of posterior to anterior leaflet length and mobility. Circulation. 1998;98:856–865. doi: 10.1161/01.cir.98.9.856. [DOI] [PubMed] [Google Scholar]
- 15.Kim DH, et al. In vivo measurement of mitral leaflet surface area and subvalvular geometry in patients with asymmetrical septal hypertrophy: insights into the mechanism of outflow tract obstruction. Circulation. 2010;122:1298–1307. doi: 10.1161/CIRCULATIONAHA.109.935551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hagege AA, et al. The mitral valve in hypertrophic cardiomyopathy: old versus new concepts. J Cardiovasc Transl Res. 2011;4:757–766. doi: 10.1007/s12265-011-9319-6. [DOI] [PubMed] [Google Scholar]
- 17.Dal-Bianco JP, Levine RA. Anatomy of the mitral valve apparatus: role of 2D and 3D echocardiography. Cardiol Clin. 2013;31:151–164. doi: 10.1016/j.ccl.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper T, et al. Structural basis of cardiac valvar function. Arch Surg. 1966;93:767–771. doi: 10.1001/archsurg.1966.01330050071010. [DOI] [PubMed] [Google Scholar]
- 19.Wit AL, Fenoglio JJ, Jr, Hordof AJ, Reemtsma K. Ultrastructure and transmembrane potentials of cardiac muscle in the human anterior mitral valve leaflet. Circulation. 1979;59:1284–1292. doi: 10.1161/01.cir.59.6.1284. [DOI] [PubMed] [Google Scholar]
- 20.Nordrum IS, Skallerud B. Smooth muscle in the human mitral valve: extent and implications for dynamic modelling. APMIS. 2012;120:484–494. doi: 10.1111/j.1600-0463.2011.02860.x. [DOI] [PubMed] [Google Scholar]
- 21.Sonnenblick EH, Napolitano LM, Daggett WM, Cooper T. An intrinsic neuromuscular basis for mitral valve motion in the dog. Circ Res. 1967;21:9–15. doi: 10.1161/01.res.21.1.9. [DOI] [PubMed] [Google Scholar]
- 22.Marron K, et al. Innervation of human atrioventricular and arterial valves. Circulation. 1996;94:368–375. doi: 10.1161/01.cir.94.3.368. [DOI] [PubMed] [Google Scholar]
- 23.Wit AL, Fenoglio JJ, Jr, Wagner BM, Bassett AL. Electrophysiological properties of cardiac muscle in the anterior mitral valve leaflet and the adjacent atrium in the dog Possible implications for the genesis of atrial dysrhythmias. Circ Res. 1973;32:731–745. doi: 10.1161/01.res.32.6.731. [DOI] [PubMed] [Google Scholar]
- 24.Fenoglio JJ, Jr, Tuan Duc P, Wit AL, Bassett AL, Wagner BM. Canine mitral complex. Ultrastructure and electromechanical properties. Circ Res. 1972;31:417–430. doi: 10.1161/01.res.31.3.417. [DOI] [PubMed] [Google Scholar]
- 25.Swanson JC, et al. Characterization of mitral valve anterior leaflet perfusion patterns. J Heart Valve Dis. 2009;18:488–495. [PMC free article] [PubMed] [Google Scholar]
- 26.Filip DA, Radu A, Simionescu M. Interstitial cells of the heart valves possess characteristics similar to smooth muscle cells. Circ Res. 1986;59:310–320. doi: 10.1161/01.res.59.3.310. [DOI] [PubMed] [Google Scholar]
- 27.Grande-Allen KJ, et al. Glycosaminoglycans and proteoglycans in normal mitral valve leaflets and chordae: association with regions of tensile and compressive loading. Glycobiology. 2004;14:621–633. doi: 10.1093/glycob/cwh076. [DOI] [PubMed] [Google Scholar]
- 28.Itoh A, et al. Active stiffening of mitral valve leaflets in the beating heart. Am J Physiol Heart Circ Physiol. 2009;296:H1766–H1773. doi: 10.1152/ajpheart.00120.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sacks MS, Yoganathan AP. Heart valve function: a biomechanical perspective. Philos Trans R Soc Lond B Biol Sci. 2007;362:1369–1391. doi: 10.1098/rstb.2007.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Padala M, et al. Mechanics of the mitral valve strut chordae insertion region. J Biomech Eng. 2010;132:081004. doi: 10.1115/1.4001682. [DOI] [PubMed] [Google Scholar]
- 31.Lim KO, Boughner DR, Smith CA. Dynamic elasticity of human mitral valve chorade tendinease. Can J Physiol Pharmacol. 1977;55:413–418. doi: 10.1139/y77-058. [DOI] [PubMed] [Google Scholar]
- 32.Lim KO, Boughner DR. Mechanical properties of human mitral valve chordae tendineae: variation with size and strain rate. Can J Physiol Pharmacol. 1975;53:330–339. doi: 10.1139/y75-048. [DOI] [PubMed] [Google Scholar]
- 33.Butcher JT, Nerem RM. Valvular endothelial cells and the mechanoregulation of valvular pathology. Philos Trans R Soc Lond B Biol Sci. 2007;362:1445–1457. doi: 10.1098/rstb.2007.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. 2007;171:1407–1418. doi: 10.2353/ajpath.2007.070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Lange FJ, et al. Lineage and morphogenetic analysis of the cardiac valves. Circ Res. 2004;95:645–654. doi: 10.1161/01.RES.0000141429.13560.cb. [DOI] [PubMed] [Google Scholar]
- 36.Lincoln J, Alfieri CM, Yutzey KE. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev Dyn. 2004;230:239–250. doi: 10.1002/dvdy.20051. [DOI] [PubMed] [Google Scholar]
- 37.Tao G, Kotick JD, Lincoln J. Heart valve development, maintenance, and disease: the role of endothelial cells. Curr Top Dev Biol. 2012;100:203–232. doi: 10.1016/B978-0-12-387786-4.00006-3. [DOI] [PubMed] [Google Scholar]
- 38.von Gise A, Pu WT. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ Res. 2012;110:1628–1645. doi: 10.1161/CIRCRESAHA.111.259960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hakuno D, et al. Periostin advances atherosclerotic and rheumatic cardiac valve degeneration by inducing angiogenesis and MMP production in humans and rodents. J Clin Invest. 2010;120:2292–2306. doi: 10.1172/JCI40973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lie-Venema H, et al. Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. Scientific World Journal. 2007;7:1777–1798. doi: 10.1100/tsw.2007.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wessels A, et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev Biol. 2012;366:111–124. doi: 10.1016/j.ydbio.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paruchuri S, et al. Human pulmonary valve progenitor cells exhibit endothelial/mesenchymal plasticity in response to vascular endothelial growth factor-A and transforming growth factor-beta2. Circ Res. 2006;99:861–869. doi: 10.1161/01.RES.0000245188.41002.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aikawa E, et al. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation. 2006;113:1344–1352. doi: 10.1161/CIRCULATIONAHA.105.591768. [DOI] [PubMed] [Google Scholar]
- 44.Fornes P, et al. Correlation between clinical and histologic patterns of degenerative mitral valve insufficiency: a histomorphometric study of 130 excised segments. Cardiovasc Pathol. 1999;8:81–92. doi: 10.1016/s1054-8807(98)00021-0. [DOI] [PubMed] [Google Scholar]
- 45.Shapero K, Wylie-Sears J, Levine RA, Mayer JE, Jr, Bischoff J. Reciprocal interactions between mitral valve endothelial and interstitial cells reduce endothelial-to-mesenchymal transition and myofibroblastic activation. J Mol Cell Cardiol. 2015;80C:175–185. doi: 10.1016/j.yjmcc.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Timek TA, et al. Ablation of mitral annular and leaflet muscle: effects on annular and leaflet dynamics. Am J Physiol Heart Circ Physiol. 2003;285:H1668–H1674. doi: 10.1152/ajpheart.00179.2003. [DOI] [PubMed] [Google Scholar]
- 47.Jensen MO, et al. Saddle-shaped mitral valve annuloplasty rings improve leaflet coaptation geometry. J Thorac Cardiovasc Surg. 2011;142:697–703. doi: 10.1016/j.jtcvs.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salgo IS, et al. Effect of annular shape on leaflet curvature in reducing mitral leaflet stress. Circulation. 2002;106:711–717. doi: 10.1161/01.cir.0000025426.39426.83. [DOI] [PubMed] [Google Scholar]
- 49.Itoh A, et al. Contribution of myocardium overlying the anterolateral papillary muscle to left ventricular deformation. Am J Physiol Heart Circ Physiol. 2012;302:H180–H187. doi: 10.1152/ajpheart.00687.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jensen MO, et al. Saddle-shaped mitral valve annuloplasty rings experience lower forces compared with flat rings. Circulation. 2008;118:S250–S255. doi: 10.1161/CIRCULATIONAHA.107.746776. [DOI] [PubMed] [Google Scholar]
- 51.Rausch MK, et al. In vivo dynamic strains of the ovine anterior mitral valve leaflet. J Biomech. 2011;44:1149–1157. doi: 10.1016/j.jbiomech.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grashow JS, Sacks MS, Liao J, Yoganathan AP. Planar biaxial creep and stress relaxation of the mitral valve anterior leaflet. Ann Biomed Eng. 2006;34:1509–1518. doi: 10.1007/s10439-006-9183-8. [DOI] [PubMed] [Google Scholar]
- 53.Grashow JS, Yoganathan AP, Sacks MS. Biaixal stress-stretch behavior of the mitral valve anterior leaflet at physiologic strain rates. Ann Biomed Eng. 2006;34:315–325. doi: 10.1007/s10439-005-9027-y. [DOI] [PubMed] [Google Scholar]
- 54.Padala M, et al. Saddle shape of the mitral annulus reduces systolic strains on the P2 segment of the posterior mitral leaflet. Ann Thorac Surg. 2009;88:1499–1504. doi: 10.1016/j.athoracsur.2009.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Niwa K, Ikeda F, Miyamoto H, Nakajima H, Ando M. Absent aortic valve with normally related great arteries. Heart Vessels. 1987;3:104–107. doi: 10.1007/BF02058528. [DOI] [PubMed] [Google Scholar]
- 56.Gupta V, Werdenberg JA, Blevins TL, Grande-Allen KJ. Synthesis of glycosaminoglycans in differently loaded regions of collagen gels seeded with valvular interstitial cells. Tissue Eng. 2007;13:41–49. doi: 10.1089/ten.2006.0091. [DOI] [PubMed] [Google Scholar]
- 57.Ku CH, et al. Collagen synthesis by mesenchymal stem cells and aortic valve interstitial cells in response to mechanical stretch. Cardiovasc Res. 2006;71:548–556. doi: 10.1016/j.cardiores.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 58.Merryman WD. Insights into (the interstitium of) degenerative aortic valve disease. J Am Coll Cardiol. 2008;51:1415. doi: 10.1016/j.jacc.2007.11.068. [DOI] [PubMed] [Google Scholar]
- 59.Li C, Gotlieb AI. Transforming growth factor-beta regulates the growth of valve interstitial cells in vitro. Am J Pathol. 2011;179:1746–1755. doi: 10.1016/j.ajpath.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Balachandran K, et al. Cyclic strain induces dual-mode endothelial-mesenchymal transformation of the cardiac valve. Proc Natl Acad Sci USA. 2011;108:19943–19948. doi: 10.1073/pnas.1106954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.El-Hamamsy I, et al. Rate of progression and functional significance of aortic root calcification after homograft versus freestyle aortic root replacement. Circulation. 2009;120:S269–S275. doi: 10.1161/CIRCULATIONAHA.108.843748. [DOI] [PubMed] [Google Scholar]
- 62.Mahler GJ, Farrar EJ, Butcher JT. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:121–130. doi: 10.1161/ATVBAHA.112.300504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kilner PJ, et al. Asymmetric redirection of flow through the heart. Nature. 2000;404:759–761. doi: 10.1038/35008075. [DOI] [PubMed] [Google Scholar]
- 64.Ro R, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol. 2014;64:1984–1995. doi: 10.1016/j.jacc.2014.04.090. [DOI] [PubMed] [Google Scholar]
- 65.Eriksson J, et al. Semi-automatic quantification of 4D left ventricular blood flow. J Cardiovasc Magn Reson. 2010;12:9. doi: 10.1186/1532-429X-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bolger AF, et al. Transit of blood flow through the human left ventricle mapped by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2007;9:741–747. doi: 10.1080/10976640701544530. [DOI] [PubMed] [Google Scholar]
- 67.Markwald RR, Fitzharris TP, Manasek FJ. Structural development of endocardial cushions. Am J Anat. 1977;148:85–119. doi: 10.1002/aja.1001480108. [DOI] [PubMed] [Google Scholar]
- 68.Markwald RR, Norris RA, Moreno-Rodriguez R, Levine RA. Developmental basis of adult cardiovascular diseases: valvular heart diseases. Ann N Y Acad Sci. 2010;1188:177–183. doi: 10.1111/j.1749-6632.2009.05098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Vlaming A, et al. Atrioventricular valve development: new perspectives on an old theme. Differentiation. 2012;84:103–116. doi: 10.1016/j.diff.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hinton RB, Yutzey KE. Heart valve structure and function in development and disease. Annu Rev Physiol. 2011;73:29–46. doi: 10.1146/annurev-physiol-012110-142145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Armstrong EJ, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circ Res. 2004;95:459–470. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Norris RA, et al. Periostin regulates atrioventricular valve maturation. Dev Biol. 2008;316:200–213. doi: 10.1016/j.ydbio.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Milgrom-Hoffman M, et al. The heart endocardium is derived from vascular endothelial progenitors. Development. 2011;138:4777–4787. doi: 10.1242/dev.061192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Puceat M. Embryological origin of the endocardium and derived valve progenitor cells: from developmental biology to stem cell-based valve repair. Biochim Biophys Acta. 2013;1833:917–922. doi: 10.1016/j.bbamcr.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 75.Inai K, Norris RA, Hoffman S, Markwald RR, Sugi Y. BMP-2 induces cell migration and periostin expression during atrioventricular valvulogenesis. Dev Biol. 2008;315:383–396. doi: 10.1016/j.ydbio.2007.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu B, et al. Nfatc1 coordinates valve endocardial cell lineage development required for heart valve formation. Circ Res. 2011;109:183–192. doi: 10.1161/CIRCRESAHA.111.245035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang JH, Wylie-Sears J, Bischoff J. Opposing actions of Notch1 and VEGF in postnatal cardiac valve endothelial cells. Biochem Biophys Res Commun. 2008;374:512–516. doi: 10.1016/j.bbrc.2008.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sell S, Scully RE. Aging changes in the aortic and mitral valves. Histologic and histochemical studies, with observations on the pathogenesis of calcific aortic stenosis and calcification of the mitral annulus. Am J Pathol. 1965;46:345–365. [PMC free article] [PubMed] [Google Scholar]
- 79.Pomerance A. Ageing changes in human heart valves. Br Heart J. 1967;29:222–231. doi: 10.1136/hrt.29.2.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pham T, Sun W. Material properties of aged human mitral valve leaflets. J Biomed Mater Res A. 2014;102:2692–2703. doi: 10.1002/jbm.a.34939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Freed LA, et al. Prevalence and clinical outcome of mitral-valve prolapse. N Engl J Med. 1999;341:1–7. doi: 10.1056/NEJM199907013410101. [DOI] [PubMed] [Google Scholar]
- 82.Waller BF, et al. Etiology of clinically isolated, severe, chronic, pure mitral regurgitation: analysis of 97 patients over 30 years of age having mitral valve replacement. Am Heart J. 1982;104:276–288. doi: 10.1016/0002-8703(82)90204-6. [DOI] [PubMed] [Google Scholar]
- 83.Freed LA, et al. Mitral valve prolapse in the general population: the benign nature of echocardiographic features in the Framingham Heart Study. J Am Coll Cardiol. 2002;40:1298–1304. doi: 10.1016/s0735-1097(02)02161-7. [DOI] [PubMed] [Google Scholar]
- 84.Avierinos JF, et al. Natural history of asymptomatic mitral valve prolapse in the community. Circulation. 2002;106:1355–1361. doi: 10.1161/01.cir.0000028933.34260.09. [DOI] [PubMed] [Google Scholar]
- 85.Procacci PM, Savran SV, Schreiter SL, Bryson AL. Prevalence of clinical mitral-valve prolapse in 1169 young women. N Engl J Med. 1976;294:1086–1088. doi: 10.1056/NEJM197605132942004. [DOI] [PubMed] [Google Scholar]
- 86.Flack JM, et al. Anthropometric and physiologic correlates of mitral valve prolapse in a biethnic cohort of young adults: the CARDIA study. Am Heart J. 1999;138:486–492. doi: 10.1016/s0002-8703(99)70151-1. [DOI] [PubMed] [Google Scholar]
- 87.Levine RA, et al. Three-dimensional echocardiographic reconstruction of the mitral valve, with implications for the diagnosis of mitral valve prolapse. Circulation. 1989;80:589–598. doi: 10.1161/01.cir.80.3.589. [DOI] [PubMed] [Google Scholar]
- 88.Levine RA, Stathogiannis E, Newell JB, Harrigan P, Weyman AE. Reconsideration of echocardiographic standards for mitral valve prolapse: lack of association between leaflet displacement isolated to the apical four chamber view and independent echocardiographic evidence of abnormality. J Am Coll Cardiol. 1988;11:1010–1019. doi: 10.1016/s0735-1097(98)90059-6. [DOI] [PubMed] [Google Scholar]
- 89.Levine RA, Triulzi MO, Harrigan P, Weyman AE. The relationship of mitral annular shape to the diagnosis of mitral valve prolapse. Circulation. 1987;75:756–767. doi: 10.1161/01.cir.75.4.756. [DOI] [PubMed] [Google Scholar]
- 90.Otani K, et al. Evidence of a vicious cycle in mitral regurgitation with prolapse: secondary tethering attributed to primary prolapse demonstrated by three-dimensional echocardiography exacerbates regurgitation. Circulation. 2012;126:S214–S221. doi: 10.1161/CIRCULATIONAHA.111.084178. [DOI] [PubMed] [Google Scholar]
- 91.Dal-Bianco JP, Beaudoin J, Handschumacher MD, Levine RA. Basic mechanisms of mitral regurgitation. Can J Cardiol. 2014;30:971–981. doi: 10.1016/j.cjca.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Slaugenhaupt S, et al. Genetic mechanisms of mitral valve prolapse. Curr Cardiovasc Risk Rep. 2008;2:463–467. [Google Scholar]
- 93.Addetia K, Mor-Avi V, Weinert L, Salgo IS, Lang RM. A new definition for an old entity: improved definition of mitral valve prolapse using three-dimensional echocardiography and color-coded parametric models. J Am Soc Echocardiogr. 2014;27:8–16. doi: 10.1016/j.echo.2013.08.025. [DOI] [PubMed] [Google Scholar]
- 94.Ling LH, et al. Early surgery in patients with mitral regurgitation due to flail leaflets: a long-term outcome study. Circulation. 1997;96:1819–1825. doi: 10.1161/01.cir.96.6.1819. [DOI] [PubMed] [Google Scholar]
- 95.Ling LH, et al. Clinical outcome of mitral regurgitation due to flail leaflet. N Engl J Med. 1996;335:1417–1423. doi: 10.1056/NEJM199611073351902. [DOI] [PubMed] [Google Scholar]
- 96.Carabello BA. The current therapy for mitral regurgitation. J Am Coll Cardiol. 2008;52:319–326. doi: 10.1016/j.jacc.2008.02.084. [DOI] [PubMed] [Google Scholar]
- 97.Grigioni F, et al. Outcomes in mitral regurgitation due to flail leaflets a multicenter European study. JACC Cardiovasc Imaging. 2008;1:133–141. doi: 10.1016/j.jcmg.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 98.Le Tourneau T, et al. Right ventricular systolic function in organic mitral regurgitation: impact of biventricular impairment. Circulation. 2013;127:1597–1608. doi: 10.1161/CIRCULATIONAHA.112.000999. [DOI] [PubMed] [Google Scholar]
- 99.Urabe Y, et al. Cellular and ventricular contractile dysfunction in experimental canine mitral regurgitation. Circ Res. 1992;70:131–147. doi: 10.1161/01.res.70.1.131. [DOI] [PubMed] [Google Scholar]
- 100.Chiechi MA, Lees WM, Thompson R. Functional anatomy of the normal mitral valve. J Thorac Surg. 1956;32:378–398. [PubMed] [Google Scholar]
- 101.Rusted IE, Scheifley CH, Edwards JE. Studies of the mitral valve. I. Anatomic features of the normal mitral valve and associated structures. Circulation. 1952;6:825–831. doi: 10.1161/01.cir.6.6.825. [DOI] [PubMed] [Google Scholar]
- 102.Ormiston JA, Shah PM, Tei C, Wong M. Size and motion of the mitral valve annulus in man. I. A two-dimensional echocardiographic method and findings in normal subjects. Circulation. 1981;64:113–120. doi: 10.1161/01.cir.64.1.113. [DOI] [PubMed] [Google Scholar]
- 103.Gorgulu S, et al. Influence of different echocardiographic imaging modes on the assessment of anterior mitral leaflet thickness. J Heart Valve Dis. 2005;14:204–208. [PubMed] [Google Scholar]
- 104.Louie EK, et al. Transesophageal echocardiographic assessment of the contribution of intrinsic tissue thickness to the appearance of a thick mitral valve in patients with mitral valve prolapse. J Am Coll Cardiol. 1996;28:465–471. doi: 10.1016/0735-1097(96)00160-X. [DOI] [PubMed] [Google Scholar]
- 105.Marks AR, Choong CY, Sanfilippo AJ, Ferre M, Weyman AE. Identification of high-risk and low-risk subgroups of patients with mitral-valve prolapse. N Engl J Med. 1989;320:1031–1036. doi: 10.1056/NEJM198904203201602. [DOI] [PubMed] [Google Scholar]
- 106.Grayburn PA, et al. Relation of echocardiographic morphology of the mitral apparatus to mitral regurgitation in mitral valve prolapse: assessment by Doppler color flow imaging. Am Heart J. 1990;119:1095–1102. doi: 10.1016/s0002-8703(05)80240-6. [DOI] [PubMed] [Google Scholar]
- 107.Schwammenthal E, et al. Dynamics of mitral regurgitant flow and orifice area. Physiologic application of the proximal flow convergence method: clinical data and experimental testing. Circulation. 1994;90:307–322. doi: 10.1161/01.cir.90.1.307. [DOI] [PubMed] [Google Scholar]
- 108.Enriquez-Sarano M, Sinak LJ, Tajik AJ, Bailey KR, Seward JB. Changes in effective regurgitant orifice throughout systole in patients with mitral valve prolapse. A clinical study using the proximal isovelocity surface area method. Circulation. 1995;92:2951–295. doi: 10.1161/01.cir.92.10.2951. [DOI] [PubMed] [Google Scholar]
- 109.Chapman DW. The cumulative risks of prolapsing mitral valve. 40 years of follow-up. Tex Heart Inst J. 1994;21:267–271. [PMC free article] [PubMed] [Google Scholar]
- 110.Avierinos JF, Detaint D, Messika-Zeitoun D, Mohty D, Enriquez-Sarano M. Risk, determinants, and outcome implications of progression of mitral regurgitation after diagnosis of mitral valve prolapse in a single community. Am J Cardiol. 2008;101:662–667. doi: 10.1016/j.amjcard.2007.10.029. [DOI] [PubMed] [Google Scholar]
- 111.Lee AP, et al. Quantitative analysis of mitral valve morphology in mitral valve prolapse with real-time 3-dimensional echocardiography: importance of annular saddle shape in the pathogenesis of mitral regurgitation. Circulation. 2013;127:832–841. doi: 10.1161/CIRCULATIONAHA.112.118083. [DOI] [PubMed] [Google Scholar]
- 112.Jensen MO, Hagege AA, Otsuji Y, Levine RA, Leducq Transatlantic MITRAL Network The unsaddled annulus: biomechanical culprit in mitral valve prolapse? Circulation. 2013;127:766–768. doi: 10.1161/CIRCULATIONAHA.112.000628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Delling FN, Vasan RS. Epidemiology and pathophysiology of mitral valve prolapse: new insights into disease progression, genetics, and molecular basis. Circulation. 2014;129:2158–2170. doi: 10.1161/CIRCULATIONAHA.113.006702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Delling FN, et al. Mild expression of mitral valve prolapse in the Framingham offspring: expanding the phenotypic spectrum. J Am Soc Echocardiogr. 2014;27:17–23. doi: 10.1016/j.echo.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nesta F, et al. New locus for autosomal dominant mitral valve prolapse on chromosome 13: clinical insights from genetic studies. Circulation. 2005;112:2022–2030. doi: 10.1161/CIRCULATIONAHA.104.516930. [DOI] [PubMed] [Google Scholar]
- 116.Zuppiroli A, Roman MJ, O’Grady M, Devereux RB. A family study of anterior mitral leaflet thickness and mitral valve prolapse. Am J Cardiol. 1998;82:823–826. doi: 10.1016/s0002-9149(98)00454-8. [DOI] [PubMed] [Google Scholar]
- 117.Flameng W, Meuris B, Herijgers P, Herregods MC. Durability of mitral valve repair in Barlow disease versus fibroelastic deficiency. J Thorac Cardiovasc Surg. 2008;135:274–282. doi: 10.1016/j.jtcvs.2007.06.040. [DOI] [PubMed] [Google Scholar]
- 118.Matsumaru I, et al. Clinical and pathological features of degenerative mitral valve disease: billowing mitral leaflet versus fibroelastic deficiency. Ann Thorac Cardiovasc Surg. 2014;20:987–994. doi: 10.5761/atcs.oa.13-00168. [DOI] [PubMed] [Google Scholar]
- 119.Chandra S, et al. Characterization of degenerative mitral valve disease using morphologic analysis of real-time three-dimensional echocardiographic images: objective insight into complexity and planning of mitral valve repair. Circ Cardiovasc Imaging. 2011;4:24–32. doi: 10.1161/CIRCIMAGING.109.924332. [DOI] [PubMed] [Google Scholar]
- 120.Anyanwu AC, Adams DH. Etiologic classification of degenerative mitral valve disease: Barlow’s disease and fibroelastic deficiency. Semin Thorac Cardiovasc Surg. 2007;19:90–96. doi: 10.1053/j.semtcvs.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 121.Akhtar S, Meek KM, James V. Ultrastructure abnormalities in proteoglycans, collagen fibrils, and elastic fibers in normal and myxomatous mitral valve chordae tendineae. Cardiovasc Pathol. 1999;8:191–201. doi: 10.1016/s1054-8807(99)00004-6. [DOI] [PubMed] [Google Scholar]
- 122.Rabkin E, et al. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. 2001;104:2525–2532. doi: 10.1161/hc4601.099489. [DOI] [PubMed] [Google Scholar]
- 123.Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J Heart Valve Dis. 2004;13:841–847. [PubMed] [Google Scholar]
- 124.Grande-Allen KJ, Griffin BP, Ratliff NB, Cosgrove DM, Vesely I. Glycosaminoglycan profiles of myxomatous mitral leaflets and chordae parallel the severity of mechanical alterations. J Am Coll Cardiol. 2003;42:271–277. doi: 10.1016/s0735-1097(03)00626-0. [DOI] [PubMed] [Google Scholar]
- 125.Dainese L, et al. Fine characterization of mitral valve glycosaminoglycans and their modification with degenerative disease. Clin Chem Lab Med. 2007;45:361–366. doi: 10.1515/CCLM.2007.061. [DOI] [PubMed] [Google Scholar]
- 126.Gupta V, et al. Abundance and location of proteoglycans and hyaluronan within normal and myxomatous mitral valves. Cardiovasc Pathol. 2009;18:191–197. doi: 10.1016/j.carpath.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mills WR, et al. Biomechanical and echocardiographic characterization of flail mitral leaflet due to myxomatous disease: further evidence for early surgical intervention. Am Heart J. 2004;148:144–150. doi: 10.1016/j.ahj.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 128.Barlow JB, Bosman CK. Aneurysmal protrusion of the posterior leaflet of the mitral valve. An auscultatory-electrocardiographic syndrome. Am Heart J. 1966;71:166–178. doi: 10.1016/0002-8703(66)90179-7. [DOI] [PubMed] [Google Scholar]
- 129.Carpentier A, et al. Reconstructive surgery of mitral valve incompetence: ten-year appraisal. J Thorac Cardiovasc Surg. 1980;79:338–348. [PubMed] [Google Scholar]
- 130.McKusick VA. Heritable Disorders of Connective Tissue. C. V. Mosby Company; 1972. [Google Scholar]
- 131.Disse S, et al. Mapping of a first locus for autosomal dominant myxomatous mitral-valve prolapse to chromosome 16p11.2-p12.1. Am J Hum Genet. 1999;65:1242–1251. doi: 10.1086/302624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Freed LA, et al. A locus for autosomal dominant mitral valve prolapse on chromosome 11p15.4. Am J Hum Genet. 2003;72:1551–1559. doi: 10.1086/375452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Oceandy D, Yusoff R, Baudoin FM, Neyses L, Ray SG. Promoter polymorphism of the matrix metalloproteinase 3 gene is associated with regurgitation and left ventricular remodelling in mitral valve prolapse patients. Eur J Heart Fail. 2007;9:1010–1017. doi: 10.1016/j.ejheart.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 134.Delling FN, et al. Familial clustering of mitral valve prolapse in the community. Circulation. 2015;131:263–268. doi: 10.1161/CIRCULATIONAHA.114.012594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pini R, Greppi B, Kramer-Fox R, Roman MJ, Devereux RB. Mitral valve dimensions and motion and familial transmission of mitral valve prolapse with and without mitral leaflet billowing. J Am Coll Cardiol. 1988;12:1423–1431. doi: 10.1016/s0735-1097(88)80005-6. [DOI] [PubMed] [Google Scholar]
- 136.Devereux RB, Brown WT, Kramer-Fox R, Sachs I. Inheritance of mitral valve prolapse: effect of age and sex on gene expression. Ann Intern Med. 1982;97:826–832. doi: 10.7326/0003-4819-97-6-826. [DOI] [PubMed] [Google Scholar]
- 137.Levine RA, Slaugenhaupt SA. Molecular genetics of mitral valve prolapse. Curr Opin Cardiol. 2007;22:171–175. doi: 10.1097/HCO.0b013e3280f3bfcd. [DOI] [PubMed] [Google Scholar]
- 138.Loardi C, et al. Biology of mitral valve prolapse: the harvest is big, but the workers are few. Int J Cardiol. 2011;151:129–135. doi: 10.1016/j.ijcard.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 139.Dietz HC, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 140.Pyeritz RE, Wappel MA. Mitral valve dysfunction in the Marfan syndrome. Clinical and echocardiographic study of prevalence and natural history. Am J Med. 1983;74:797–807. doi: 10.1016/0002-9343(83)91070-7. [DOI] [PubMed] [Google Scholar]
- 141.Taub CC, et al. Mitral valve prolapse in Marfan syndrome: an old topic revisited. Echocardiography. 2009;26:357–364. doi: 10.1111/j.1540-8175.2008.00825.x. [DOI] [PubMed] [Google Scholar]
- 142.Montgomery RA, et al. Multiple molecular mechanisms underlying subdiagnostic variants of Marfan syndrome. Am J Hum Genet. 1998;63:1703–1711. doi: 10.1086/302144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Glesby MJ, Pyeritz RE. Association of mitral valve prolapse and systemic abnormalities of connective tissue. A phenotypic continuum. JAMA. 1989;262:523–528. [PubMed] [Google Scholar]
- 144.Judge DP, Rouf R, Habashi J, Dietz HC. Mitral valve disease in Marfan syndrome and related disorders. J Cardiovasc Transl Res. 2011;4:741–747. doi: 10.1007/s12265-011-9314-y. [DOI] [PubMed] [Google Scholar]
- 145.Habashi JP, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Neptune ER, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 147.Isogai Z, et al. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem. 2003;278:2750–2757. doi: 10.1074/jbc.M209256200. [DOI] [PubMed] [Google Scholar]
- 148.Brooke BS, et al. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cohn RD, et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13:204–210. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Geirsson A, et al. Modulation of transforming growth factor-beta signaling and extracellular matrix production in myxomatous mitral valves by angiotensin II receptor blockers. Circulation. 2012;126:S189–S197. doi: 10.1161/CIRCULATIONAHA.111.082610. [DOI] [PubMed] [Google Scholar]
- 151.Loeys BL, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 152.Lindsay ME, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44:922–927. doi: 10.1038/ng.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Attias D, et al. Comparison of clinical presentations and outcomes between patients with TGFBR2 and FBN1 mutations in Marfan syndrome and related disorders. Circulation. 2009;120:2541–2549. doi: 10.1161/CIRCULATIONAHA.109.887042. [DOI] [PubMed] [Google Scholar]
- 154.van de Laar IM, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43:121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 155.van de Laar IM, et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J Med Genet. 2012;49:47–57. doi: 10.1136/jmedgenet-2011-100382. [DOI] [PubMed] [Google Scholar]
- 156.Habashi JP, et al. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;332:361–365. doi: 10.1126/science.1192152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Holm TM, et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358–361. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011;473:308–316. doi: 10.1038/nature10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Monteleone PL, Fagan LF. Possible X-linked congenital heart disease. Circulation. 1969;39:611–614. doi: 10.1161/01.cir.39.5.611. [DOI] [PubMed] [Google Scholar]
- 160.Kyndt F, et al. Mapping of X-linked myxomatous valvular dystrophy to chromosome Xq28. Am J Hum Genet. 1998;62:627–632. doi: 10.1086/301747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kyndt F, et al. Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation. 2007;115:40–49. doi: 10.1161/CIRCULATIONAHA.106.622621. [DOI] [PubMed] [Google Scholar]
- 162.Aalberts JJ, et al. Screening of TGFBR1, TGFBR2, and FLNA in familial mitral valve prolapse. Am J Med Genet A. 2014;164A:113–119. doi: 10.1002/ajmg.a.36211. [DOI] [PubMed] [Google Scholar]
- 163.Nakamura F, Stossel TP, Hartwig JH. The filamins: organizers of cell structure and function. Cell Adh Migr. 2011;5:160–169. doi: 10.4161/cam.5.2.14401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhou AX, Hartwig JH, Akyurek LM. Filamins in cell signaling, transcription and organ development. Trends Cell Biol. 2010;20:113–123. doi: 10.1016/j.tcb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 165.Baldassarre M, et al. Filamins regulate cell spreading and initiation of cell migration. PLoS ONE. 2009;4:e7830. doi: 10.1371/journal.pone.0007830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Norris RA, et al. Expression of the familial cardiac valvular dystrophy gene, filamin-A, during heart morphogenesis. Dev Dyn. 2010;239:2118–2127. doi: 10.1002/dvdy.22346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Duval D, et al. Valvular dystrophy associated filamin A mutations reveal a new role of its first repeats in small-GTPase regulation. Biochim Biophys Acta. 2014;1843:234–244. doi: 10.1016/j.bbamcr.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Feng Y, et al. Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc Natl Acad Sci USA. 2006;103:19836–19841. doi: 10.1073/pnas.0609628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Stossel TP, et al. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001;2:138–145. doi: 10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- 170.Sauls K, et al. Developmental basis for filamin-A-associated myxomatous mitral valve disease. Cardiovasc Res. 2012;96:109–119. doi: 10.1093/cvr/cvs238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Grau JB, Pirelli L, Yu PJ, Galloway AC, Ostrer H. The genetics of mitral valve prolapse. Clin Genet. 2007;72:288–295. doi: 10.1111/j.1399-0004.2007.00865.x. [DOI] [PubMed] [Google Scholar]
- 172.Padang R, Bagnall RD, Semsarian C. Genetic basis of familial valvular heart disease. Circ Cardiovasc Genet. 2012;5:569–580. doi: 10.1161/CIRCGENETICS.112.962894. [DOI] [PubMed] [Google Scholar]
- 173.Kolibash AJ, Jr, et al. Evidence for progression from mild to severe mitral regurgitation in mitral valve prolapse. Am J Cardiol. 1986;58:762–767. doi: 10.1016/0002-9149(86)90352-8. [DOI] [PubMed] [Google Scholar]
- 174.Jimenez JH, Soerensen DD, He Z, He S, Yoganathan AP. Effects of a saddle shaped annulus on mitral valve function and chordal force distribution: an in vitro study. Ann Biomed Eng. 2003;31:1171–1181. doi: 10.1114/1.1616929. [DOI] [PubMed] [Google Scholar]
- 175.Hagler MA, et al. TGF-beta signalling and reactive oxygen species drive fibrosis and matrix remodelling in myxomatous mitral valves. Cardiovasc Res. 2013;99:175–184. doi: 10.1093/cvr/cvt083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hulin A, et al. Emerging pathogenic mechanisms in human myxomatous mitral valve: lessons from past and novel data. Cardiovasc Pathol. 2013;22:245–250. doi: 10.1016/j.carpath.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 177.Sainger R, et al. Human myxomatous mitral valve prolapse: role of bone morphogenetic protein 4 in valvular interstitial cell activation. J Cell Physiol. 2012;227:2595–2604. doi: 10.1002/jcp.22999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Madsen MB, et al. Identification of 2 loci associated with development of myxomatous mitral valve disease in Cavalier King Charles Spaniels. J Hered. 2011;102(Suppl 1):S62–S67. doi: 10.1093/jhered/esr041. [DOI] [PubMed] [Google Scholar]
- 179.Obayashi K, et al. Effects of transforming growth factor-beta3 and matrix metalloproteinase-3 on the pathogenesis of chronic mitral valvular disease in dogs. Am J Vet Res. 2011;72:194–202. doi: 10.2460/ajvr.72.2.194. [DOI] [PubMed] [Google Scholar]
- 180.Spirito P, Maron BJ. Patterns of systolic anterior motion of the mitral valve in hypertrophic cardiomyopathy: assessment by two-dimensional echocardiography. Am J Cardiol. 1984;54:1039–1046. doi: 10.1016/s0002-9149(84)80141-1. [DOI] [PubMed] [Google Scholar]
- 181.Maron MS, et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003;348:295–303. doi: 10.1056/NEJMoa021332. [DOI] [PubMed] [Google Scholar]
- 182.Maron MS, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. 2006;114:2232–2239. doi: 10.1161/CIRCULATIONAHA.106.644682. [DOI] [PubMed] [Google Scholar]
- 183.Maron BJ, Epstein SE. Hypertrophic cardiomyopathy. Recent observations regarding the specificity of three hallmarks of the disease: asymmetric septal hypertrophy, septal disorganization and systolic anterior motion of the anterior mitral leaflet. Am J Cardiol. 1980;45:141–154. doi: 10.1016/0002-9149(80)90232-5. [DOI] [PubMed] [Google Scholar]
- 184.Levine RA, Schwammenthal E, Song JK. Diastolic leading to systolic anterior motion: new technology reveals physiology. J Am Coll Cardiol. 2014;64:1996–1999. doi: 10.1016/j.jacc.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Jiang L, Levine RA, King ME, Weyman AE. An integrated mechanism for systolic anterior motion of the mitral valve in hypertrophic cardiomyopathy based on echocardiographic observations. Am Heart J. 1987;113:633–644. doi: 10.1016/0002-8703(87)90701-0. [DOI] [PubMed] [Google Scholar]
- 186.Lefebvre XP, He S, Levine RA, Yoganathan AP. Systolic anterior motion of the mitral valve in hypertrophic cardiomyopathy: an in vitro pulsatile flow study. J Heart Valve Dis. 1995;4:422–438. [PubMed] [Google Scholar]
- 187.Levine RA, et al. Papillary muscle displacement causes systolic anterior motion of the mitral valve. Experimental validation and insights into the mechanism of subaortic obstruction. Circulation. 1995;91:1189–1195. doi: 10.1161/01.cir.91.4.1189. [DOI] [PubMed] [Google Scholar]
- 188.Sherrid MV, Gunsburg DZ, Moldenhauer S, Pearle G. Systolic anterior motion begins at low left ventricular outflow tract velocity in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2000;36:1344–1354. doi: 10.1016/s0735-1097(00)00830-5. [DOI] [PubMed] [Google Scholar]
- 189.Cape EG, et al. Chordal geometry determines the shape and extent of systolic anterior mitral motion: in vitro studies. J Am Coll Cardiol. 1989;13:1438–1448. doi: 10.1016/0735-1097(89)90326-4. [DOI] [PubMed] [Google Scholar]
- 190.Nakatani S, et al. New insights into the reduction of mitral valve systolic anterior motion after ventricular septal myectomy in hypertrophic obstructive cardiomyopathy. Am Heart J. 1996;131:294–300. doi: 10.1016/s0002-8703(96)90357-9. [DOI] [PubMed] [Google Scholar]
- 191.Yoganathan AP, Lemmon JD, Jr, Kim YH, Levine RA, Vesier CC. A three-dimensional computational investigation of intraventricular fluid dynamics: examination into the initiation of systolic anterior motion of the mitral valve leaflets. J Biomech Eng. 1995;117:94–102. doi: 10.1115/1.2792276. [DOI] [PubMed] [Google Scholar]
- 192.Maron BJ, Harding AM, Spirito P, Roberts WC, Waller BF. Systolic anterior motion of the posterior mitral leaflet: a previously unrecognized cause of dynamic subaortic obstruction in patients with hypertrophic cardiomyopathy. Circulation. 1983;68:282–293. doi: 10.1161/01.cir.68.2.282. [DOI] [PubMed] [Google Scholar]
- 193.Delling FN, et al. Frequency and mechanism of persistent systolic anterior motion and mitral regurgitation after septal ablation in obstructive hypertrophic cardiomyopathy. Am J Cardiol. 2007;100:1691–1695. doi: 10.1016/j.amjcard.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 194.Kaple RK, et al. Mitral valve abnormalities in hypertrophic cardiomyopathy: echocardiographic features and surgical outcomes. Ann Thorac Surg. 2008;85:1527–1535.e2. doi: 10.1016/j.athoracsur.2008.01.061. [DOI] [PubMed] [Google Scholar]
- 195.Bryant R, 3rd, Smedira NG. Papillary muscle realignment for symptomatic left ventricular outflow tract obstruction. J Thorac Cardiovasc Surg. 2008;135:223–224. doi: 10.1016/j.jtcvs.2007.08.034. [DOI] [PubMed] [Google Scholar]
- 196.McIntosh CL, Maron BJ, Cannon RO, 3rd, Klues HG. Initial results of combined anterior mitral leaflet plication and ventricular septal myotomy-myectomy for relief of left ventricular outflow tract obstruction in patients with hypertrophic cardiomyopathy. Circulation. 1992;86(Suppl II):II60–II67. [PubMed] [Google Scholar]
- 197.Ross RE, Sherrid MV, Casey MM, Swistel DG, Balaram SK. Does surgical relief of obstruction improve prognosis for hypertrophic cardiomyopathy? Prog Cardiovasc Dis. 2012;54:529–534. doi: 10.1016/j.pcad.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 198.Schwammenthal E, Levine RA. Dynamic subaortic obstruction: a disease of the mitral valve suitable for surgical repair? J Am Coll Cardiol. 1996;28:203–20. doi: 10.1016/0735-1097(96)00213-6. [DOI] [PubMed] [Google Scholar]
- 199.Seeburger J, Passage J, Borger MA, Mohr FW. A new concept for correction of systolic anterior motion and mitral valve regurgitation in patients with hypertrophic obstructive cardiomyopathy. J Thorac Cardiovasc Surg. 2010;140:481–483. doi: 10.1016/j.jtcvs.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 200.van der Lee C, Kofflard MJ, van Herwerden LA, Vletter WB, ten Cate FJ. Sustained improvement after combined anterior mitral leaflet extension and myectomy in hypertrophic obstructive cardiomyopathy. Circulation. 2003;108:2088–2092. doi: 10.1161/01.CIR.0000092912.57140.14. [DOI] [PubMed] [Google Scholar]
- 201.He S, et al. Importance of leaflet elongation in causing systolic anterior motion of the mitral valve. J Heart Valve Dis. 1997;6:149–159. [PubMed] [Google Scholar]
- 202.Klues HG, Roberts WC, Maron BJ. Anomalous insertion of papillary muscle directly into anterior mitral leaflet in hypertrophic cardiomyopathy. Significance in producing left ventricular outflow obstruction. Circulation. 1991;84:1188–1197. doi: 10.1161/01.cir.84.3.1188. [DOI] [PubMed] [Google Scholar]
- 203.Musumeci B, Spirito P, Parodi MI, Assenza GE, Autore C. Congenital accessory mitral valve tissue anomaly in a patient with genetically confirmed hypertrophic cardiomyopathy. J Am Soc Echocardiogr. 2011;24:592.e5–e6. doi: 10.1016/j.echo.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 204.Zhu WX, Oh JK, Kopecky SL, Schaff HV, Tajik AJ. Mitral regurgitation due to ruptured chordae tendineae in patients with hypertrophic obstructive cardiomyopathy. J Am Coll Cardiol. 1992;20:242–247. doi: 10.1016/0735-1097(92)90166-k. [DOI] [PubMed] [Google Scholar]
- 205.Petrone RK, Klues HG, Panza JA, Peterson EE, Maron BJ. Coexistence of mitral valve prolapse in a consecutive group of 528 patients with hypertrophic cardiomyopathy assessed with echocardiography. J Am Coll Cardiol. 1992;20:55–61. doi: 10.1016/0735-1097(92)90137-c. [DOI] [PubMed] [Google Scholar]
- 206.Maron MS, et al. Mitral valve abnormalities identified by cardiovascular magnetic resonance represent a primary phenotypic expression of hypertrophic cardiomyopathy. Circulation. 2011;124:40–47. doi: 10.1161/CIRCULATIONAHA.110.985812. [DOI] [PubMed] [Google Scholar]
- 207.Klues HG, Roberts WC, Maron BJ. Morphological determinants of echocardiographic patterns of mitral valve systolic anterior motion in obstructive hypertrophic cardiomyopathy. Circulation. 1993;87:1570–1579. doi: 10.1161/01.cir.87.5.1570. [DOI] [PubMed] [Google Scholar]
- 208.Jebara VA, et al. Left ventricular outflow tract obstruction after mitral valve repair. Results of the sliding leaflet technique. Circulation. 1993;88(Suppl II):II30–II34. [PubMed] [Google Scholar]
- 209.Klues HG, Maron BJ, Dollar AL, Roberts WC. Diversity of structural mitral valve alterations in hypertrophic cardiomyopathy. Circulation. 1992;85:1651–1660. doi: 10.1161/01.cir.85.5.1651. [DOI] [PubMed] [Google Scholar]
- 210.Hagege AA, et al. Familial hypertrophic cardiomyopathy. Cardiac ultrasonic abnormalities in genetically affected subjects without echocardiographic evidence of left ventricular hypertrophy. Eur Heart J. 1998;19:490–499. doi: 10.1053/euhj.1997.0735. [DOI] [PubMed] [Google Scholar]
- 211.Nematalla H, et al. Targeted Mybpc3 knock-out mice with non-obstructive hypertrophic cardiomyopathy exhibit structural mitral valve abnormalities [abstract] J Am Coll Cardiol. 2011;57:E1397. [Google Scholar]
- 212.Dal-Bianco JP, et al. Active adaptation of the tethered mitral valve: insights into a compensatory mechanism for functional mitral regurgitation. Circulation. 2009;120:334–342. doi: 10.1161/CIRCULATIONAHA.108.846782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Butcher JT, Markwald RR. Valvulogenesis: the moving target. Philos Trans R Soc Lond B Biol Sci. 2007;362:1489–1503. doi: 10.1098/rstb.2007.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Olivotto I, Cecchi F, Poggesi C, Yacoub MH. Developmental origins of hypertrophic cardiomyopathy phenotypes: a unifying hypothesis. Nat Rev Cardiol. 2009;6:317–321. doi: 10.1038/nrcardio.2009.9. [DOI] [PubMed] [Google Scholar]
- 215.Gittenberger-de Groot AC, Vrancken Peeters MP, Mentink MM, Gourdie RG, Poelmann RE. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ Res. 1998;82:1043–1052. doi: 10.1161/01.res.82.10.1043. [DOI] [PubMed] [Google Scholar]
- 216.Prabhakar R, et al. A mouse model of familial hypertrophic cardiomyopathy caused by a alpha-tropomyosin mutation. Mol Cell Biochem. 2003;251:33–42. [PubMed] [Google Scholar]
- 217.Rajan S, et al. Microarray analysis of gene expression during early stages of mild and severe cardiac hypertrophy. Physiol Genomics. 2006;27:309–317. doi: 10.1152/physiolgenomics.00072.2006. [DOI] [PubMed] [Google Scholar]
- 218.Teekakirikul P, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J Clin Invest. 2010;120:3520–3529. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Niu Z, et al. Serum response factor orchestrates nascent sarcomerogenesis and silences the biomineralization gene program in the heart. Proc Natl Acad Sci USA. 2008;105:17824–17829. doi: 10.1073/pnas.0805491105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Norris RA, et al. Identification and detection of the periostin gene in cardiac development. Anat Rec A Discov Mol Cell Evol Biol. 2004;281:1227–1233. doi: 10.1002/ar.a.20135. [DOI] [PubMed] [Google Scholar]
- 221.Levine RA, Hung J. Ischemic mitral regurgitation, the dynamic lesion: clues to the cure. J Am Coll Cardiol. 2003;42:1929–1932. doi: 10.1016/j.jacc.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 222.Godley RW, Wann LS, Rogers EW, Feigenbaum H, Weyman AE. Incomplete mitral leaflet closure in patients with papillary muscle dysfunction. Circulation. 1981;63:565–571. doi: 10.1161/01.cir.63.3.565. [DOI] [PubMed] [Google Scholar]
- 223.Otsuji Y, et al. Mechanism of ischemic mitral regurgitation with segmental left ventricular dysfunction: three-dimensional echocardiographic studies in models of acute and chronic progressive regurgitation. J Am Coll Cardiol. 2001;37:641–648. doi: 10.1016/s0735-1097(00)01134-7. [DOI] [PubMed] [Google Scholar]
- 224.Otsuji Y, et al. Insights from three-dimensional echocardiography into the mechanism of functional mitral regurgitation: direct in vivo demonstration of altered leaflet tethering geometry. Circulation. 1997;96:1999–2008. doi: 10.1161/01.cir.96.6.1999. [DOI] [PubMed] [Google Scholar]
- 225.Yiu SF, Enriquez-Sarano M, Tribouilloy C, Seward JB, Tajik AJ. Determinants of the degree of functional mitral regurgitation in patients with systolic left ventricular dysfunction: A quantitative clinical study. Circulation. 2000;102:1400–1406. doi: 10.1161/01.cir.102.12.1400. [DOI] [PubMed] [Google Scholar]
- 226.Kalra K, et al. Temporal changes in interpapillary muscle dynamics as an active indicator of mitral valve and left ventricular interaction in ischemic mitral regurgitation. J Am Coll Cardiol. 2014;64:1867–1879. doi: 10.1016/j.jacc.2014.07.988. [DOI] [PubMed] [Google Scholar]
- 227.Otsuji Y, et al. Isolated annular dilation does not usually cause important functional mitral regurgitation: comparison between patients with lone atrial fibrillation and those with idiopathic or ischemic cardiomyopathy. J Am Coll Cardiol. 2002;39:1651–1656. doi: 10.1016/s0735-1097(02)01838-7. [DOI] [PubMed] [Google Scholar]
- 228.Meris A, et al. Mechanisms and predictors of mitral regurgitation after high-risk myocardial infarction. J Am Soc Echocardiogr. 2012;25:535–542. doi: 10.1016/j.echo.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Beaudoin J, et al. Severe ischemic mitral regurgitation despite normally contracting subpapillary myocardium. Circulation. 2012;126:138–141. doi: 10.1161/CIRCULATIONAHA.111.064253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Chinitz JS, et al. Mitral apparatus assessment by delayed enhancement CMR: relative impact of infarct distribution on mitral regurgitation. JACC Cardiovasc Imaging. 2013;6:220–234. doi: 10.1016/j.jcmg.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Gorman JH, 3rd, et al. Infarct size and location determine development of mitral regurgitation in the sheep model. J Thorac Cardiovasc Surg. 1998;115:615–622. doi: 10.1016/S0022-5223(98)70326-5. [DOI] [PubMed] [Google Scholar]
- 232.Kumanohoso T, et al. Mechanism of higher incidence of ischemic mitral regurgitation in patients with inferior myocardial infarction: quantitative analysis of left ventricular and mitral valve geometry in 103 patients with prior myocardial infarction. J Thorac Cardiovasc Surg. 2003;125:135–143. doi: 10.1067/mtc.2003.78. [DOI] [PubMed] [Google Scholar]
- 233.Yosefy C, et al. Mitral regurgitation after anteroapical myocardial infarction: new mechanistic insights. Circulation. 2011;123:1529–1536. doi: 10.1161/CIRCULATIONAHA.110.977843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kim K, et al. Mechanism of asymmetric leaflet tethering in ischemic mitral regurgitation: 3D analysis with multislice CT. JACC Cardiovasc Imaging. 2012;5:230–232. doi: 10.1016/j.jcmg.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Ryan LP, et al. Mitral valve tenting index for assessment of subvalvular remodeling. Ann Thorac Surg. 2007;84:1243–1249. doi: 10.1016/j.athoracsur.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 236.Padala M, Gyoneva LI, Thourani VH, Yoganathan AP. Impact of mitral valve geometry on hemodynamic efficacy of surgical repair in secondary mitral regurgitation. J Heart Valve Dis. 2014;23:79–87. [PubMed] [Google Scholar]
- 237.Zeng X, et al. Asymmetric versus symmetric tethering patterns in ischemic mitral regurgitation: geometric differences from three-dimensional transesophageal echocardiography. J Am Soc Echocardiogr. 2014;27:367–375. doi: 10.1016/j.echo.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 238.Barzilai B, Gessler C, Jr, Perez JE, Schaab C, Jaffe AS. Significance of Doppler-detected mitral regurgitation in acute myocardial infarction. Am J Cardiol. 1988;61:220–223. doi: 10.1016/0002-9149(88)90919-8. [DOI] [PubMed] [Google Scholar]
- 239.Grigioni F, et al. Contribution of ischemic mitral regurgitation to congestive heart failure after myocardial infarction. J Am Coll Cardiol. 2005;45:260–267. doi: 10.1016/j.jacc.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 240.Grigioni F, Enriquez-Sarano M, Zehr KJ, Bailey KR, Tajik AJ. Ischemic mitral regurgitation: long-term outcome and prognostic implications with quantitative Doppler assessment. Circulation. 2001;103:1759–1764. doi: 10.1161/01.cir.103.13.1759. [DOI] [PubMed] [Google Scholar]
- 241.Lamas GA, et al. Clinical significance of mitral regurgitation after acute myocardial infarction. Survival and Ventricular Enlargement Investigators. Circulation. 1997;96:827–833. doi: 10.1161/01.cir.96.3.827. [DOI] [PubMed] [Google Scholar]
- 242.Okura H, et al. Functional mitral regurgitation predicts prognosis independent of left ventricular systolic and diastolic indices in patients with ischemic heart disease. J Am Soc Echocardiogr. 2008;21:355–360. doi: 10.1016/j.echo.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 243.Tcheng JE, et al. Outcome of patients sustaining acute ischemic mitral regurgitation during myocardial infarction. Ann Intern Med. 1992;117:18–24. doi: 10.7326/0003-4819-117-1-18. [DOI] [PubMed] [Google Scholar]
- 244.Szymanski C, et al. Impact of mitral regurgitation on exercise capacity and clinical outcomes in patients with ischemic left ventricular dysfunction. Am J Cardiol. 2011;108:1714–1720. doi: 10.1016/j.amjcard.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Beeri R, et al. Gene delivery of sarcoplasmic reticulum calcium ATPase inhibits ventricular remodeling in ischemic mitral regurgitation. Circ Heart Fail. 2010;3:627–634. doi: 10.1161/CIRCHEARTFAILURE.109.891184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Beeri R, et al. Early repair of moderate ischemic mitral regurgitation reverses left ventricular remodeling: a functional and molecular study. Circulation. 2007;116(Suppl I):I288–I293. doi: 10.1161/CIRCULATIONAHA.106.681114. [DOI] [PubMed] [Google Scholar]
- 247.Beeri R, et al. Mitral regurgitation augments post-myocardial infarction remodeling failure of hypertrophic compensation. J Am Coll Cardiol. 2008;51:476–486. doi: 10.1016/j.jacc.2007.07.093. [DOI] [PubMed] [Google Scholar]
- 248.Beaudoin J, et al. Late repair of ischemic mitral regurgitation does not prevent left ventricular remodeling: importance of timing for beneficial repair. Circulation. 2013;128:S248–S252. doi: 10.1161/CIRCULATIONAHA.112.000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Soleimani M, et al. Moderate mitral regurgitation accelerates left ventricular remodeling after posterolateral myocardial infarction. Ann Thorac Surg. 2011;92:1614–1620. doi: 10.1016/j.athoracsur.2011.05.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Hung J, et al. Mechanism of recurrent ischemic mitral regurgitation after annuloplasty: continued LV remodeling as a moving target. Circulation. 2004;110(Suppl II):II85–II90. doi: 10.1161/01.CIR.0000138192.65015.45. [DOI] [PubMed] [Google Scholar]
- 251.Levine RA, Messas E, Nathan NS, Rudski LG. New understanding of ischemic mitral regurgitation: the marionette and its masters. Eur J Echocardiogr. 2004;5:313–317. doi: 10.1016/j.euje.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 252.Qin JX, et al. Importance of mitral valve repair associated with left ventricular reconstruction for patients with ischemic cardiomyopathy: a real-time three-dimensional echocardiographic study. Circulation. 2003;108(Suppl II):II241–II246. doi: 10.1161/01.cir.0000087653.99527.50. [DOI] [PubMed] [Google Scholar]
- 253.Levine RA. Dynamic mitral regurgitation— more than meets the eye. N Engl J Med. 2004;351:1681–1684. doi: 10.1056/NEJMe048165. [DOI] [PubMed] [Google Scholar]
- 254.Lancellotti P, Lebrun F, Pierard LA. Determinants of exercise-induced changes in mitral regurgitation in patients with coronary artery disease and left ventricular dysfunction. J Am Coll Cardiol. 2003;42:1921–1928. doi: 10.1016/j.jacc.2003.04.002. [DOI] [PubMed] [Google Scholar]
- 255.Lancellotti P, Troisfontaines P, Toussaint AC, Pierard LA. Prognostic importance of exercise-induced changes in mitral regurgitation in patients with chronic ischemic left ventricular dysfunction. Circulation. 2003;108:1713–1717. doi: 10.1161/01.CIR.0000087599.49332.05. [DOI] [PubMed] [Google Scholar]
- 256.Sheikh KH, Bengtson JR, Rankin JS, de Bruijn NP, Kisslo J. Intraoperative transesophageal Doppler color flow imaging used to guide patient selection and operative treatment of ischemic mitral regurgitation. Circulation. 1991;84:594–604. doi: 10.1161/01.cir.84.2.594. [DOI] [PubMed] [Google Scholar]
- 257.Grewal KS, et al. Effect of general anesthesia on the severity of mitral regurgitation by transesophageal echocardiography. Am J Cardiol. 2000;85:199–203. doi: 10.1016/s0002-9149(99)00644-x. [DOI] [PubMed] [Google Scholar]
- 258.Tei C, Horikiri Y, Park JC, Jeong JW, Chang KS, Toyama Y, Tanaka N. Acute hemodynamic improvement by thermal vasodilation in congestive heart failure. Circulation. 1995;91:2582–2590. doi: 10.1161/01.cir.91.10.2582. [DOI] [PubMed] [Google Scholar]
- 259.Pierard LA, Lancellotti P. The role of ischemic mitral regurgitation in the pathogenesis of acute pulmonary edema. N Engl J Med. 2004;351:1627–1634. doi: 10.1056/NEJMoa040532. [DOI] [PubMed] [Google Scholar]
- 260.Lancellotti P, et al. Recommendations for the echocardiographic assessment of native valvular regurgitation: an executive summary from the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2013;14:611–644. doi: 10.1093/ehjci/jet105. [DOI] [PubMed] [Google Scholar]
- 261.Grossi EA, et al. Late results of isolated mitral annuloplasty for “functional” ischemic mitral insufficiency. J Card Surg. 2001;16:328–332. doi: 10.1111/j.1540-8191.2001.tb00530.x. [DOI] [PubMed] [Google Scholar]
- 262.McGee EC, et al. Recurrent mitral regurgitation after annuloplasty for functional ischemic mitral regurgitation. J Thorac Cardiovasc Surg. 2004;128:916–924. doi: 10.1016/j.jtcvs.2004.07.037. [DOI] [PubMed] [Google Scholar]
- 263.Cheng A, et al. Undersized mitral annuloplasty inhibits left ventricular basal wall thickening but does not affect equatorial wall cardiac strains. J Heart Valve Dis. 2007;16:349–358. [PubMed] [Google Scholar]
- 264.Acker MA, et al. Mitral-valve repair versus replacement for severe ischemic mitral regurgitation. N Engl J Med. 2014;370:23–32. doi: 10.1056/NEJMoa1312808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Kron IL, Green GR, Cope JT. Surgical relocation of the posterior papillary muscle in chronic ischemic mitral regurgitation. Ann Thorac Surg. 2002;74:600–601. doi: 10.1016/s0003-4975(02)03749-9. [DOI] [PubMed] [Google Scholar]
- 266.Zhu F, et al. Mechanism of persistent ischemic mitral regurgitation after annuloplasty: importance of augmented posterior mitral leaflet tethering. Circulation. 2005;112(Suppl I):I396–I401. doi: 10.1161/CIRCULATIONAHA.104.524561. [DOI] [PubMed] [Google Scholar]
- 267.Kubota K, et al. Functional mitral stenosis after surgical annuloplasty for ischemic mitral regurgitation: importance of subvalvular tethering in the mechanism and dynamic deterioration during exertion. J Thorac Cardiovasc Surg. 2010;140:617–623. doi: 10.1016/j.jtcvs.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Bertrand PB, et al. Mitral valve area during exercise after restrictive mitral valve annuloplasty: importance of diastolic anterior leaflet tethering. J Am Coll Cardiol. 2015;65:452–461. doi: 10.1016/j.jacc.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Schwammenthal E. Undersized and overstretched: mitral mechanics after restrictive annuloplasty. J Am Coll Cardiol. 2015;65:462–464. doi: 10.1016/j.jacc.2014.10.068. [DOI] [PubMed] [Google Scholar]
- 270.Langer F, et al. Subvalvular repair: the key to repairing ischemic mitral regurgitation? Circulation. 2005;112(Suppl I):I383–I389. doi: 10.1161/CIRCULATIONAHA.104.523464. [DOI] [PubMed] [Google Scholar]
- 271.Einstein DR, et al. Fluid-structure interactions of the mitral valve and left heart: comprehensive strategies, past, present and future. Int J Numer Methods Eng. 2010;26:348–380. doi: 10.1002/cnm.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kunzelman KS, Reimink MS, Cochran RP. Annular dilatation increases stress in the mitral valve and delays coaptation: a finite element computer model. Cardiovasc Surg. 1997;5:427–434. doi: 10.1177/096721099700500416. [DOI] [PubMed] [Google Scholar]
- 273.Wenk JF, et al. First finite element model of the left ventricle with mitral valve: insights into ischemic mitral regurgitation. Ann Thorac Surg. 2010;89:1546–1553. doi: 10.1016/j.athoracsur.2010.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Calafiore AM, et al. Echocardiographically based treatment of chronic ischemic mitral regurgitation. J Thorac Cardiovasc Surg. 2011;1411:1150–1156e. doi: 10.1016/j.jtcvs.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 275.Hung J, et al. Reverse ventricular remodeling reduces ischemic mitral regurgitation: echo-guided device application in the beating heart. Circulation. 2002;106:2594–2600. doi: 10.1161/01.cir.0000038363.83133.6d. [DOI] [PubMed] [Google Scholar]
- 276.Messas E, et al. Relief of mitral leaflet tethering following chronic myocardial infarction by chordal cutting diminishes left ventricular remodeling. Circ Cardiovasc Imaging. 2010;3:679–686. doi: 10.1161/CIRCIMAGING.109.931840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Moainie SL, et al. Infarct restraint attenuates remodeling and reduces chronic ischemic mitral regurgitation after postero-lateral infarction. Ann Thorac Surg. 2002;74:444–449. doi: 10.1016/s0003-4975(02)03747-5. [DOI] [PubMed] [Google Scholar]
- 278.Szymanski C, et al. Comprehensive annular and subvalvular repair of chronic ischemic mitral regurgitation improves long-term results with the least ventricular remodeling. Circulation. 2012;126:2720–2727. doi: 10.1161/CIRCULATIONAHA.111.033472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Hung J, et al. A novel approach for reducing ischemic mitral regurgitation by injection of a polymer to reverse remodel and reposition displaced papillary muscles. Circulation. 2008;118:S263–S269. doi: 10.1161/CIRCULATIONAHA.107.756502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Hung J, et al. Persistent reduction of ischemic mitral regurgitation by papillary muscle repositioning: structural stabilization of the papillary muscle-ventricular wall complex. Circulation. 2007;116(Suppl I):I259–I263. doi: 10.1161/CIRCULATIONAHA.106.679951. [DOI] [PubMed] [Google Scholar]
- 281.Liel-Cohen N, et al. Design of a new surgical approach for ventricular remodeling to relieve ischemic mitral regurgitation: insights from 3-dimensional echocardiography. Circulation. 2000;101:2756–2763. doi: 10.1161/01.cir.101.23.2756. [DOI] [PubMed] [Google Scholar]
- 282.Solis J, et al. Polymer injection therapy to reverse remodel the papillary muscles: efficacy in reducing mitral regurgitation in a chronic ischemic model. Circ Cardiovasc Interven. 2010;3:499–505. doi: 10.1161/CIRCINTERVENTIONS.109.850255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Langer F, Schafers HJ. RING plus STRING: papillary muscle repositioning as an adjunctive repair technique for ischemic mitral regurgitation. J Thorac Cardiovasc Surg. 2007;133:247–249. doi: 10.1016/j.jtcvs.2006.04.059. [DOI] [PubMed] [Google Scholar]
- 284.Wakiyama H, et al. Chordal cutting for the treatment of ischemic mitral regurgitation: two case reports [Japanese] J Cardiol. 2004;44:113–117. [PubMed] [Google Scholar]
- 285.Messas E, et al. Chordal cutting: a new therapeutic approach for ischemic mitral regurgitation. Circulation. 2001;104:1958–1963. doi: 10.1161/hc4201.097135. [DOI] [PubMed] [Google Scholar]
- 286.Messas E, et al. Efficacy of chordal cutting to relieve chronic persistent ischemic mitral regurgitation. Circulation. 2003;108(Suppl II):II111–II115. doi: 10.1161/01.cir.0000087658.47544.7f. [DOI] [PubMed] [Google Scholar]
- 287.Messas E, et al. Chordal cutting does not adversely affect left ventricle contractile function. Circulation. 2006;114(Suppl I):I524–I528. doi: 10.1161/CIRCULATIONAHA.105.000612. [DOI] [PubMed] [Google Scholar]
- 288.Yamamoto H, Iguro Y, Sakata R, Arata K, Yotsumoto G. Effectively treating ischemic mitral regurgitation with chordal cutting in combination with ring annuloplasty and left ventricular reshaping approach. J Thorac Cardiovasc Surg. 2005;130:589–590. doi: 10.1016/j.jtcvs.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 289.Borger MA, et al. Initial results of the chordal-cutting operation for ischemic mitral regurgitation. J Thorac Cardiovasc Surg. 2007;133:1483–1492. doi: 10.1016/j.jtcvs.2007.01.064. [DOI] [PubMed] [Google Scholar]
- 290.Jassar AS, et al. Posterior leaflet augmentation in ischemic mitral regurgitation increases leaflet coaptation and mobility. Ann Thorac Surg. 2012;94:1438–1445. doi: 10.1016/j.athoracsur.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Kanzaki H, et al. A mechanism for immediate reduction in mitral regurgitation after cardiac resynchronization therapy: insights from mechanical activation strain mapping. J Am Coll Cardiol. 2004;44:1619–1625. doi: 10.1016/j.jacc.2004.07.036. [DOI] [PubMed] [Google Scholar]
- 292.Smith PK, et al. Surgical treatment of moderate ischemic mitral regurgitation. N Engl J Med. 2014;371:2178–2188. doi: 10.1056/NEJMoa1410490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Volo SC, et al. Effect of myocardial perfusion pattern on frequency and severity of mitral regurgitation in patients with known or suspected coronary artery disease. Am J Cardiol. 2014;114:355–361. doi: 10.1016/j.amjcard.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Nielsen SL, et al. Mechanism of incomplete mitral leaflet coaptation—interaction of chordal restraint and changes in mitral leaflet coaptation geometry. Insight from in vitro validation of the premise of force equilibrium. J Biomech Eng. 2002;124:596–608. doi: 10.1115/1.1500741. [DOI] [PubMed] [Google Scholar]
- 295.Stephens EH, Chu CK, Grande-Allen KJ. Valve proteoglycan content and glycosaminoglycan fine structure are unique to microstructure, mechanical load and age: Relevance to an age-specific tissue-engineered heart valve. Acta Biomater. 2008;4:1148–1160. doi: 10.1016/j.actbio.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Timek TA, et al. Mitral leaflet remodeling in dilated cardiomyopathy. Circulation. 2006;114(Suppl I):I518–I523. doi: 10.1161/CIRCULATIONAHA.105.000554. [DOI] [PubMed] [Google Scholar]
- 297.Kunzelman KS, Quick DW, Cochran RP. Altered collagen concentration in mitral valve leaflets: biochemical and finite element analysis. Ann Thorac Surg. 1998;66:S198–S205. doi: 10.1016/s0003-4975(98)01106-0. [DOI] [PubMed] [Google Scholar]
- 298.Quick DW, Kunzelman KS, Kneebone JM, Cochran RP. Collagen synthesis is upregulated in mitral valves subjected to altered stress. ASAIO J. 1997;43:181–186. [PubMed] [Google Scholar]
- 299.Carceller-Blanchard AM, Fouron JC. Determinants of the Doppler flow velocity profile through the mitral valve of the human fetus. Br Heart J. 1993;70:457–460. doi: 10.1136/hrt.70.5.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Schoen FJ. Evolving concepts of cardiac valve dynamics: the continuum of development, functional structure, pathobiology, and tissue engineering. Circulation. 2008;118:1864–1880. doi: 10.1161/CIRCULATIONAHA.108.805911. [DOI] [PubMed] [Google Scholar]
- 301.Rausch MK, Tibayan FA, Miller DC, Kuhl E. Evidence of adaptive mitral leaflet growth. J Mech Behav Biomed Mater. 2012;15:208–217. doi: 10.1016/j.jmbbm.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Stephens EH, et al. Significant changes in mitral valve leaflet matrix composition and turnover with tachycardia-induced cardiomyopathy. Circulation. 2009;120:S112–S119. doi: 10.1161/CIRCULATIONAHA.108.844159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Saito K, et al. Influence of chronic tethering of the mitral valve on mitral leaflet size and coaptation in functional mitral regurgitation. JACC Cardiovasc Imaging. 2012;5:337–345. doi: 10.1016/j.jcmg.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 304.Wylie-Sears J, Aikawa E, Levine RA, Yang JH, Bischoff J. Mitral valve endothelial cells with osteogenic differentiation potential. Arterioscl Thromb Vasc Biol. 2011;31:598–607. doi: 10.1161/ATVBAHA.110.216184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Grande-Allen KJ, et al. Apparently normal mitral valves in patients with heart failure demonstrate biochemical and structural derangements: an extracellular matrix and echocardiographic study. J Am Coll Cardiol. 2005;45:54–61. doi: 10.1016/j.jacc.2004.06.079. [DOI] [PubMed] [Google Scholar]
- 306.Beaudoin J, et al. Mitral valve enlargement in chronic aortic regurgitation as a compensatory mechanism to prevent functional mitral regurgitation in the dilated left ventricle. J Am Coll Cardiol. 2013;61:1809–1816. doi: 10.1016/j.jacc.2013.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Enriquez-Sarano M, et al. Valve repair improves the outcome of surgery for mitral regurgitation. A multivariate analysis. Circulation. 1995;91:1022–1028. doi: 10.1161/01.cir.91.4.1022. [DOI] [PubMed] [Google Scholar]
- 308.Gillam LD, Marcoff L, Shames S. Timing of surgery in valvular heart disease: prophylactic surgery vs watchful waiting in the asymptomatic patient. Can J Cardiol. 2014;30:1035–1045. doi: 10.1016/j.cjca.2014.06.019. [DOI] [PubMed] [Google Scholar]
- 309.Kuwahara E, et al. Mechanism of recurrent/persistent ischemic/functional mitral regurgitation in the chronic phase after surgical annuloplasty: importance of augmented posterior leaflet tethering. Circulation. 2006;114(Suppl I):I529–I534. doi: 10.1161/CIRCULATIONAHA.105.000729. [DOI] [PubMed] [Google Scholar]
- 310.Agricola E, et al. Long-term prognosis of medically treated patients with functional mitral regurgitation and left ventricular dysfunction. Eur J Heart Fail. 2009;11:581–587. doi: 10.1093/eurjhf/hfp051. [DOI] [PubMed] [Google Scholar]
- 311.Kizilbash AM, Willett DL, Brickner ME, Heinle SK, Grayburn PA. Effects of afterload reduction on vena contracta width in mitral regurgitation. J Am Coll Cardiol. 1998;32:427–431. doi: 10.1016/s0735-1097(98)00236-8. [DOI] [PubMed] [Google Scholar]
- 312.Pu M, Gao Z, Pu DK, Davidson WR., Jr Effects of early, late, and long-term nonselective beta-blockade on left ventricular remodeling, function, and survival in chronic organic mitral regurgitation. Circ Heart Fail. 2013;6:756–762. doi: 10.1161/CIRCHEARTFAILURE.112.000196. [DOI] [PubMed] [Google Scholar]
- 313.Borer JS. Mitral regurgitation: has another magic bullet bitten the dust? Circ Heart Fail. 2013;6:624–626. doi: 10.1161/CIRCHEARTFAILURE.113.000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Eldadah ZA, et al. The benefit of upgrading chronically right ventricle-paced heart failure patients to resynchronization therapy demonstrated by strain rate imaging. Heart Rhythm. 2006;3:435–442. doi: 10.1016/j.hrthm.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 315.Ypenburg C, et al. Acute effects of initiation and withdrawal of cardiac resynchronization therapy on papillary muscle dyssynchrony and mitral regurgitation. J Am Coll Cardiol. 2007;50:2071–2077. doi: 10.1016/j.jacc.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 316.Nury D, Neri T, Puceat M. Human embryonic stem cells and cardiac cell fate. J Cell Physiol. 2009;218:455–459. doi: 10.1002/jcp.21631. [DOI] [PubMed] [Google Scholar]
- 317.Leschik J, Stefanovic S, Brinon B, Puceat M. Cardiac commitment of primate embryonic stem cells. Nat Protoc. 2008;3:1381–1387. doi: 10.1038/nprot.2008.116. [DOI] [PubMed] [Google Scholar]
- 318.Ferreira LS, et al. Vascular progenitor cells isolated from human embryonic stem cells give rise to endothelial and smooth muscle like cells and form vascular networks in vivo. Circ Res. 2007;101:286–294. doi: 10.1161/CIRCRESAHA.107.150201. [DOI] [PubMed] [Google Scholar]