

CHAPTER SUMMARY
Intracellular bacterial pathogens have evolved to exploit the protected niche provided within the boundaries of a eukaryotic host cell. Upon entering a host cell, some bacteria can evade the adaptive immune response of its host, and replicate in a relatively nutrient-rich environment devoid of competition from other host flora. Growth within a host cell is not without its hazards, however. Many pathogens enter their hosts through receptor-mediated endocytosis or phagocytosis, two intracellular trafficking pathways that terminate in a highly degradative organelle, the phagolysosome. This usually deadly compartment is maintained at a low pH, and contains degradative enzymes, and reactive oxygen species resulting in an environment to which few bacterial species are adapted. Some intracellular pathogens, like Shigella, Listeria, Francisella, and Rickettsia escape the phagosome to replicate within the cytosol of the host cell. Bacteria that remain within a vacuole either alter the trafficking of their initial phagosomal compartment or adapt to survive within the harsh environment it will soon become. In this chapter, we focus on the mechanisms by which different vacuolar pathogens either evade lysosomal fusion, as in the case of Mycobacterium and Chlamydia, or allow interaction with lysosomes to varying degrees, such as Brucella and Coxiella, and their specific adaptations to inhabit a replicative niche.
Keywords: Mycobacterium, Chlamydia, Brucella, Coxiella, endocytosis, phagocytosis, intracellular trafficking
INTRODUCTION
Bacterial pathogens that adopt an intracellular lifestyle often avoid many challenges that are faced by their extracellular counterparts. However, upon entering the host cell, they have a new set of trials with which to contend. Upon phagocytic uptake or entry by receptor-mediated endocytosis, they immediately traffic to a degradative subcellular compartment. Successful intracellular pathogens have adopted different strategies to avoid trafficking of their initial phagosome along the endocytic pathway to fusion with the lysosome, a subcellular compartment that has specifically evolved to degrade them. In the following chapter, we will compare the different molecular mechanisms employed by four intracellular pathogens that have adopted distinct vacuolar niches and lifestyles. Many vacuolar pathogens alter their initial phagosomal compartment in order to stall or exit the endocytic pathway, and thereby avoid elimination. Yet, a few species require at least some interaction with lysosomes for completion of their infectious cycle. In this chapter, we will compare the different strategies employed by Mycobacterium and Chlamydia to avoid lysosomal fusion with those of Brucella, whose vacuole interacts with lysosomes transiently and Coxiella, a bacterium adapted to growth in a compartment that closely resembles a terminal phagolysosome.
The Benefits of an Intracellular Lifestyle
Pathogens that live within a eukaryotic host cell obtain several benefits of a relatively protected niche. One important advantage to living within a host cell is evasion of acquired immunity. Once inside the confines of a eukaryotic cell, the pathogen is no longer susceptible to complement or neutralizing antibodies. Another benefit of this lifestyle derives from the fact that few bacterial species have evolved to live within a host cell, even fewer within a professional phagocytic cell, like a macrophage. Pathogens that have adapted to this environment experience limited competition from other resident bacteria for nutrients. In fact, the intracellular environment can provide so many vital nutrients to a pathogen that they often have minimal genomes, saving coding capacity and energy for the expression of essential “housekeeping” genes, and virulence factors promoting its infectivity.
The Hazards of an Intracellular Lifestyle
Once a bacterium is taken up into a cell either by phagocytosis or receptor-mediated endocytosis, it begins to traffic along the endocytic pathway towards lysosomal fusion and destruction. There are two main strategies intracellular pathogens have adopted to avoid the mature phagolysosome: modify the vacuole to create a replicative niche, or escape and replicate within the cytosol. Whether a bacterium remains in the phagosome or escapes, it must contend with the host's defenses. Cytosolic pathogens like Listeria, Shigella, Rickettsia, and Francisella rapidly escape the initial phagosome and thereby avoid lysosomal fusion (1). However, once in the cytosol, a pathogen must evade xenophagy, a selective form of autophagy that serves as an innate immune defense against cytosolic pathogens (2). Bacteria that remain within the phagosome can avoid xenophagic killing, provided their vacuole remains intact. Taking up residence in a pathogen-tailored vacuole therefore provides a comfortable and protected compartment within a host cell, if phagocytic maturation is quickly curtailed.
The mature phagosome is a dangerous place for most intracellular pathogens
In eukaryotic cells, the specific mechanism of particle internalization depends on the size of the individual item that is consumed. Phagocytosis generally refers to particles >0.5 μm in size, while smaller objects are taken up by receptor-mediated endocytosis, or pinocytosis. Phagocytosis requires the extrusion of membrane for capture, whereas endocytosed particles submerge into the host cell in response to the binding of a ligand to a specific host receptor. Although the initial signaling events and structural changes at the membrane differ between these uptake processes, there are many common features of the phagocytic maturation pathway that follows (3).
The phagosomal maturation pathway is characterized by membrane changes in lipid content and the small GTPases that associate with them (Figure 1). After a phagosome separates from the membrane, the small GTPase, Rab5, and vacuolar-sorting protein-34 (VPS-34) are recruited to its membrane. VPS-34, a class III PI3K, generates phagosomal phosphatidylinositol-3-phosphate (PtdIns3P) (4), identifying the vacuole as a phagosome/early endosome, distinct from the PtdIns(4,5)P2- and PtdIns(4)P-enriched plasma membrane (5). Early endosome antigen-1 (EEA1), a Rab5 effector, binds to the PtdIns3P as well as Rab5, and facilitates endosome fusion (6). The early endosome is slightly acidified (7), but a further reduction in pH is achieved later with the recruitment of V-type ATPases. Rab5 is then exchanged for Rab7, in a process that appears to be regulated by SAND-1/Mon1 (8, 9) and requires lysosome-associated membrane proteins (LAMP) 1 and 2 (10).
Fig 1. Endosomal trafficking is coordinated by exchanges of lipids and Rab-GTPases.
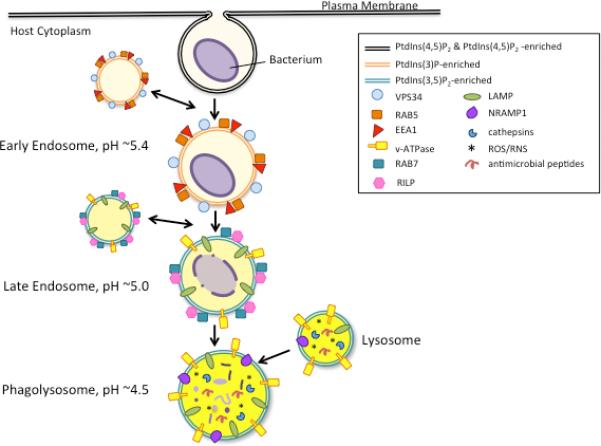
The normal endosomal pathway is illustrated here with major regulatory factors highlighted at each step. After uptake of a cargo, the phagosome fuses with early endosomes, acquiring Rab5, its effector EEA1, and VPS34, which coordinate the change in lipid profile of the endosomal membrane. Soon after, the compartment fuses with late endsomes, and Rab5 is exchanged for Rab7 and its effector RILP. LAMP and v-ATPases are also characteristic of the late endosome. Finally, the late endosome fuses with lysosomes, at which point the compartment is fully matured and highly degradative.
With the swap of Rab5 for Rab7, the early endosome transitions to a late endosome. The late endosome acquires V-ATPases, which pump protons across the phagosomal membrane, dropping the vacuolar pH to approximately 5.0, making it an increasingly less hospitable place. The vacuoles also acquire RILP (Rab7-interacting lysosomal protein), a Rab7 effector that is required for trafficking of late endosomes to the perinuclear region and subsequent fusion with lysosomes (11). In addition to changes in associated Rabs and their effectors, the lipid profile of late endosomes is distinct from that of early endosomes. Late endosomes and lysosomes are enriched in PtdIns(3,5)P2, the result of conversion of PtdIns3P by PtdIns(3)P5-kinase.
Upon maturation of the phagosome into a phagolysosome, the pH drops to approximately 4.5, activating proteolytic cathepsins, and contributing to the generation of deadly reactive oxygen species (ROS) (12). In addition to these microbicidal contents, the lysosome also harbors antimicrobial peptides, NO−, and natural resistance–associated macrophage protein 1 (NRAMP1), which serves to eject Zn+ and Mn+ from the lysosome to preserve its low pH and deprive any microbes from essential cofactors (13). Most pathogens that have adapted to life within a host vacuole have to act quickly to halt trafficking or otherwise tailor their environment in order to avoid destruction; maturation can occur within 18 minutes of uptake (14).
To Fuse, or Not to Fuse
As mentioned above, many intracellular pathogens have successfully adopted a vacuolar lifestyle. However, the points at which these pathogen-tailored vacuoles depart from the endocytic pathway are diverse and species-specific. Most vacuolar pathogens have evolved mechanisms to avoid lysosomal fusion by arresting trafficking through the endocytic pathway, as exemplified by Mycobacterium tuberculosis. The Chlamydia trachomatis inclusion diverges from the pathway early on, modifying its vacuole so that it resembles an exocytic vacuole. Several intracellular bacteria allow acidification. Brucella require a transient drop in vacuolar pH to activate the expression and secretion of key virulence factors and complete its infection cycle. In contrast, Coxiella burnetii has evolved to require sustained interaction with the lysosome in order to complete its developmental cycle. Over the course of this chapter, we will discuss the specific adaptations these four different pathogens have evolved to create a hospitable niche inside a host vacuole.
MYCOBACTERIUM TUBERCULOSIS
Tuberculosis (Tb) is a leading cause of death in the world, with 2-3 million fatal cases annually. These deaths really represent global burden on public health, since over one billion people are latently infected with the etiologic agent of Tb, Mycobacterium tuberculosis (Mtb) (15). While the vast majority of these individuals are asymptomatic for much of their life, they have a small chance (about 10%) of eventually developing active disease. The morbidity and mortality associated with Mtb has been amplified by the pandemic of HIV/AIDS patients with MTBC (Mycobacterium tuberculosis Complex, of which Mtb is a member) infection (16). Additionally, the slow-growing and chronic nature of the infection has lead to widespread hypervirulent multidrug-resistant (MDR and XDR-Tb, extremely drug resistant) cases that are challenging to treat and strain public health systems world-wide (17). The MTBC also includes M. bovis, from which the Calmette and Guerin vaccine is derived (BCG). By evolution through a common ancestor, MTBC members can be compared genomically with respect to 14 regions of difference (RD-14) the help to identify specific virulence-associated regions (16). For the majority of comparisons across intracellular bacterial models, we highlight findings from the virulent isolate, H37Rv for lifestyle and intracellular trafficking studies.
Mtb colonize macrophages and persist inside structurally complex granulomas
Macrophages and monocytes are the principally infected cell during Mtb infection, and have served as the most common cell type for intracellular trafficking studies. Once inside the macrophage, Mtb arrests phagosomal maturation at a very early stage, thereby avoiding degradation and enabling its intracellular replication within this compartment (18). Infected alveolar macrophages initiate cellular responses by nearby epithelial cells that recruit more macrophages to the infection site creating a granuloma within the infected lung. These infection foci function as sites of chronic inflammation, and provide a mechanism for bacterial spread as the newly arriving macrophages provide a growing niche for replication (19). Eventually, acquired immunity can control the bacterial growth, until necrosis develops. Mtb released from necrotic cells inside the granuloma can replicate rapidly in the extracellular space, and it is these bacteria that are transmitted to a new host. As a result, the dynamics of the Mtb lifestyle is complicated by stage of infection, activation status of the host cell, and the ensuing inflammatory response. For this discussion, we will focus primarily on resident macrophages as replication sites for Mtb.
Macrophage infection by M. tuberculosis is characterized by an arrest of phagosomal maturation
Mtb avoids digestion by lysosomal enzymes by halting its progression along the phagosomal pathway at the early endosome stage (Figure 2) (20, 21). In doing so, it specifically excludes V-ATPase from its vacuole and disables fusogenicity with late endosomes and lysosomes. The result is a parasitophorous vacuole of near-neutral pH that is exclusive of reactive oxygen species and degradative enzymes. While the Mycobacterium phagosomal compartment (MPC) evades fusion with degradative endosomal compartments, it retains the ability to interact with early endosomes, which provide the bacteria with essential nutrients for replication. Mtb orchestrates its trafficking arrest through the production of several key virulence factors that interfere with calcium flux, host membrane fusion events, and recruitment of inducible nitrous oxide synthase (iNOS) (18).
Fig 2. Intracellular pathogens have evolved distinct trafficking pathways to arrive within their ideal replicative niche.

Intracelluar trafficking pathways are shown for Mycobacterium tuberculosis (Mtb), Chlamydia (Chl), Brucella (Br), and Coxiella burnetii (Cb). Key regulatory factors, such as Rab-GTPases, lipids, and bacterial factors are shown. Mtb fuses with early endosomes (EE), but inhibits fusion with late endosomes (LE), and replicates in a compartment (MCP) that is stalled at an early point along the endocytic pathway. Chl traffics away from the endosomal pathway, and escapes to replicate in a Golgi-associated Inclusion. Br allows EE and LE fusion as well as limited lysosomal fusion before trafficking to the ER to replicate in rBCVs (replicative Brucella containing vacuoles). Cb interacts with EE, autophagosomes, LE, and allows lysosomal fusion to arrive in the CCV (Coxiella containing vacuole), which resembles a terminal phagolysosome.
(i) Lipoarabinomannan arrests phagosomal maturation and prevents acidification of the MCP
Mtb begin the invasion process by interaction with several distinct receptors, but uptake of complement-opsonized bacteria by complement receptors represents its main route of entry (22, 23). Cholesterol-rich lipid domains may also drive selective entry, and likely affect the signaling response to attachment as well as MPC formation (24). Soon after entry, the early endosome containing Mtb acquires Rab5, but Rab7 is altered or absent (25, 26). This block in the canonical trafficking pathway is the result of two converging signaling pathways that are regulated by Mtb lipoarabinomannan (LAM) (18). LAM is a glycosylated phophatidylinsositol that is conserved among MTBC members, with the exception of the heterogeneous nature of its sugar cap. The LAM of Mtb has a cap of mannose, and is therefore referred to as ManLAM. While ManLAM affects two different signaling pathways, the net effect of its activities is the exclusion of V-ATPase on the MCP.
As mentioned above, the maturation block coordinated by ManLAM has two arms, one that affects calcium signaling, and the other inhibits recruitment of EEA1. ManLAM contributes to the absence of VPS34 on the MCP by inhibiting an increase in cytosolic Ca2+ in response to Mtb infection (27). In macrophages, an increase cytosolic Ca2+ levels, as a response to phagocytosis, activates calmodulin to associate with Ca2+/calmodulin protein kinase CaMKII, an essential factor for phagolysosome maturation (28). This signaling cascade is responsible for the recruitment of VPS34 to the phagosome, which generates the PtdIns3P necessary for EEA1 binding (27). ManLAM may also inhibit association of EEA1 through the activation of p38 MAP kinase (29). Activation of p38 mitogen-activated protein kinase reduces levels of Rab5 on early endosomes and likewise levels of EEA1 (30). EEA1, a membrane tethering molecule, serves as a binding partner for syntaxin 6, a SNARE that delivers V-ATPase and cathepsins from the trans-Golgi network (TGN) to endosomes (31). Through the complementary effects of ManLAM on macrophage signal transduction and membrane trafficking, the MCP is protected from acidification and lysosomal hydrolases.
(ii) Mtb uses multiple strategies to impede phagosomal maturation
In addition to the expression of ManLAM, Mtb employs other virulence factors in an effort to preserve the early endosomal qualities of the MCP. Another lipid produced by Mtb called trehalose dimycolate (TDM) can delay phagosomal acidification via an unknown mechanism (32). In addition to lipids, a variety of protein “effectors” have been identified, via transposon library screens, as being involved in arresting phagosomal maturation. Ndk is a nucleoside diphosphate kinase, which is secreted into growth medium and is cytotoxic to macrophages (33). Based on in vitro and biochemical data, Ndk appears to dephosphorylate cellular Rab7-GTP and Rab5-GTP, thereby preventing Rab7-dependent heterotypic fusion of the MCP. PtpA is a low-molecular weight tyrosine phosphatase that dephosphorylates VPS33B, a host protein involved in regulation of membrane fusion in the endocytic pathway. PtpA also binds to the H subunit of V-ATPase, inhibiting its ability to acidify vacuoles (34). The above examples of Mtb products that participate in the regulation of intracellular trafficking of the MCP are only a subset of the identified bacterial factors that may play a role in this complex process.
(iii) Mtb evades iNOS through interactions with the host actin cytoskeleton
In addition to acidification and lysosomal hydrolases, Mtb also avoids the reactive oxygen and nitrogen species that normally accompany phagosomal maturation in macrophages. Inducible nitrous oxide synthase (iNOS) synthesizes NO− gas on the cytosolic face of the phagosomal membrane which then diffuses through the membrane into the phagosomal lumen. The apposition of iNOS to the phagosome is actin-dependent, and Mtb interference with the actin cytoskeleton prevented iNOS from contacting the MCP (35). Building on these observations, Davis, et al. found that the scaffolding protein EBP50, which normally links iNOS to the actin cytoskeleton and regulates its localization, was rapidly lost from the Mtb-containing phagosomes (36). Keeping iNOS at a distance from the MCP excludes NO− from the phagosome, protecting Mtb from its harmful affects.
(iv) The Mtb lipid PIM counteracts, in part, the effects of ManLAM
ManLAM significantly reduces the fusogenicity of the MCP by the mechanisms detailed above. However, the Mtb is capable of bypassing the restrictions that ManLAM sets in place through the production of another bacterial glycosylated phophatidylinsositol called PIM (phophatidylinsositol mannoside). PIM specifically stimulates homotypic fusion of the MCP with early endosomes in a Rab-dependent manner (37). PIM's effect is insensitive to inhibition of PtdIns3P, and the result is a membrane fusion pathway that is independent of that particular host lipid (27). The significance of this bypass pathway is that it solves the problem of how Mtb acquires iron, via transferrin, through interaction with early endosomes (38).
Mycobacterium expresses several virulence factors that impart resistance to its host's oxidative burst
While Mtb manages to evade interaction with the lysosome of macrophages, it must still contend with the oxidative burst induced by invading its host cell. There are several proteins that either directly or indirectly contribute to the ability of Mtb to resist this form of macrophage-mediated killing. Additionally, the integrity of the mycobacterial lipid-rich cell wall may protect Mtb from reactive oxygen and nitrogen species (ROS/RNS).
(i) Several Mtb proteins directly detoxify ROS and RNS
Mtb employs many classical mediators of ROS/RNS resistance by directly deactivating them during infection. Alkyl hydroperoxidase C (AhpC), an enzyme that reduces organic peroxides, has been shown to exhibit peroxynitrite reductase activity in vitro (39). Mutants of this gene show increased susceptibility to peroxynitrite in resting macrophages compared to wildtype Mtb, however this difference was nullified upon activation with IFN-γ (40). Another detoxifying enzyme that is important for Mtb survival is KatG, which degrades hydrogen peroxide to water and oxygen. KatG mutants of Mtb are attenuated in mice and guinea pigs (41, 42). TpX, a thiol peroxidase, is required for optimal replication in both activated and resting macrophages. A tpx mutant was not attenuated in iNOS−/− mice, which exhibit a modest oxidative burst, indicating that TpX is indispensible for resistance to RNS and ROS (43). The outer membrane lipoprotein SodC that bestows Mtb with the ability to dismutate extracellular superoxide was also found to promote survival within IFN-γ activated macrophages (44). Having these enzymes in its arsenal are a requirement for Mtb virulence.
(ii) Resistance to ROS/RNS is indirectly supplied by other Mtb virulence factors
Several Mtb proteins or other virulence factors, such as the lipid-rich cell wall, provide resistance to ROS and RNS without directly detoxifying them. Acr2 is one such protein whose expression is up-regulated by the bacterium under various conditions of stress, including oxidative stress (16). While the molecular chaperone Acr2 is not absolutely essential to the pathogenic program of Mtb, deletion mutants exhibit a protracted disease progression (45). The role of the hypothetical gene Rv2136c in resistance to low pH, heat shock, and ROS, among other stressors, was identified as part of a transposon mutant screen (46). The transposon mutant also displayed attenuation in a mouse infection with regard to its ability to persist in the lung and spleen. The specific function of this protein is unknown, but it has homology to a peptidoglycan synthesis gene in Escherichia coli. Other genes of interest identified in the screen include Rv2224c and ponA2, which had more modest infection defects, but are also predicted to play a role in cell wall synthesis or maintenance. These proteins that impart oxidative stress relief via prevention or repair of damage also play an important role in the survival and therefore pathogenesis of Mtb.
The primary survival strategy of Mycobacterium tuberculosis is to quickly stall the progression of its internalized phagosome before it becomes too hostile to inhabit. It orchestrates a delicate balance between blocking of phagosomal fusogenicity with the ability to interact with early endosomes as a source of essential nutrients. While the arrested phagosome is free of degradative cathepsins and excludes acidifying V-ATPase, the niche that Mtb occupies is not devoid of host defenses. Mtb counters the vigorous oxidative burst of the infected macrophage by direct detoxification and indirectly through the expression of protective chaperones. These extremely effective pathogenic attributes have been fine-tuned over the course of millennia, as the relationship M. tuberculosis has with its host, is truly ancient.
CHLAMYDIA
There are two main species of Chlamydiae that cause the majority of human disease. Chlamydia trahomatis is the causative agent of the most commonly reported sexually transmitted bacterial infection in the United States (47). It is also responsible for the blinding disease of trachoma, common in the developing world. Chlamydophila pneumoniae frequently causes community-acquired pneumonia, and has been identified as a risk factor for atherosclerosis and asthma (48, 49). Another species, Chlamydophila psittaci, is known to cause a zoonotic fever carried by psittacine birds that is transmitted by aerosol and contrasts as an intracellular model. Psittacosis is a severe pulmonary infection that is difficult to treat, and therefore it has been proposed to have potential for use as a biological weapon (50).
Chlamydia has a biphasic developmental cycle
These Gram-negative obligate intracellular parasites have a characteristic developmental cycle that alternates between two disparate morphologies. The small, extracellular, infectious but metabolically inactive form is termed the elementary body (EB) (Figure 2). The metabolically active reticulate body (RB) is larger and non-infectious, and is found exclusively inside host cells. Upon attachment to its host cell, the elementary body induces its uptake by injection of a Type III secreted effector called TARP, which remodels host actin at the site of entry (51). Once inside the host cell, the EB is contained within a specialized parasitophorous vacuole, called the inclusion. EBs rapidly differentiate into RBs, and begin to replicate by binary fission within the inclusion. The bacteria continue to replicate while actively modifying the inclusion by inserting bacterial proteins (Incs) and incorporating host lipids as it traffics to the microtubule organizing center (MTOC) of the cell. The infection cycle completes between 48 and 72 hours later when the RBs asynchronously differentiate back into their infectious form and are released from the host cell by either lysis or a process called extrusion (52).
Over the course of the intracellular phase of chlamydial development, the bacteria are known to subvert a wide variety of host cell functions. Actin is remodeled at the entry site of EBs (51, 53, 54), and the host cytoskeleton is used to build a supporting scaffold for the growing inclusion (55). The lipid composition of the inclusion membrane is changed quickly after the infection is initiated, and the chlamydiae themselves incorporate host lipids into their membranes (56, 57). Rab GTPases that identify recycling endosomes and ER-Golgi trafficking pathways are recruited to the inclusion, and interactions with many host cell organelles have been documented, including the Golgi apparatus, mitochondria, lipid droplets, and multivesicular bodies (MVBs) (58). Chlamydiae are also known to interfere with host cell signaling to inhibit apoptosis and promote survival, and block innate immune signaling (59). All of these functions serve to create a parasitophorous vacuole that is separate from the endocytic pathway, thereby evading lysosomal fusion while at the same time, allowing acquisition of essential nutrients and replication within a protected niche.
Chlamydiae depart the endocytic pathway early in infection
In contrast to Mycobacterium, which arrests phagocytic trafficking at an early point in the pathway, the chlamydial inclusion diverges from it completely in order to evade destruction and create a hospitable environment for replication. Nascent inclusions do not acquire transferrin receptor, EEA1, mannose 6 phosphate receptor, or LAMP1 following EB uptake. In fact, the vesicles containing endocytosed EBs are nonfusogenic with endocytic vacuoles, including recycling endosomes (60). Inclusions do not acidify, and do not acquire V-ATPases (61, 62), despite the fact that they can be found in close apposition to lysosomes (63). Regulation of these early trafficking events of the chlamydial inclusion requires de novo protein synthesis, as chloramphenicol treatment results in an increase in lysosomal fusion. Orchestrating the inclusion's escape involves the co-opting of the host's microtubule network, changing the lipid content of the vacuolar membrane, and recruiting specific Rab GTPases associated with recycling endosomes and ER-Gogi transport. The end result is a parasitophorous vacuole that operates as a unique pathogen-constructed organelle.
(i) The host cytoskeleton is co-opted by Chlamydia for the duration of its infectious cycle
As soon as an EB is irreversibly attached to its host cell, it begins to use the host cytoskeleton to its advantage. EBs translocate the Type III secretion effector TARP (translocated actin-recruiting phosphoprotein) across the host cell membrane to nucleate actin polymerization at the site of entry (51, 53, 54). C. trachomatis TARP is also known to activate Rac1, a master regulator of lamellipodium formation (64, 65). This initial actin remodeling induced by TARP is transient, and while the mechanisms governing the subsequent actin depolymerization are not understood, Chlamydia expresses two potential regulators for this process: CT166, an inhibitor of Rac1 (66, 67), and CT694, which results in an AHNAK-dependent loss of actin stress-fibers (68).
While the actin rearrangements induced by EB attachment are quickly undone, Chlamydiae continue to appropriate the host cytoskeleton to support inclusion development. Following entry, the nascent inclusion is trafficked in a dynein-dependent, but dynactin-independent manner to the peri-Golgi region of the host cell (69). Once there, the inclusion maintains an intimate association with the MTOC. Mital, et al. identified cholesterol-rich microdomains on the inclusion membrane that appear to serve as platforms for signaling through Src-family kinases and interaction with centrosomes and dynein (70). Once at the MTOC, the growing inclusion requires a dynamic cytoskeletal scaffold for stabilization (55, 71). The structural integrity maintained by this framework may prevent leakage of inclusion contents into the host cytosol, which might otherwise alert the host cell's innate immune system.
The actin cytoskeleton has a final role to play in the infection cycle as it facilitates extrusion, one of two egress processes described for Chlamydia (52). During extrusion, the unbroken inclusion is ejected from the host cell through the plasma membrane, leaving the host cell intact. Pharmacological inhibitors revealed that extrusion requires Wiskott-Aldrich syndrome protein (WASP), myosin II, Rho GTPase, and actin polymerization (52). A recent study also demonstrated that the myosin phosphatase pathway is also involved in extrusion, as siRNA depletion of myosin IIA/IIB, MLC2, and MLCK significantly reduced detectable extrusion events (72). The fate of an extruded inclusion is unknown, but it has been speculated that this release method may facilitate chlamydial persistence or spread to more distant sites of infection (52)
(ii) The inclusion incorporates host lipids that typify exocytic compartments
As discussed previously, the lipid content of a eukaryotic vacuole is highly regulated and helps to define the identity of that specific subcellular compartment. In contrast to Mycobacterium, which halts endocytic trafficking by retaining the PtdIns(3)P on its phagosome, Chlamydia rapidly incorporates other host-derived lipids into its inclusion membrane, helping to identify it as an organelle separate from the endocytic pathway (73).
The first study to demonstrate host lipid acquisition by the inclusion membrane demonstrated that sphingomyelin (SM) generated from a fluorescent precursor (C6-NBD-ceramide) in the Golgi accumulated in the parasitophorous vacuole as soon as 2 hours post-infection, and the lipid was also incorporated into the bacteria themselves (57). Since then, it has been shown that Chlamydia intercept SM trafficking from the Golgi to the plasma membrane (56, 74, 75), as well as from vesicles trafficking basolaterally (76). Additionally, Chlamydia co-opts CERT, a host protein that regulates ceramide traffic from the ER to the Golgi, through binding to chlamydial inclusion membrane protein IncD. CERT interacts with the inclusion membrane to regulate SM incorporation (77, 78).
Additional host lipids have been detected in the inclusion membrane, including cholesterol (79), phosphatidic acid (80), and PtdIns(4)P (81). PtdIns(4)P is an identifying lipid component of the Golgi apparatus, and two of its associated regulatory proteins, OCRL1 and PIK4Iiα, are also inclusion-localized. Interestingly, depletion of either of these proteins in host cells results in decreased chlamydial infection (81). Other pathways for lipid acquisition include interactions with mutlivesicular bodies, and lipid droplets (82, 83). By changing the lipid profile of the inclusion from that of a PtdIns(3)P-rich endosome to that resembling a Golgi-derived vesicle, the inclusion can avoid termination within a lysosome.
(iii) The chlamydial inclusion interacts with many host Rab GTPases
In addition to remodeling its lipid profile, Chlamydia recruits Rab GTPases to the inclusion that are characteristic of recycling endosomes and vesicles involved in transport to and from the Golgi apparatus. Rab GTPases, like membrane lipid content, also have an important role in organelle identification. As mentioned earlier, Rabs tightly control maturation of phagosomes, with Rab5 and Rab7 being among the most important players in the process. Rab5 regulates fusion of early endosomes and early phagosomes, and is replaced as phagosomes mature by Rab7, which regulates fusion of late endosomes with lysosomes (12). As Chlamydia quickly exits the phagocytic pathway, the inclusion membrane does not associate with either Rab5 or Rab7. Rather, the Rabs of recycling endosomes (Rab4 and Rab11), and ER to Golgi transport (Rab1) are recruited the inclusion (84). While the association of Rabs 1, 4, and 11 with the inclusion are universal to Chlamydiae, some species-specific interactions have also been described. Golgi-associated Rabs 6 and 14 appear only on the inclusions of C. trachomatis. Rab10, which is a regulator of ER dynamics and post-Golgi transport, is found only on the inclusions of C. pneumoniae (84, 85). The mechanisms by which Rab GTPases are recruited to the inclusion membrane are not well defined, however, a few chlamydial inclusion membrane proteins (Incs) have been shown to bind Rabs. C. trachomatis CT229, a member of the Inc family of chlamydial proteins, interacts directly with Rab4-GTP in a number of assays, including two-hybrid binding and pull-down experiments (86). A C. pneumoniae Inc, Cpn0585, is capable of interacting with Rabs 1, 10, and 11 (87).
The specific Rabs that have been observed on the inclusion membrane not only identify the vacuole as a compartment apart from the endocytic pathway, but also enable interaction with host vesicles and organelles that support chlamydial growth and replication. Rabs 6, 11, and 14 coordinate acquisition of Golgi-derived lipids from the host cell (88, 89). Chlamydiae depend on host cell transferrin as a source of iron for growth. Concurrent inhibition of Rab4 and Rab11, which coordinate transferrin recycling to the host plasma membrane, resulted in decreased chlamydial replication and an accumulation of transferrin-containing vesicles adjacent to the inclusion (90). Rab11 is involved in the generation of multivesicular bodies, which have also been observed interacting with inclusions. Inhibition of their trafficking to the inclusion decreases lipid incorporation and bacterial replication (80, 82, 91). Mitochondria have long been associated with chlamydial inclusions (92, 93), and at least in the case of C. caviae, interaction with them is essential for replication (94). This is not surprising, as Chlamydia rely on the host cell for ATP (95, 96).
The chlamydial replicative niche is thus characterized by its early departure from the endocytic pathway to escape the terminal lysosome, and rapid association with the Golgi in order to acquire nutrients. While much is known about how Chlamydia actively remodels the inclusion to create a replicative niche, there is still much to discover, as many of the predicted Incs encoded within the chlamydial genome lack functional assignments (97, 98). The co-opting of host cytoskeleton by the inclusion provides structural integrity as it quickly takes up the bulk of the host cell's volume, while at the same time provides a network for trafficking away from destruction and reception of host cell cargoes that provide necessary nutrients. Modification of the inclusion membrane further disguises the vacuole as a host organelle that is separate from the endocytic pathway. It is an elegant example of how to successfully hide from the host within one of its cells.
BRUCELLA
While Brucellae are known to infect a wide range of host species, there are three that cause the majority of human infection: Brucella melitensis (primarily associated with goats and sheep), Brucella suis (swine) and Brucella abortus (cattle). Humans are incidental hosts for these pathogens, however brucellosis is among the world's most common zoonoses, with over an estimated 500,000 new cases each year (99). The disease is highly infectious, requiring only 10-100 organisms to establish an infection via aerosol (100). While inhalation of the pathogen is certainly an important source of human infection, it is also commonly acquired through ingestion of contaminated animal products, particularly unpasteurized dairy (101). After passing through the mucus membranes to infect phagocytic cells, Brucella establishes a protected intracellular niche. As mentioned earlier, an intracellular lifestyle allows a pathogen to evade immune surveillance, and this is particularly important for Brucella, which often becomes chronically associated with its host (101, 102). The infection begins with flu-like symptoms, and is characterized by an undulant fever and night sweats. If left untreated, Brucella can disseminate via infected phagocytic cells to many different tissues and organs within the body.
Brucella transitions between three subcellular compartments to complete its infectious cycle
Upon entry, Brucella begins an intracellular journey in which it will traffic through the endocytic pathway to the ER for replication, and then an autophagosome-like compartment for egress. First, intracellular trafficking of Brucella to a replicative compartment is dependent on its LPS status, as only the smooth (virulent) variants with full-length O-antigen can avoid phagocytic maturation and destruction. However, it should be noted that even virulent Brucella are often killed within the phagolysosomes of macrophages, as fewer than 10% of the internalized bacteria survive to establish a replicative Brucella-containing vacuole (rBCV) (Figure 2) (103). In contrast to Mycobacterium and Chlamydia, once internalized, the parasitophorous vacuole traffics along the endocytic pathway, acquiring early (104), as well as late endosomal markers (105). There is even some limited interaction with host lysosomes and the BCV acidifies transiently (106). After about 8 hours post-infection, the acidification of the BCV activates expression of the VirB-dependent type 4 secretion system (107) directing the vacuole's escape from the endocytic pathway to set up an ER-associated rBCV (108, 109). It is within this compartment that Brucella replicates for up to 48 hours post-infection. Following replication, a subpopulation of Brucella traffic to a new subcellular compartment resembling an autophagosome, the aBCV (105). The precise signals that induce aBCV formation are not understood, but these tertiary vacuoles appear to enhance spread of Brucella to neighboring cells for a second round of infection. Brucella may also exit host cells as rough mutants that sporadically arise within the rBCV causing host cell cytotoxicity and lysis. The released bacteria, an assortment of smooth and rough Brucella, then seed a new round of infection (110).
Trafficking to the ER requires the coordination of a number of bacterial and host cell factors
Many different host factors required for efficient Brucella replication have been identified, including Rho-family GTPases, SNARES, PtdIns(3) kinases, cytoskeletal proteins, and IRE1α, a kinase that regulates the unfolded protein response (UPR) and autophagosome formation (111). Brucella coordinates its trafficking through the secretion of a number of virulence factors, and expression of proteins to resist the transient acidification of the BCV. Once it reaches the confines of ER and Brucella has successfully evaded the terminal phagolysosome, it is safe to begin replication.
(i) Endosomal trafficking of Brucella ends with acidification of the BCV
Unlike the other intracellular pathogens discussed in the first part of this chapter, Brucella stays within the endocytic trafficking pathway until it reaches a late stage. Smooth, nonopsonized Brucella entry into murine macrophages is dependent on lipid rafts (112), PtdIns(3) kinase (113) and TLR4 (114), and fails to activate macrophages. Rough mutants, which lack O-antigen, enter cells independently of lipid rafts and are rapidly killed within activated macrophages (115-117). However, naturally rough B. ovis and B. canis are capable of lipid raft-dependent entry (118), so there must be additional factors that regulate Brucella entry. Similarly to Chlamydia, entry is also dependent on the host actin cytoskeleton. The small GTPase, Cdc42, which regulates actin polymerization through interaction with WASP, is recruited to and activated at the entry site (113).
Endosomal trafficking of the BCV proceeds with acquisition of early endosomal markers EEA1 and Rab5 (104, 108). Their association with the BCV is transient, as they are rapidly exchanged for the late endosomal markers Rab7, RILP, and LAMP1 (106). Acquisition of Rab7 and RILP are required for normal intracellular trafficking of Brucella, as expression of a dominant-negative Rab7 or overexpression of RILP interfered with conversion of the BCV to an ER-derived vacuole. As they traffic along the endosomal pathway, the BCV allow limited lysosomal fusion. Fusion with lysosomes can be observed up to about 12 hours post-infection, at which point the BCV looses its late endosomal markers. This study also confirmed that Brucella require acidification for completion of their infectious cycle, as pharmacological inhibition of v-ATPases resulted in a dramatic decrease in viable bacteria (106, 108). This is likely because acidification is required for the activation of type IV secretion system gene expression, which is crucial for setting up the rBCV (106-109, 119, 120).
(ii) Adaptations to resist degradation upon lysosomal fusion
Like Chlamydia, Brucella has evolved to escape destruction by trafficking away from the endocytic pathway before it can be degraded, but not before the BCV fuses with lysosomes. First, it should be noted that lysosomal fusion of the BCV is well controlled by the bacteria, and significantly less fusion occurs with virulent Brucella than with heat-killed bacteria (106). Cyclic-β-1,2-glucan, a critical virulence factor, has a role in decreasing lysosomal fusion by depletion of BCV membrane cholesterol (121). Nevertheless, the lumen of the BCV is acidified and does contain degradative enzymes. To mitigate the risk of destruction during this transition, Brucellae express several proteins to aid in resistance to the drop in pH, oxidative and nitrosative damage, and antimicrobial peptides (122).
Multiple Brucella proteins have been identified as having a role to play in acid resistance. HdeA, a homolog of the E. coli acid-resisting periplasmic chaperone, imparts acid resistance in vitro (123). CydB was found to contribute to both acid tolerance and resistance to reactive oxygen species (124). Disruption of the cydB gene demonstrated that cytochrome bd ubiquinol oxidase is required for intracellular survival and virulence. Brucellae also express a functional urease that protects the bacteria from extremely low pH and is presumed to allow safe passage of the bacteria through the gastrointestinal tract after ingestion, but it is not required for virulence in cultured cells (125).
Reduced pH is certainly not the only hazard that intracellular Brucella encounter, as reactive oxygen (ROS) and nitrogen (RNS) species are key defense mechanisms employed by infected phagocytic cells. In fact, ROS and nitric oxide production have been shown to be important for microbicidal activity against Brucella in macrophages (126). SodC, a Cu/Zn superoxide dismutase that counteracts ROS production, is required by B. abortus for survival within macrophages and virulence in a mouse model of infection (127). To resist the deleterious effects of RNS, Brucella generate nitric oxide reductase to detoxify NO within macrophages (128, 129). Brucella also enlist AhpC, a peroxiredoxin that is primarily involved in scavenging endogeneous ROS generated by Brucella metabolism (130), to impart some resistance to peroxynitrite (122).
Finally, the lysosomes of macrophages contain cationic antimicrobial peptides called defensins that pose a threat to bacteria by creating pores in their outer membranes (12). The BvRS two-component regulatory system controls the expression of genes responsible for the acylation status of lipid A (131). Without this important modification of LPS, BvRS mutants are more susceptible to destruction by cationic antimicrobial peptides due to their altered membrane properties. It is evident that Brucellae have evolved many solutions to the problem of requiring lysosomal fusion for replication by expressing proteins that impart resistance to the myriad hazards encountered during endocytic trafficking.
(iii) The carefully coordinated transition to the Endoplasmic Reticulum begins the replicative phase of the Brucella infection
In epithelial cells, prior to ER fusion, the BCV has a short-lived association with autophagosomes. The BCVs acquire the autophagosome marker monodansylcadaverine, and have a multilamellar appearance (104, 132). An autophagy-related protein IRE1α, is required for Brucella replication at this stage (111). IRE1α is a kinase that regulates the host unfolded protein response (UPR) as well as the generation of ER-derived autophagosomes. Its role is autophagy induction was highlighted as part of the ER-fusion process, as other kinases involved in the UPR were not found to be important for Brucella replication in that study (111). However, this autophagic transition prior to ER fusion is not observed in macrophages. Interestingly, the UPR is induced in Brucella-infected macrophages, and is required for efficient replication (133). The microtubule-associated Brucella protein TcpB has been identified as playing an important role in UPR induction in these cells. It is not surprising that the UPR is induced by Brucella infection, as a major reorganization of the ER is required for replication, but the idea that Brucella may actively induce the process is intriguing.
Brucella-ER fusion, which is required for bacterial replication, is carefully coordinated by secreted virulence factors. BCV-ER fusion requires the small GTPase Rab2, which is part of a multi-protein complex that regulates vesicular trafficking from the Golgi to the ER. Inhibition of Rab2 results in retention of LAMP1 on BCVs (134). A Brucella type IV secretion effector RicA has been implicated in initiating Rab2 recruitment through its interactions with the small GTPase (135, 136). Another Brucella secreted protein, CstA, which interacts with the ERES-associated protein Sec24A, has recently been discovered as having role in ER fusion of the BCV. Deletion mutants of cstA have delayed trafficking to the ER relative to wildtype B. abortus (137). Secretion of CstA into cell culture supernatants appears to be dependent on flagellar genes, not type IV secretion, but more work must be done to confirm its secretion in infected cells. Several other type IV substrates have been identified as ER-localized during infection. Brucella effectors BspA, BspB, and BspF disrupt the host secretory pathway and inhibit host protein secretion (138). While deletion of these individual effectors does not significantly affect Brucella replication, the triple deletion mutant ΔbspABF displays reduced intracellular replication and virulence in a mouse model of chronic infection (138).
As BCVs lose fusogenicity with lysosomes, they progressively merge with the ER and acquire markers of this new subcellular compartment that is pH-neutral and represents a safe haven for replication. BCVs initially contact the ER at ER exit sites (ERES) through interactions with the SarI/COPII complex (139). Inhibition of Sar1 prevents BCV-ER fusion and results in a block in replication. Other ER-specific proteins found on rBCVs include calnexin, calreticulin, and Sec61 (104, 108). The rBCV membrane is also studded with ribosomes. The ER-targeted RNA-interference screen that identified IRE1α also found 29 other novel host ER proteins to be important for virulence, including important regulators of vesicular trafficking and host protein biosynthesis, as well as cytoskeletal proteins (111). As Brucella replicates in this new compartment, the rBCV membrane septates with the bacteria, so they continue to reside singly in tight vacuoles as the host cell fills with bacteria.
Following the replicative phase, egress from the host cells is poorly characterized, but two complementary hypotheses describing an exit route for Brucella have been proposed. Starr, et al. described the formation of the late autophagosome-like aBCV, which appears to promote spread of Brucella to neighboring uninfected cells (105). aBCV formation was observed at 72 hours post-infection, when rBCVs appear to transition back into a LAMP1-positive subcellular compartment and lose the ER marker calreticulin. Although the cells that harbor aBCVs make up a subset of the infected cell population, those cells had a tendency to form re-infection foci within the monolayer, suggesting that aBCVs offer a mode of egress for Brucella (105). Another method of host cell exit may involve a change in the LPS status of replicating Brucella. Spontaneous dissociation of smooth Brucella to rough mutants with truncated LPS O-antigen has been observed during infection of murine macrophages. Rough Brucella are cytotoxic to macrophages, and cause host cell lysis. A recent report demonstrated that the subpopulation of rough mutants that arises during infection with B. melitensis enhances cell-to-cell spread of the bacteria by causing host cell lysis and release of smooth Brucella along with the sporadic rough mutants (110). Neither exit mechanism excludes the other, nor would it be surprising to find that like Chlamydia, Brucella uses more than one exit strategy.
Brucella has a unique strategy for setting up its replicative niche. Although trafficking to ER-derived replicative vacuoles is not specific to Brucella, the fact that it allows lysosomal fusion prior to arriving in this protected compartment is fairly distinctive. Utilizing the acidic environment of the late endosome as a signal to activate virulence genes and coordinate its escape to the ER is not without risk. Recall that over 90% of the bacteria that attempt to colonize macrophages meet an untimely demise. For the remaining 10%, the tools that Brucella employs to resist destruction are effective, and allow it to hide within phagocytic cells for protracted periods of time, making brucellosis a particularly unpleasant disease to contract.
COXIELLA
Coxiella burnetii is a Gram-negative naturally obligate intracellular pathogen that causes Q fever in humans, a flu-like illness that is generally self-limiting and readily resolves without antibiotics. The organism has an extremely wide zoonotic host range that spans mammals, arthropods, fish and avian species. t, a clonal isolate that is avirulent and has a rough LPS phenotype (phase II), which can be studied in a biosafety level 2 environment. C. burnetii isolates cause either acute or chronic disease, and phylogenetic studies show that they cluster into three clades corresponding to their clinical presentation. Experimental animal models support the hypothesis that there are three specific pathotypes of Coxiella (140). Recent outbreaks have highlighted the continually emerging nature of this pathogen (141), and the significant impact of Coxiella on U.S. warfighters in Iraq and Afghanistan demonstrates the endemic nature of the disease (142).
Coxiella alternates between two morphologies during its developmental lifecycle
The highly infectious nature of C. burnetii is partially due to its ability to survive in the environment for long periods of time in its metabolically inactive, small cell variant (SCV) form. Replication in a range of developmental stages is similar in design, but not in mechanism, to that described for Chlamydia. In the current model, SCV are predominantly extracellular, and while they are environmentally resistant, they are incapable of replication. After SCV invade host cells, they transition to metabolically active large cell variant (LCV) form following the acidification of the Coxiella-containing vacuole (CCV). The percentage of Coxiella in the CCV that have transitioned to LCVs approaches 80% after 1 hour of infection, and by 16 hours the CCV contains exclusively LCV (143, 144). LCV replicate in an expanding CCV for 4-7 days, transitioning back to SCV at later stages of the infection cycle, generating stable extracellular forms for future rounds of infection.
Completion of the Coxiella infection cycle requires replication in a phagolysosome-like compartment
Unlike the vacuolar pathogens discussed to this point, Coxiella allows phagosomal trafficking to proceed all the way to lysosomal fusion. Development of the CCV involves a combination of normal phagosomal trafficking and pathogen-direction manipulation of host processes. The end result is a niche that represents a hostile environment to most other pathogens, and is therefore free of competitors.
(i) Adhesion and invasion
After aerosol transmission, C. burnetii targets alveolar macrophages and passively enters these cells by actin-dependent phagocytosis (145, 146). Recent studies using type IV secretion system (T4SS) mutants that have no defect in their ability to infect either phagocytic or non-phagocytic cells (147, 148) support the hypothesis that entry into phagocytic cells proceeds via host-driven actin-dependent phagocytosis. Virulent C. burnetii bind to phagocytic cells (monocytes/macrophages) using αVβ3 integrin as the main receptor and are taken up into the cell via a Rac1-dependent phagocytosis that depends on membrane ruffling (149, 150).
Histopathologic analysis in both animal models of pneumonia and in humans with atypical pneumonia caused by C. burnetii primarily identified monocyte/macrophage infection, but epithelial and endothelial cell infection has also been reported (151). Non-phagocytic cells do not usually engulf large particles, however, intracellular pathogens can invade after direct contact through a zippering of bacterial ligands to host cell receptors. While the identities of the host receptors for Coxiella on non-professional phagocytes remain to be determined, the mechanism of entry also appears to be dependent on actin rearrangement (145). Recently, a Coxiella protein that is required for invasion of epithelial cells has been identified. Mutants in CBU1260, which encodes the invasin OmpA, exhibit an epithelial cell-specific entry defect (Martinez, 2014). Interestingly, attachment to epithelial cells is unaffected by mutation of CBU1260, indicating that binding and entry of Coxiella are independent processes. Despite the invasion defect observed in non-phagocytic cells, actin rearrangement is unaffected by mutation of CBU1260 in THP-1 macrophages, highlighting again the host-driven nature of phagocytic uptake of Coxiella.
In a variety of tissue culture models C. burnetii phase variants exhibit distinct uptake kinetics whereby avirulent phase II bacteria are more readily internalized than fully virulent phase I bacteria (145, 146). However, both variants replicate with similar kinetics within an indistinguishable, proteolytically active CCV in a variety of tissue culture cell lines (152), which is important to note, as the majority of studies that examine intracellular trafficking have used the avirulent phase II C. burnetii (153).
(ii) Trafficking from the phagosome to the CCV
Phagocytosis results in the formation of the phagosome, which matures into a phagolysosome following a series of highly ordered and regulated fusion and fission events (12). The maturation of the CCV essentially follows the general phagocytic pathway terminating in a compartment that strongly resembles a mature phagolysosome (140). After internalization, the nascent CCV is similar in size to an endosome, and it traffics through the pathway described above as indicated by the sequential recruitment of Rab5 and Rab7 (Figure 2) (153). A hallmark of Coxiella infection is a marked expansion in CCV size during maturation, prior to when the majority of LCV replication begins, resulting in an extremely spacious vacuole at early stages of infection. Functional inhibition of both Rab5 and Rab7 by overexpression of dominant negative forms of these proteins impairs large vacuole formation (153). Furthermore, formation of a spacious CCV is dependent on de novo C. burnetii protein synthesis, as well as secretion of T4SS effectors across the CCV membrane into the host cytoplasm (147, 154). Indeed, a growing number of Coxiella T4SS substrates are known to be required for CCV expansion and several effectors have recently been shown to localize the CCV membrane during infection (155). However, in the absence of protein synthesis, small CCVs still acquire LAMP proteins and acidify, suggesting that acquisition of LAMPs and acidification are a consequence of the normal progression of phagosomal trafficking (156).
Coxiella is unlike the intracellular pathogens discussed so far, which disrupt the endosomal cascade, or arrest maturation of the phagosome, thereby avoiding lysosomal fusion (157). In the case of C. burnetii infection, lysosomal enzymes including cathepsin D and acid phosphatase, accumulate within the CCV by 1 to 2 hours post infection (144), a protracted time course compared to the 15 minutes required for phagosomes harboring inert particles. The observed delay is dependent upon C. burnetii protein synthesis, suggesting it is a pathogen-induced process (144, 158, 159). While the delay in lysosomal fusion of the CCV requires bacterial protein synthesis, the T4SS an unlikely mediator of the process, as secretion has not been detected until at least 8 hours post-infection (148). There is some evidence that interaction with the autophagy pathway contributes to this delayed phagosomal maturation (153). Autophagy serves as an innate host defense against many intracellular pathogens (160), and as early as 5 minutes after internalization, the phase II CCV is decorated with the autophagy marker microtubule-associated protein light-chain 3 (LC3) (Figure 2) (153, 161). However, C. burnetii may benefit from this early interaction with autophagosomes as autophagy induction in infected cells increases CCV size (162).
(iii) Survival inside a degradative vacuole
Coxiella burnetii has adapted to replicate in the very compartment that Mycobacterium, Chlamydia, and Brucella have evolved to avoid. In fact, the metabolic activity of Coxiella is activated in response the reduced pH of the nascent CCV (143, 144). As mentioned above, the CCV acquires degradative cathepsin D as well as acid phosphatase soon after infection (144). Other normally bactericidal contents of a lysosome include ROS and RNS, and Coxiella may employ various strategies to resist these deadly host defenses against infection (163). Upon host cell invasion, Coxiella upregulates transcription of stress response genes, which may serve to protect its DNA from the damaging effects of ROS (164). To supplement its SOS response, the Coxiella genome encodes several genes that enable the bacterium to resist the damaging effects of oxidative stress. An acid phosphatase, encoded by CBU0335, can suppress ROS production in neutrophils by preventing the assembly of host NADPH phagocyte oxidase (165), a major generator of superoxide radicals. Coxiella also expresses ROS scavenging systems for the direct detoxification of superoxide. C. burnetii encodes a functional cytoplasmic FeSOD (CBU1708) that complemented a double sodA sodC mutant strain of E. coli under conditions of oxidative stress (166). Other gene products that may take part in ROS scavenging include a periplasmic Cu/ZnSOD (CBU1822), and two alkyl hydroperoxide reductases, AhpC1 (CBU1706) and AhpC2D (CBU1477-1478). Resistance to acid may also be imparted by the expression of a large number of basic proteins, as close to 45% of C. burnetii proteins have a predicted pI ≥9 (167). While C. burnetii certainly has a cohort of genetic tools that may impart resistance to damaging phagolysosomal contents, they are not likely to be their first line of defense. A study by Brennan, et al. illustrated that C. burnetii infection is well-controlled by the host's oxidative burst (168). As a stealth pathogen, it is probably Coxiella's ability to infect macrophages without alerting the innate immune response that serves are its primary mechanism to avoid killing due to oxidative stress.
(iv) Expansion of the CCV into a spacious and replication competent compartment
The CCV enlarges dramatically between 8 hours and two days post infection, and can occupy nearly the entire space of the host cell (169). In the case of multiply infected cells, the formation of a large CCV is due to homotypic fusion of several smaller CCV, while the expansion of the compartment proceeds through heterotypic fusion with autophagic, endocytic, and lysosomal vesicles (154, 156). The development of the CCV not only requires bacterial protein synthesis (156), but several host proteins are also necessary. The actin cytoskeleton supports CCV development, and treatment with actin-depolymerizing agents results in the formation of only small CCV (170). Additionally, C. burnetii infection activates several host cell kinases, including protein kinase C, protein kinase A, and myosin light chain kinase, all of which are required for the establishment and maintenance of the CCV (171). The parasitophorous vacuole also interacts with the early secretory pathway, as Rab1b accumulates on the CCV membrane, an interaction which is required for the formation of the large CCV (Figure 2) (172). Interaction with the ER through the early secretory pathway may provide another source of membrane for the formation of a spacious CCV (172). The large number of cellular processes that are coordinated during Coxiella infection suggest that many virulence factors that direct the formation of the CCV remain to be identified.
Coxiella burnetii is a unique bacterium in that it is specifically adapted to infect immune cells that are employed to destroy invading pathogens. Not only does it infect a particularly restrictive cell type, but it replicates within a niche that is especially hostile towards bacteria, in general. It utilizes the low pH of the CCV to activate its metabolism, as well as its pathogenic program of type 4 secreted substrates. It has adapted to resist the toxic ROS and RNS, as well as the degradative enzymes resident within lysosomes. Life in a CCV affords Coxiella some very important benefits in the form of acquired immune evasion and reduced competition from other pathogens. However, it is little wonder why other pathogens have evolved to avoid growth under these same conditions.
CONCLUSION
Intracellular pathogens have evolved many unique and elegant strategies for the survival within their host cells. Entry via phagocytosis does not necessarily sentence a pathogen to death in a phagolysosome. Some bacteria manage to break free of their phagosome in order to replicate in the nutrient-rich cytosol. For those that spend the duration of their intracellular life inside a vacuole, there are myriad ways to customize the replicative niche. The problem of lysosomal fusion can be avoided by stalling trafficking, as in the case of M. tuberculosis, or by diverting the vacuole from the endocytic pathway entirely, like Chlamydia and Brucella. For C. burnetii, lysosomal fusion is not avoided, but required for metabolic activation and replication. Whenever a pathogen adapts to a specific intracellular niche, its lifestyle is shaped by the innate immune defenses therein. For bacteria that replicate primarily in phagocytic cells, such as Mtb and Coxiella, the oxidative burst is a major evolutionary driver. Although they each inhabit and replicate in very different subcellular compartments, Mtb, Chlamydia, Brucella, and Coxiella are all very successful pathogens that can serve as valuable tools to enlighten our knowledge of host cell trafficking and immunity.
ACKNOWLEDGEMENTS
We are grateful to Erin van Schaik for helpful discussions and critical reading of the manuscript. This review was supported by grants from the Defense Threat Reduction Agency (DTRA) and National Institute of Allergy and Infectious Diseases (NIAID) to J. Samuel. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the funding agencies.
REFERENCES
- 1.Fredlund J, Enninga J. Cytoplasmic access by intracellular bacterial pathogens. Trends in Microbiology. 2014;22:128–137. doi: 10.1016/j.tim.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Molecular Cell. 2014;54:224–233. doi: 10.1016/j.molcel.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nature Reviews. Molecular Cell Biology. 2008;9:781–795. doi: 10.1038/nrm2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, Schreiber A, Backer JM, Cantley LC, Grinstein S. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. The Journal of Cell Biology. 2001;155:19–25. doi: 10.1083/jcb.200107069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 6.Simonsen A, Lippe R, Christoforidis S, Gaullier J-M, Brech A, Callaghan J, Toh B-H, Murphy C, Zerial M, Stenmark H. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 1998;394:494–498. doi: 10.1038/28879. [DOI] [PubMed] [Google Scholar]
- 7.Hackam DJ, Rotstein OD, Zhang W-J, Demaurex N, Woodside M, Tsai O, Grinstein S. Regulation of Phagosomal Acidification: Differential targeting of Na+/H+exchangers, Na+/K+-ATPases, and vacuolar-type H+-ATPases. Journal of Biological Chemistry. 1997;272:29810–29820. doi: 10.1074/jbc.272.47.29810. [DOI] [PubMed] [Google Scholar]
- 8.Kinchen JM, Ravichandran KS. Identification of two evolutionarily conserved genes regulating processing of engulfed apoptotic cells. Nature. 2010;464:778–782. doi: 10.1038/nature08853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poteryaev D, Datta S, Ackema K, Zerial M, Spang A. Identification of the switch in early-to-late endosome transition. Cell. 2010;141:497–508. doi: 10.1016/j.cell.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 10.Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. The EMBO Journal. 2007;26(2):313–324. doi: 10.1038/sj.emboj.7601511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harrison RE, Bucci C, Vieira OV, Schroer TA, Grinstein S. Phagosomes Fuse with Late Endosomes and/or Lysosomes by Extension of Membrane Protrusions along Microtubules: Role of Rab7 and RILP. Molecular and Cellular Biology. 2003;23:6494–6506. doi: 10.1128/MCB.23.18.6494-6506.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flannagan RS, Jaumouillé V, Grinstein S. The cell biology of phagocytosis. Annual Review of Pathology: Mechanisms of Disease. 2012;7:61–98. doi: 10.1146/annurev-pathol-011811-132445. [DOI] [PubMed] [Google Scholar]
- 13.Jabado N, Jankowski A, Dougaparsad S, Picard V, Grinstein S, Gros P. Natural resistance to intracellular infections: natural resistance–associated macrophage protein 1 (Nramp1) functions as a pH-dependent manganese transporter at the phagosomal membrane. The Journal of Experimental Medicine. 2000;192:1237–1248. doi: 10.1084/jem.192.9.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yates RM, Hermetter A, Russell DG. The kinetics of phagosome maturation as a function of phagosome/lysosome fusion and acquisition of hydrolytic activity. Traffic. 2005;6:413–420. doi: 10.1111/j.1600-0854.2005.00284.x. [DOI] [PubMed] [Google Scholar]
- 15.Zumla A, Raviglione M, Hafner R, von Reyn CF. Tuberculosis. The New England Journal of Medicine. 2013;368:745–755. doi: 10.1056/NEJMra1200894. [DOI] [PubMed] [Google Scholar]
- 16.Forrellad MA, Klepp LI, Gioffre A, Sabio y Garcia J, Morbidoni HR, de la Paz Santangelo M, Cataldi AA, Bigi F. Virulence factors of the Mycobacterium tuberculosis complex. Virulence. 2013;4:3–66. doi: 10.4161/viru.22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matteelli A, Roggi A, Carvalho AC. Extensively drug-resistant tuberculosis: epidemiology and management. Clinical Epidemiology. 2014;6:111–118. doi: 10.2147/CLEP.S35839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vergne I, Chua J, Singh SB, Deretic V. Cell biology of Mycobacterium tuberculosis phagosome. Annual Review of Cell and Developmental Biology. 2004;20:367–394. doi: 10.1146/annurev.cellbio.20.010403.114015. [DOI] [PubMed] [Google Scholar]
- 19.Cambier CJ, Falkow S, Ramakrishnan L. Host evasion and exploitation schemes of Mycobacterium tuberculosis. Cell. 2014;159:1497–1509. doi: 10.1016/j.cell.2014.11.024. [DOI] [PubMed] [Google Scholar]
- 20.Clemens DL, Horwitz MA. Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. The Journal of Experimental Medicine. 1995;181:257–270. doi: 10.1084/jem.181.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Via LE, Deretic D, Ulmer RJ, Hibler NS, Huber LA, Deretic V. Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by Rab5 and Rab7. The Journal of Biological Chemistry. 1997;272:13326–13331. doi: 10.1074/jbc.272.20.13326. [DOI] [PubMed] [Google Scholar]
- 22.Kang BK, Schlesinger LS. Characterization of mannose receptor-dependent phagocytosis mediated by Mycobacterium tuberculosis lipoarabinomannan. Infection and Immunity. 1998;66:2769–2777. doi: 10.1128/iai.66.6.2769-2777.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ernst JD. Macrophage receptors for Mycobacterium tuberculosis. Infection and Immunity. 1998;66:1277–1281. doi: 10.1128/iai.66.4.1277-1281.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gatfield J, Pieters J. Essential role for cholesterol in entry of mycobacteria into macrophages. Science. 2000;288:1647–1650. doi: 10.1126/science.288.5471.1647. [DOI] [PubMed] [Google Scholar]
- 25.Clemens DL, Lee BY, Horwitz MA. Mycobacterium tuberculosis and Legionella pneumophila phagosomes exhibit arrested maturation despite acquisition of Rab7. Infection and Immunity. 2000;68:5154–5166. doi: 10.1128/iai.68.9.5154-5166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clemens DL, Lee BY, Horwitz MA. Deviant expression of Rab5 on phagosomes containing the intracellular pathogens Mycobacterium tuberculosis and Legionella pneumophila is associated with altered phagosomal fate. Infection and Immunity. 2000;68:2671–2684. doi: 10.1128/iai.68.5.2671-2684.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vergne I, Chua J, Deretic V. Tuberculosis toxin blocking phagosome maturation inhibits a novel Ca2+/calmodulin-PI3K hVPS34 cascade. The Journal of Experimental Medicine. 2003;198:653–659. doi: 10.1084/jem.20030527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malik ZA, Denning GM, Kusner DJ. Inhibition of Ca(2+) signaling by Mycobacterium tuberculosis is associated with reduced phagosome-lysosome fusion and increased survival within human macrophages. The Journal of Experimental Medicine. 2000;191:287–302. doi: 10.1084/jem.191.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fratti RA, Chua J, Deretic V. Induction of p38 mitogen-activated protein kinase reduces early endosome autoantigen 1 (EEA1) recruitment to phagosomal membranes. The Journal of Biological Chemistry. 2003;278:46961–46967. doi: 10.1074/jbc.M305225200. [DOI] [PubMed] [Google Scholar]
- 30.Cavalli V, Vilbois F, Corti M, Marcote MJ, Tamura K, Karin M, Arkinstall S, Gruenberg J. The stress-induced MAP kinase p38 regulates endocytic trafficking via the GDI:Rab5 complex. Molecular Cell. 2001;7:421–432. doi: 10.1016/s1097-2765(01)00189-7. [DOI] [PubMed] [Google Scholar]
- 31.Fratti RA, Chua J, Vergne I, Deretic V. Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proceedings of the National Acadamy of Sciences. 2003;100:5437–5442. doi: 10.1073/pnas.0737613100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Indrigo J, Hunter RL, Jr., Actor JK. Cord factor trehalose 6,6'-dimycolate (TDM) mediates trafficking events during mycobacterial infection of murine macrophages. Microbiology. 2003;149:2049–2059. doi: 10.1099/mic.0.26226-0. [DOI] [PubMed] [Google Scholar]
- 33.Sun J, Wang X, Lau A, Liao TY, Bucci C, Hmama Z. Mycobacterial nucleoside diphosphate kinase blocks phagosome maturation in murine RAW 264.7 macrophages. PLoS One. 2010;5:e8769. doi: 10.1371/journal.pone.0008769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proceedings of the National Academy of Sciences. 2011;108:19371–19376. doi: 10.1073/pnas.1109201108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller BH, Fratti RA, Poschet JF, Timmins GS, Master SS, Burgos M, Marletta MA, Deretic V. Mycobacteria inhibit nitric oxide synthase recruitment to phagosomes during macrophage infection. Infection and Immunity. 2004;72:2872–2878. doi: 10.1128/IAI.72.5.2872-2878.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis AS, Vergne I, Master SS, Kyei GB, Chua J, Deretic V. Mechanism of inducible nitric oxide synthase exclusion from mycobacterial phagosomes. PLoS Pathogens. 2007;3:e186. doi: 10.1371/journal.ppat.0030186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vergne I, Fratti RA, Hill PJ, Chua J, Belisle J, Deretic V. Mycobacterium tuberculosis phagosome maturation arrest: mycobacterial phosphatidylinositol analog phosphatidylinositol mannoside stimulates early endosomal fusion. Molecular Biology of the Cell. 2004;15:751–760. doi: 10.1091/mbc.E03-05-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clemens DL, Horwitz MA. The Mycobacterium tuberculosis phagosome interacts with early endosomes and is accessible to exogenously administered transferrin. The Journal of Experimental Medicine. 1996;184:1349–1355. doi: 10.1084/jem.184.4.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chauhan R, Mande SC. Characterization of the Mycobacterium tuberculosis H37Rv alkyl hydroperoxidase AhpC points to the importance of ionic interactions in oligomerization and activity. The Biochemical Journal. 2001;354:209–215. doi: 10.1042/0264-6021:3540209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Master SS, Springer B, Sander P, Boettger EC, Deretic V, Timmins GS. Oxidative stress response genes in Mycobacterium tuberculosis: role of ahpC in resistance to peroxynitrite and stage-specific survival in macrophages. Microbiology. 2002;148:3139–3144. doi: 10.1099/00221287-148-10-3139. [DOI] [PubMed] [Google Scholar]
- 41.Heym B, Stavropoulos E, Honore N, Domenech P, Saint-Joanis B, Wilson TM, Collins DM, Colston MJ, Cole ST. Effects of overexpression of the alkyl hydroperoxide reductase AhpC on the virulence and isoniazid resistance of Mycobacterium tuberculosis. Infection and Immunity. 1997;65:1395–1401. doi: 10.1128/iai.65.4.1395-1401.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Z, Kelley C, Collins F, Rouse D, Morris S. Expression of katG in Mycobacterium tuberculosis is associated with its growth and persistence in mice and guinea pigs. The Journal of Infectious Diseases. 1998;177:1030–1035. doi: 10.1086/515254. [DOI] [PubMed] [Google Scholar]
- 43.Hu Y, Coates AR. Acute and persistent Mycobacterium tuberculosis infections depend on the thiol peroxidase TpX. PLoS One. 2009;4:e5150. doi: 10.1371/journal.pone.0005150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piddington DL, Fang FC, Laessig T, Cooper AM, Orme IM, Buchmeier NA. Cu,Zn superoxide dismutase of Mycobacterium tuberculosis contributes to survival in activated macrophages that are generating an oxidative burst. Infection and Immunity. 2001;69:4980–4987. doi: 10.1128/IAI.69.8.4980-4987.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stewart GR, Newton SM, Wilkinson KA, Humphreys IR, Murphy HN, Robertson BD, Wilkinson RJ, Young DB. The stress-responsive chaperone alpha-crystallin 2 is required for pathogenesis of Mycobacterium tuberculosis. Molecular Microbiology. 2005;55:1127–1137. doi: 10.1111/j.1365-2958.2004.04450.x. [DOI] [PubMed] [Google Scholar]
- 46.Vandal OH, Roberts JA, Odaira T, Schnappinger D, Nathan CF, Ehrt S. Acid-susceptible mutants of Mycobacterium tuberculosis share hypersusceptibility to cell wall and oxidative stress and to the host environment. Journal of Bacteriology. 2009;191:625–631. doi: 10.1128/JB.00932-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson NB, Hayes LD, Brown K, Hoo EC, Ethier KA, Centers for Disease C. CDC National Health Report: leading causes of morbidity and mortality and associated behavioral risk and protective factors--United States, 2005-2013. Morbidity and Mortality Weekly Report. Surveillance Summaries. 2014;63(Suppl 4):3–27. Prevention. [PubMed] [Google Scholar]
- 48.Hahn DL, Schure A, Patel K, Childs T, Drizik E, Webley W. Chlamydia pneumoniae-specific IgE is prevalent in asthma and is associated with disease severity. PLoS One. 2012;7:e35945. doi: 10.1371/journal.pone.0035945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Honarmand H. Atherosclerosis induced by Chlamydophila pneumoniae: a controversial theory. Interdisciplinary Perspectives on Infectious Diseases. 2013;2013:941392. doi: 10.1155/2013/941392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harkinezhad T, Geens T, Vanrompay D. Chlamydophila psittaci infections in birds: a review with emphasis on zoonotic consequences. Veterinary Microbiology. 2009;135:68–77. doi: 10.1016/j.vetmic.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 51.Clifton DR, Fields KA, Grieshaber SS, Dooley CA, Fischer ER, Mead DJ, Carabeo RA, Hackstadt T. A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proceedings of the National Academy of Sciences. 2004;101:10166–10171. doi: 10.1073/pnas.0402829101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hybiske K, Stephens RS. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proceedings of the National Academy of Sciences. 2007;104:11430–11435. doi: 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clifton DR, Dooley CA, Grieshaber SS, Carabeo RA, Fields KA, Hackstadt T. Tyrosine phosphorylation of the chlamydial effector protein Tarp is species specific and not required for recruitment of actin. Infection and Immunity. 2005;73:3860–3868. doi: 10.1128/IAI.73.7.3860-3868.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jewett TJ, Fischer ER, Mead DJ, Hackstadt T. Chlamydial TARP is a bacterial nucleator of actin. Proceedings of the National Academy of Sciences. 2006;103:15599–15604. doi: 10.1073/pnas.0603044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumar Y, Valdivia RH. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host and Microbe. 2008;4:159–169. doi: 10.1016/j.chom.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hackstadt T, Rockey D, Heinzen R, Scidmore M. Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. The EMBO Journal. 1996;15:964–977. [PMC free article] [PubMed] [Google Scholar]
- 57.Hackstadt T, Scidmore MA, Rockey DD. Lipid metabolism in Chlamydia trachomatis-infected cells: directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proceedings of the National Academy of Sciences. 1995;92:4877–4881. doi: 10.1073/pnas.92.11.4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Damiani MT, Gambarte Tudela J, Capmany A. Targeting eukaryotic Rab proteins: a smart strategy for chlamydial survival and replication. Cellular Microbiology. 2014;16:1329–1338. doi: 10.1111/cmi.12325. [DOI] [PubMed] [Google Scholar]
- 59.Bastidas RJ, Elwell CA, Engel JN, Valdivia RH. Chlamydial intracellular survival strategies. Cold Spring Harbor Perspectives in Medicine. 2013;3:a010256. doi: 10.1101/cshperspect.a010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scidmore MA, Fischer ER, Hackstadt T. Restricted fusion of Chlamydia trachomatis vesicles with endocytic compartments during the initial stages of infection. Infection and Immunity. 2003;71:973–984. doi: 10.1128/IAI.71.2.973-984.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heinzen RA, Scidmore MA, Rockey DD, Hackstadt T. Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infection and Immunity. 1996;64:796–809. doi: 10.1128/iai.64.3.796-809.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schramm N, Bagnell CR, Wyrick PB. Vesicles containing Chlamydia trachomatis serovar L2 remain above pH 6 within HEC-1B cells. Infection and Immunity. 1996;64:1208–1214. doi: 10.1128/iai.64.4.1208-1214.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ouellette SP, Dorsey FC, Moshiach S, Cleveland JL, Carabeo RA. Chlamydia species-dependent differences in the growth requirement for lysosomes. PLoS One. 2011;6:e16783. doi: 10.1371/journal.pone.0016783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carabeo RA, Dooley CA, Grieshaber SS, Hackstadt T. Rac interacts with Abi-1 and WAVE2 to promote an Arp2/3-dependent actin recruitment during chlamydial invasion. Cellular Microbiology. 2007;9:2278–2288. doi: 10.1111/j.1462-5822.2007.00958.x. [DOI] [PubMed] [Google Scholar]
- 65.Lane BJ, Mutchler C, Al Khodor S, Grieshaber SS, Carabeo RA. Chlamydial entry involves TARP binding of guanine nucleotide exchange factors. PLoS Pathogens. 2008;4:e1000014. doi: 10.1371/journal.ppat.1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Belland RJ, Scidmore MA, Crane DD, Hogan DM, Whitmire W, McClarty G, Caldwell HD. Chlamydia trachomatis cytotoxicity associated with complete and partial cytotoxin genes. Proceedings of the National Academy of Sciences. 2001;98:13984–13989. doi: 10.1073/pnas.241377698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thalmann J, Janik K, May M, Sommer K, Ebeling J, Hofmann F, Genth H, Klos A. Actin re-organization induced by Chlamydia trachomatis serovar D--evidence for a critical role of the effector protein CT166 targeting Rac. PLoS One. 2010;5:e9887. doi: 10.1371/journal.pone.0009887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hower S, Wolf K, Fields KA. Evidence that CT694 is a novel Chlamydia trachomatis T3S substrate capable of functioning during invasion or early cycle development. Molecular Microbiology. 2009;72:1423–1437. doi: 10.1111/j.1365-2958.2009.06732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grieshaber SS, Grieshaber NA, Hackstadt T. Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process. Journal of Cell Science. 2003;116:3793–3802. doi: 10.1242/jcs.00695. [DOI] [PubMed] [Google Scholar]
- 70.Mital J, Miller NJ, Fischer ER, Hackstadt T. Specific chlamydial inclusion membrane proteins associate with active Src family kinases in microdomains that interact with the host microtubule network. Cellular Microbiology. 2010;12:1235–1249. doi: 10.1111/j.1462-5822.2010.01465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Campbell S, Richmond SJ, Yates PS. The effect of Chlamydia trachomatis infection on the host cell cytoskeleton and membrane compartments. Journal of General Microbiology. 1989;135:2379–2386. doi: 10.1099/00221287-135-9-2379. [DOI] [PubMed] [Google Scholar]
- 72.Lutter EI, Barger AC, Nair V, Hackstadt T. Chlamydia trachomatis inclusion membrane protein CT228 recruits elements of the myosin phosphatase pathway to regulate release mechanisms. Cell Reports. 2013;3:1921–1931. doi: 10.1016/j.celrep.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Elwell CA, Engel JN. Lipid acquisition by intracellular Chlamydiae. Cellular Microbiology. 2012;14:1010–1018. doi: 10.1111/j.1462-5822.2012.01794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rockey DD, Fischer ER, Hackstadt T. Temporal analysis of the developing Chlamydia psittaci inclusion by use of fluorescence and electron microscopy. Infection and Immunity. 1996;64:4269–4278. doi: 10.1128/iai.64.10.4269-4278.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wolf K, Hackstadt T. Sphingomyelin trafficking in Chlamydia pneumoniae-infected cells. Cellular Microbiology. 2001;3:145–152. doi: 10.1046/j.1462-5822.2001.00098.x. [DOI] [PubMed] [Google Scholar]
- 76.Moore ER, Fischer ER, Mead DJ, Hackstadt T. The chlamydial inclusion preferentially intercepts basolaterally directed sphingomyelin-containing exocytic vacuoles. Traffic. 2008;9:2130–2140. doi: 10.1111/j.1600-0854.2008.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Derre I, Swiss R, Agaisse H. The lipid transfer protein CERT interacts with the Chlamydia inclusion protein IncD and participates to ER-Chlamydia inclusion membrane contact sites. PLoS Pathogens. 2011;7:e1002092. doi: 10.1371/journal.ppat.1002092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Elwell CA, Jiang S, Kim JH, Lee A, Wittmann T, Hanada K, Melancon P, Engel JN. Chlamydia trachomatis co-opts GBF1 and CERT to acquire host sphingomyelin for distinct roles during intracellular development. PLoS Pathogens. 2011;7:e1002198. doi: 10.1371/journal.ppat.1002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carabeo RA, Mead DJ, Hackstadt T. Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proceedings of the National Academy of Sciences. 2003;100:6771–6776. doi: 10.1073/pnas.1131289100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beatty WL. Late endocytic multivesicular bodies intersect the chlamydial inclusion in the absence of CD63. Infection and Immunity. 2008;76:2872–2881. doi: 10.1128/IAI.00129-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moorhead AM, Jung JY, Smirnov A, Kaufer S, Scidmore MA. Multiple host proteins that function in phosphatidylinositol-4-phosphate metabolism are recruited to the chlamydial inclusion. Infection and Immunity. 2010;78:1990–2007. doi: 10.1128/IAI.01340-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beatty WL. Trafficking from CD63-positive late endocytic multivesicular bodies is essential for intracellular development of Chlamydia trachomatis. Journal of Cell Science. 2006;119:350–359. doi: 10.1242/jcs.02733. [DOI] [PubMed] [Google Scholar]
- 83.Kumar Y, Cocchiaro J, Valdivia RH. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Current Biology. 2006;16:1646–1651. doi: 10.1016/j.cub.2006.06.060. [DOI] [PubMed] [Google Scholar]
- 84.Rzomp KA, Scholtes LD, Briggs BJ, Whittaker GR, Scidmore MA. Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infection and Immunity. 2003;71:5855–5870. doi: 10.1128/IAI.71.10.5855-5870.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brumell JH, Scidmore MA. Manipulation of rab GTPase function by intracellular bacterial pathogens. Microbiology and Molecular Biology Reviews. 2007;71:636–652. doi: 10.1128/MMBR.00023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rzomp KA, Moorhead AR, Scidmore MA. The GTPase Rab4 interacts with Chlamydia trachomatis inclusion membrane protein CT229. Infection and Immunity. 2006;74:5362–5373. doi: 10.1128/IAI.00539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cortes C, Rzomp KA, Tvinnereim A, Scidmore MA, Wizel B. Chlamydia pneumoniae inclusion membrane protein Cpn0585 interacts with multiple Rab GTPases. Infection and Immunity. 2007;75:5586–5596. doi: 10.1128/IAI.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Capmany A, Leiva N, Damiani MT. Golgi-associated Rab14, a new regulator for Chlamydia trachomatis infection outcome. Communicative and Integrative Biology. 2011;4:590–593. doi: 10.4161/cib.4.5.16594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rejman Lipinski A, Heymann J, Meissner C, Karlas A, Brinkmann V, Meyer TF, Heuer D. Rab6 and Rab11 regulate Chlamydia trachomatis development and golgin-84-dependent Golgi fragmentation. PLoS Pathogens. 2009;5:e1000615. doi: 10.1371/journal.ppat.1000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ouellette SP, Carabeo RA. A functional slow recycling pathway of transferrin is required for growth of Chlamydia. Frontiers in Microbiology. 2010;1:112. doi: 10.3389/fmicb.2010.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Robertson DK, Gu L, Rowe RK, Beatty WL. Inclusion biogenesis and reactivation of persistent Chlamydia trachomatis requires host cell sphingolipid biosynthesis. PLoS Pathogens. 2009;5:e1000664. doi: 10.1371/journal.ppat.1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Matsumoto A, Bessho H, Uehira K, Suda T. Morphological studies of the association of mitochondria with chlamydial inclusions and the fusion of chlamydial inclusions. Journal of Electron Microscopy. 1991;40:356–363. [PubMed] [Google Scholar]
- 93.Peterson EM, de la Maza LM. Chlamydia parasitism: ultrastructural characterization of the interaction between the chlamydial cell envelope and the host cell. Journal of Bacteriology. 1988;170:1389–1392. doi: 10.1128/jb.170.3.1389-1392.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Derre I, Pypaert M, Dautry-Varsat A, Agaisse H. RNAi screen in Drosophila cells reveals the involvement of the Tom complex in Chlamydia infection. PLoS Pathogens. 2007;3:1446–1458. doi: 10.1371/journal.ppat.0030155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McClarty G, Tipples G. In situ studies on incorporation of nucleic acid precursors into Chlamydia trachomatis DNA. Journal of Bacteriology. 1991;173:4922–4931. doi: 10.1128/jb.173.16.4922-4931.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McClarty G, Fan H. Purine metabolism by intracellular Chlamydia psittaci. Journal of Bacteriology. 1993;175:4662–4669. doi: 10.1128/jb.175.15.4662-4669.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bannantine JP, Griffiths RS, Viratyosin W, Brown WJ, Rockey DD. A secondary structure motif predictive of protein localization to the chlamydial inclusion membrane. Cellular Microbiology. 2000;2:35–47. doi: 10.1046/j.1462-5822.2000.00029.x. [DOI] [PubMed] [Google Scholar]
- 98.Dehoux P, Flores R, Dauga C, Zhong G, Subtil A. Multi-genome identification and characterization of chlamydiae-specific type III secretion substrates: the Inc proteins. BMC Genomics. 2011;12:109. doi: 10.1186/1471-2164-12-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Franco MP, Mulder M, Gilman RH, Smits HL. Human brucellosis. The Lancet Infectious Diseases. 2007;7:775–786. doi: 10.1016/S1473-3099(07)70286-4. [DOI] [PubMed] [Google Scholar]
- 100.Bossi P, Tegnell A, Baka A, Van Loock F, Hendriks J, Werner A, Maidhof H, Gouvras G, Task Force on B, Chemical Agent Threats PHDECL Bichat guidelines for the clinical management of brucellosis and bioterrorism-related brucellosis. Euro Surveillance: European Communicable Disease Bulletin. 2004;9:E15–16. [PubMed] [Google Scholar]
- 101.Seleem MN, Boyle SM, Sriranganathan N. Brucellosis: a re-emerging zoonosis. Veterinary Microbiology. 2010;140:392–398. doi: 10.1016/j.vetmic.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 102.Gomez G, Adams LG, Rice-Ficht A, Ficht TA. Host-Brucella interactions and the Brucella genome as tools for subunit antigen discovery and immunization against brucellosis. Frontiers in Cellular and Infection Microbiology. 2013;3:17. doi: 10.3389/fcimb.2013.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.von Bargen K, Gorvel JP, Salcedo SP. Internal affairs: investigating the Brucella intracellular lifestyle. FEMS Microbiology Reviews. 2012;36:533–562. doi: 10.1111/j.1574-6976.2012.00334.x. [DOI] [PubMed] [Google Scholar]
- 104.Pizarro-Cerda J, Meresse S, Parton RG, van der Goot G, Sola-Landa A, Lopez-Goni I, Moreno E, Gorvel JP. Brucella abortus transits through the autophagic pathway and replicates in the endoplasmic reticulum of nonprofessional phagocytes. Infection and Immunity. 1998;66:5711–5724. doi: 10.1128/iai.66.12.5711-5724.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Starr T, Child R, Wehrly TD, Hansen B, Hwang S, Lopez-Otin C, Virgin HW, Celli J. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host and Microbe. 2012;11:33–45. doi: 10.1016/j.chom.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Starr T, Ng TW, Wehrly TD, Knodler LA, Celli J. Brucella intracellular replication requires trafficking through the late endosomal/lysosomal compartment. Traffic. 2008;9:678–694. doi: 10.1111/j.1600-0854.2008.00718.x. [DOI] [PubMed] [Google Scholar]
- 107.Boschiroli ML, Ouahrani-Bettache S, Foulongne V, Michaux-Charachon S, Bourg G, Allardet-Servent A, Cazevieille C, Lavigne JP, Liautard JP, Ramuz M, O'Callaghan D. Type IV secretion and Brucella virulence. Veterinary Microbiology. 2002;90:341–348. doi: 10.1016/s0378-1135(02)00219-5. [DOI] [PubMed] [Google Scholar]
- 108.Celli J, de Chastellier C, Franchini DM, Pizarro-Cerda J, Moreno E, Gorvel JP. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. The Journal of Experimental Medicine. 2003;198:545–556. doi: 10.1084/jem.20030088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Comerci DJ, Martinez-Lorenzo MJ, Sieira R, Gorvel JP, Ugalde RA. Essential role of the VirB machinery in the maturation of the Brucella abortus-containing vacuole. Cellular Microbiology. 2001;3:159–168. doi: 10.1046/j.1462-5822.2001.00102.x. [DOI] [PubMed] [Google Scholar]
- 110.Pei J, Kahl-McDonagh M, Ficht TA. Brucella dissociation is essential for macrophage egress and bacterial dissemination. Frontiers in Cellular and Infection Microbiology. 2014;4:23. doi: 10.3389/fcimb.2014.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Qin QM, Pei J, Ancona V, Shaw BD, Ficht TA, de Figueiredo P. RNAi screen of endoplasmic reticulum-associated host factors reveals a role for IRE1α in supporting Brucella replication. PLoS Pathogens. 2008;4:e1000110. doi: 10.1371/journal.ppat.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Naroeni A, Porte F. Role of cholesterol and the ganglioside GM(1) in entry and short-term survival of Brucella suis in murine macrophages. Infection and Immunity. 2002;70:1640–1644. doi: 10.1128/IAI.70.3.1640-1644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guzman-Verri C, Chaves-Olarte E, von Eichel-Streiber C, Lopez-Goni I, Thelestam M, Arvidson S, Gorvel JP, Moreno E. GTPases of the Rho subfamily are required for Brucella abortus internalization in nonprofessional phagocytes: direct activation of Cdc42. The Journal of Biological Chemistry. 2001;276:44435–44443. doi: 10.1074/jbc.M105606200. [DOI] [PubMed] [Google Scholar]
- 114.Pei J, Turse JE, Ficht TA. Evidence of Brucella abortus OPS dictating uptake and restricting NF-kappaB activation in murine macrophages. Microbes and Infection. 2008;10:582–590. doi: 10.1016/j.micinf.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pei J, Ficht TA. Brucella abortus rough mutants are cytopathic for macrophages in culture. Infection and Immunity. 2004;72:440–450. doi: 10.1128/IAI.72.1.440-450.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Porte F, Naroeni A, Ouahrani-Bettache S, Liautard JP. Role of the Brucella suis lipopolysaccharide O antigen in phagosomal genesis and in inhibition of phagosome-lysosome fusion in murine macrophages. Infection and Immunity. 2003;71:1481–1490. doi: 10.1128/IAI.71.3.1481-1490.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rittig MG, Kaufmann A, Robins A, Shaw B, Sprenger H, Gemsa D, Foulongne V, Rouot B, Dornand J. Smooth and rough lipopolysaccharide phenotypes of Brucella induce different intracellular trafficking and cytokine/chemokine release in human monocytes. Journal of Leukocyte Biology. 2003;74:1045–1055. doi: 10.1189/jlb.0103015. [DOI] [PubMed] [Google Scholar]
- 118.Martin-Martin AI, Vizcaino N, Fernandez-Lago L. Cholesterol, ganglioside GM1 and class A scavenger receptor contribute to infection by Brucella ovis and Brucella canis in murine macrophages. Microbes and Infection. 2010;12:246–251. doi: 10.1016/j.micinf.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 119.Hong PC, Tsolis RM, Ficht TA. Identification of genes required for chronic persistence of Brucella abortus in mice. Infection and Immunity. 2000;68:4102–4107. doi: 10.1128/iai.68.7.4102-4107.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sieira R, Comerci DJ, Sanchez DO, Ugalde RA. A homologue of an operon required for DNA transfer in Agrobacterium is required in Brucella abortus for virulence and intracellular multiplication. Journal of Bacteriology. 2000;182:4849–4855. doi: 10.1128/jb.182.17.4849-4855.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arellano-Reynoso B, Lapaque N, Salcedo S, Briones G, Ciocchini AE, Ugalde R, Moreno E, Moriyon I, Gorvel JP. Cyclic beta-1,2-glucan is a Brucella virulence factor required for intracellular survival. Nature Immunology. 2005;6:618–625. doi: 10.1038/ni1202. [DOI] [PubMed] [Google Scholar]
- 122.Roop RM, 2nd, Gaines JM, Anderson ES, Caswell CC, Martin DW. Survival of the fittest: how Brucella strains adapt to their intracellular niche in the host. Medical Microbiology and Immunology. 2009;198:221–238. doi: 10.1007/s00430-009-0123-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Valderas MW, Alcantara RB, Baumgartner JE, Bellaire BH, Robertson GT, Ng WL, Richardson JM, Winkler ME, Roop RM., 2nd. Role of HdeA in acid resistance and virulence in Brucella abortus 2308. Veterinary Microbiology. 2005;107:307–312. doi: 10.1016/j.vetmic.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 124.Endley S, McMurray D, Ficht TA. Interruption of the cydB locus in Brucella abortus attenuates intracellular survival and virulence in the mouse model of infection. Journal of Bacteriology. 2001;183:2454–2462. doi: 10.1128/JB.183.8.2454-2462.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bandara AB, Contreras A, Contreras-Rodriguez A, Martins AM, Dobrean V, Poff-Reichow S, Rajasekaran P, Sriranganathan N, Schurig GG, Boyle SM. Brucella suis urease encoded by ure1 but not ure2 is necessary for intestinal infection of BALB/c mice. BMC Microbiology. 2007;7:57. doi: 10.1186/1471-2180-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jimenez de Bagues MP, Dudal S, Dornand J, Gross A. Cellular bioterrorism: how Brucella corrupts macrophage physiology to promote invasion and proliferation. Clinical Immunology. 2005;114:227–238. doi: 10.1016/j.clim.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 127.Gee JM, Valderas MW, Kovach ME, Grippe VK, Robertson GT, Ng WL, Richardson JM, Winkler ME, Roop RM., 2nd. The Brucella abortus Cu,Zn superoxide dismutase is required for optimal resistance to oxidative killing by murine macrophages and wild-type virulence in experimentally infected mice. Infection and Immunity. 2005;73:2873–2880. doi: 10.1128/IAI.73.5.2873-2880.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Haine V, Dozot M, Dornand J, Letesson JJ, De Bolle X. NnrA is required for full virulence and regulates several Brucella melitensis denitrification genes. Journal of Bacteriology. 2006;188:1615–1619. doi: 10.1128/JB.188.4.1615-1619.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Loisel-Meyer S, Jimenez de Bagues MP, Basseres E, Dornand J, Kohler S, Liautard JP, Jubier-Maurin V. Requirement of norD for Brucella suis virulence in a murine model of in vitro and in vivo infection. Infection and Immunity. 2006;74:1973–1976. doi: 10.1128/IAI.74.3.1973-1976.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Steele KH, Baumgartner JE, Valderas MW, Roop RM., 2nd. Comparative study of the roles of AhpC and KatE as respiratory antioxidants in Brucella abortus 2308. Journal of Bacteriology. 2010;192:4912–4922. doi: 10.1128/JB.00231-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Manterola L, Moriyon I, Moreno E, Sola-Landa A, Weiss DS, Koch MH, Howe J, Brandenburg K, Lopez-Goni I. The lipopolysaccharide of Brucella abortus BvrS/BvrR mutants contains lipid A modifications and has higher affinity for bactericidal cationic peptides. Journal of Bacteriology. 2005;187:5631–5639. doi: 10.1128/JB.187.16.5631-5639.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pizarro-Cerda J, Moreno E, Sanguedolce V, Mege JL, Gorvel JP. Virulent Brucella abortus prevents lysosome fusion and is distributed within autophagosome-like compartments. Infection and Immunity. 1998;66:2387–2392. doi: 10.1128/iai.66.5.2387-2392.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Smith JA, Khan M, Magnani DD, Harms JS, Durward M, Radhakrishnan GK, Liu YP, Splitter GA. Brucella induces an unfolded protein response via TcpB that supports intracellular replication in macrophages. PLoS Pathogens. 2013;9:e1003785. doi: 10.1371/journal.ppat.1003785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fugier E, Salcedo SP, de Chastellier C, Pophillat M, Muller A, Arce-Gorvel V, Fourquet P, Gorvel JP. The glyceraldehyde-3-phosphate dehydrogenase and the small GTPase Rab 2 are crucial for Brucella replication. PLoS Pathogens. 2009;5:e1000487. doi: 10.1371/journal.ppat.1000487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.de Barsy M, Jamet A, Filopon D, Nicolas C, Laloux G, Rual JF, Muller A, Twizere JC, Nkengfac B, Vandenhaute J, Hill DE, Salcedo SP, Gorvel JP, Letesson JJ, De Bolle X. Identification of a Brucella spp. secreted effector specifically interacting with human small GTPase Rab2. Cellular Microbiology. 2011;13:1044–1058. doi: 10.1111/j.1462-5822.2011.01601.x. [DOI] [PubMed] [Google Scholar]
- 136.Nkengfac B, Pouyez J, Bauwens E, Vandenhaute J, Letesson JJ, Wouters J, De Bolle X. Structural analysis of Brucella abortus RicA substitutions that do not impair interaction with human Rab2 GTPase. BMC Biochemistry. 2012;13:16. doi: 10.1186/1471-2091-13-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.de Barsy M, Mirabella A, Letesson JJ, De Bolle X. A Brucella abortus cstA mutant is defective for association with endoplasmic reticulum exit sites and displays altered trafficking in HeLa cells. Microbiology. 2012;158:2610–2618. doi: 10.1099/mic.0.060509-0. [DOI] [PubMed] [Google Scholar]
- 138.Myeni S, Child R, Ng TW, Kupko JJ, 3rd, Wehrly TD, Porcella SF, Knodler LA, Celli J. Brucella modulates secretory trafficking via multiple type IV secretion effector proteins. PLoS Pathogens. 2013;9:e1003556. doi: 10.1371/journal.ppat.1003556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Celli J, Salcedo SP, Gorvel JP. Brucella coopts the small GTPase Sar1 for intracellular replication. Proceedings of the National Academy of Sciences. 2005;102:1673–1678. doi: 10.1073/pnas.0406873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.van Schaik EJ, Chen C, Mertens K, Weber MM, Samuel JE. Molecular pathogenesis of the obligate intracellular bacterium Coxiella burnetii. Nature Reviews. Microbiology. 2013;11:561–573. doi: 10.1038/nrmicro3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Enserink M. Infectious diseases. Questions abound in Q-fever explosion in the Netherlands. Science. 2010;327:266–267. doi: 10.1126/science.327.5963.266-a. [DOI] [PubMed] [Google Scholar]
- 142.White B, Brooks T, Seaton RA. Q fever in military and paramilitary personnel in conflict zones: case report and review. Travel Medicine and Infectious Disease. 2013;11:134–137. doi: 10.1016/j.tmaid.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 143.Coleman SA, Fischer ER, Howe D, Mead DJ, Heinzen RA. Temporal analysis of Coxiella burnetii morphological differentiation. Journal of Bacteriology. 2004;186:7344–7352. doi: 10.1128/JB.186.21.7344-7352.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Howe D, Mallavia LP. Coxiella burnetii exhibits morphological change and delays phagolysosomal fusion after internalization by J774A.1 cells. Infection and Immunity. 2000;68:3815–3821. doi: 10.1128/iai.68.7.3815-3821.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Baca OG, Klassen DA, Aragon AS. Entry of Coxiella burnetii into host cells. Acta Virologica. 1993;37:143–155. [PubMed] [Google Scholar]
- 146.Tujulin E, Macellaro A, Lilliehook B, Norlander L. Effect of endocytosis inhibitors on Coxiella burnetii interaction with host cells. Acta Virologica. 1998;42:125–131. [PubMed] [Google Scholar]
- 147.Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Cockrell DC, Howe D, Voth DE, Heinzen RA. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. MBio. 2011;2:e00175–00111. doi: 10.1128/mBio.00175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Carey KL, Newton HJ, Luhrmann A, Roy CR. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of Ttype IV effectors into host cells and is required for intracellular replication. PLoS Pathogens. 2011;7:e1002056. doi: 10.1371/journal.ppat.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Capo C, Lindberg FP, Meconi S, Zaffran Y, Tardei G, Brown EJ, Raoult D, Mege JL. Subversion of monocyte functions by Coxiella burnetii: impairment of the cross-talk between alphavbeta3 integrin and CR3. Journal of Immunology. 1999;163:6078–6085. [PubMed] [Google Scholar]
- 150.Dellacasagrande J, Ghigo E, Machergui-El S, Hammami, Toman R, Raoult D, Capo C, Mege J-L. Alpha vbeta 3 integrin and bacterial lipopolysaccharide are involved in Coxiella burnetii-stimulated production of tumor necrosis factor by human monocytes. Infection and Immunity. 2000;68:5673–5678. doi: 10.1128/iai.68.10.5673-5678.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Russell-Lodrigue KE, Zhang GQ, McMurray DN, Samuel JE. Clinical and pathologic changes in a guinea pig aerosol challenge model of acute Q fever. Infection and Immunity. 2006;74:6085–6091. doi: 10.1128/IAI.00763-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Howe D, Shannon JG, Winfree S, Dorward DW, Heinzen RA. Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infection and Immunity. 2010;78:3465–3474. doi: 10.1128/IAI.00406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Romano PS, Gutierrez MG, Beron W, Rabinovitch M, Colombo MI. The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cellular Microbiology. 2007;9:891–909. doi: 10.1111/j.1462-5822.2006.00838.x. [DOI] [PubMed] [Google Scholar]
- 154.Howe D, Melnicâakova J, Barâak I, Heinzen RA, University of Wyoming DoMBLWUSA Fusogenicity of the Coxiella burnetii parasitophorous vacuole. Annals of the New York Academy of Sciences. 2003;990:556–562. doi: 10.1111/j.1749-6632.2003.tb07426.x. [DOI] [PubMed] [Google Scholar]
- 155.Larson CL, Beare PA, Voth DE, Howe D, Cockrell DC, Bastidas RJ, Valdivia RH, Heinzen RA. Coxiella burnetii effector proteins that localize to the parasitophorous vacuole membrane promote intracellular replication. Infection and Immunity. 2015;83:661–670. doi: 10.1128/IAI.02763-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Howe D, Melnicâakovâa J, Barâak I, Heinzen RA. Maturation of the Coxiella burnetii parasitophorous vacuole requires bacterial protein synthesis but not replication. Cellular Microbiology. 2003;5(7):469–480. doi: 10.1046/j.1462-5822.2003.00293.x. [DOI] [PubMed] [Google Scholar]
- 157.Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nature Reviews Microbiology. 2009;7:355–366. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- 158.Oh YK, Alpuche-Aranda C, Berthiaume E, Jinks T, Miller SI, Swanson JA. Rapid and complete fusion of macrophage lysosomes with phagosomes containing Salmonella typhimurium. Infection and Immunity. 1996;64:3877–3883. doi: 10.1128/iai.64.9.3877-3883.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Swanson MS, Fernandez-Moreira E. A microbial strategy to multiply in macrophages: the pregnant pause. Traffic. 2002;3:170–177. doi: 10.1034/j.1600-0854.2002.030302.x. [DOI] [PubMed] [Google Scholar]
- 160.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Beron W, Gutierrez MG, Rabinovitch M, Colombo MI. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infection and Immunity. 2002;70:5816–5821. doi: 10.1128/IAI.70.10.5816-5821.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gutierrez MG, Vazquez CL, Munafo DB, Zoppino FC, Beron W, Rabinovitch M, Colombo MI. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cellular Microbiology. 2005;7:981–993. doi: 10.1111/j.1462-5822.2005.00527.x. [DOI] [PubMed] [Google Scholar]
- 163.Criscitiello MF, Dickman MB, Samuel JE, de Figueiredo P. Tripping on acid: trans-kingdom perspectives on biological acids in immunity and pathogenesis. PLoS Pathogens. 2013;9:e1003402. doi: 10.1371/journal.ppat.1003402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mertens K, Lantsheer L, Ennis DG, Samuel JE. Constitutive SOS expression and damage-inducible AddAB-mediated recombinational repair systems for Coxiella burnetii as potential adaptations for survival within macrophages. Molecular Microbiology. 2008;69:1411–1426. doi: 10.1111/j.1365-2958.2008.06373.x. [DOI] [PubMed] [Google Scholar]
- 165.Hill J, Samuel JE. Coxiella burnetii acid phosphatase inhibits the release of reactive oxygen intermediates in polymorphonuclear leukocytes. Infection and Immunity. 2011;79:414–420. doi: 10.1128/IAI.01011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Heinzen RA, Frazier ME, Mallavia LP. Coxiella burnetii superoxide dismutase gene: cloning, sequencing, and expression in Escherichia coli. Infection and Immunity. 1992;60:3814–3823. doi: 10.1128/iai.60.9.3814-3823.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Seshadri R, Paulsen IT, Eisen JA, Read TD, Nelson KE, Nelson WC, Ward NL, Tettelin H, Davidsen TM, Beanan MJ, Deboy RT, Daugherty SC, Brinkac LM, Madupu R, Dodson RJ, Khouri HM, Lee KH, Carty HA, Scanlan D, Heinzen RA, Thompson HA, Samuel JE, Fraser CM, Heidelberg JF. Complete genome sequence of the Q-fever pathogen Coxiella burnetii. Proceedings of the National Academy of Sciences. 2003;100:5455–5460. doi: 10.1073/pnas.0931379100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Brennan RE, Russell K, Zhang G, Samuel JE. Both inducible nitric oxide synthase and NADPH oxidase contribute to the control of virulent phase I Coxiella burnetii infections. Infection and Immunity. 2004;72:6666–6675. doi: 10.1128/IAI.72.11.6666-6675.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Roman MJ, Crissman HA, Samsonoff WA, Hechemy KE, Baca OG. Analysis of Coxiella burnetii isolates in cell culture and the expression of parasite-specific antigens on the host membrane surface. Acta Virologica. 1991;35:503–510. [PubMed] [Google Scholar]
- 170.Aguilera M, Salinas R, Rosales E, Carminati S, Colombo MI, Beron W. Actin dynamics and Rho GTPases regulate the size and formation of parasitophorous vacuoles containing Coxiella burnetii. Infection and Immunity. 2009;77:4609–4620. doi: 10.1128/IAI.00301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hussain SK, Broederdorf LJ, Sharma UM, Voth DE. Host kinase activity is required for Coxiella burnetii parasitophorous vacuole formation. Frontiers in Microbiology. 2011;1 doi: 10.3389/fmicb.2010.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Campoy EM, Zoppino FC, Colombo MI. The early secretory pathway contributes to the growth of the Coxiella-replicative niche. Infection and Immunity. 2011;79:402–413. doi: 10.1128/IAI.00688-10. [DOI] [PMC free article] [PubMed] [Google Scholar]