

Abstract
The cannabinoid 1 receptor (CB1R) is one of the most abundant G protein-coupled receptors (GPCRs) in the central nervous system. CB1R involvement in multiple physiological processes, especially neurotransmitter release and synaptic function, has made this GPCR a prime drug discovery target, and pharmacological CB1R activation has been demonstrated to be a tenable therapeutic modality. Accordingly, the design and profiling of novel, drug-like CB1R modulators to inform the receptor’s ligand-interaction landscape and molecular pharmacology constitute a prime contemporary research focus. For this purpose, we report utilization of AM3677, a designer endocannabinoid (anandamide) analogue derivatized with a reactive electrophilic isothiocyanate functionality, as a covalent, CB1R-selective chemical probe. The data demonstrate that reaction of AM3677 with a cysteine residue in transmembrane helix 6 of human CB1R (hCB1R), C6.47(355), is a key feature of AM3677’s ligand-binding motif. Pharmacologically, AM3677 acts as a high-affinity, lowefficacy CB1R agonist that inhibits forskolin-stimulated cellular cAMP formation and stimulates CB1R coupling to G protein. AM3677 also induces CB1R endocytosis and irreversible receptor internalization. Computational docking suggests the importance of discrete hydrogen bonding and aromatic interactions as determinants of AM3677’s topology within the ligand-binding pocket of active-state hCB1R. These results constitute the initial identification and characterization of a potent, high-affinity, hCB1R-selective covalent agonist with utility as a pharmacologically active, orthostericsite probe for providing insight into structure-function correlates of ligand-induced CB1R activation and the molecular features of that activation by the native ligand, anandamide.
Keywords: Amino acid, binding domain, central nervous system, chemical probe, cysteine, G protein-coupled receptor, homology modeling, isothiocyanate, ligand-binding motif, receptor activation, 7-transmembrane receptor, signal transduction
Graphical Abstract

The endocannabinoid system is a ubiquitous information-transducing network in mammals whose principal molecular components include the cannabinoid 1 (CB1R) and 2 (CB2R) G protein-coupled receptors (GPCRs); their naturally occurring activator ligands, the arachidonic acid-derived endocannabinoid lipid mediators 2-arachidonoylglycerol (2-AG) and arachidonoylethanolamide (anandamide) (AEA); and enzymes responsible for endocannabinoid synthesis and inactivation.1,2 Of these components, CB1R has garnered particular experimental and clinical interest since the discovery over 25 years ago that the main psychoactive cannabis constituent, the phytocannabinoid (-)-Δ9-tetrahy-drocannabinol (Δ9-THC), exerts its psychotropic and addictive effects by activating brain CB1R.3 Expressed to varying extents in peripheral organs where it plays important roles in cardiovascular, reproductive, and metabolic processes, CB1R retains a high (≥97%) degree of amino acid sequence identity across mouse, rat, and human and is one of the most abundant GPCRs in the central nervous system (CNS).2 In CNS physiology, CB1R-mediated cannabinergic signaling serves as a critical retrograde modulator of neurotransmitter release4 and influences many parameters of psychobehavioral state in mammals, including cognition, learning, memory, and emotional valence and reactivity.3,5,6 CB1R information output has also been implicated in other CNS-related processes including synaptic plasticity and neurogenesis.7,8 Although 2-AG is considered to be the primary endocannabinoid modulator of synaptic function, AEA also acts as a lipid messenger in isolated neuronal cells and in the CNS.4,9
Participation of CB1R in multiple biological functions has made this GPCR a focus of medicinal chemistry efforts to design and develop drug-like ligands that modulate its activity for therapeutic gain.10,11 Phytocannabinoids or their congeners that engage and activate CB1R have been approved as drugs to control nausea/emesis, stimulate appetite, manage pain, and reduce multiple sclerosis-related spasticity.12 Preclinical data suggest an appreciably broader utility for synthetic CB1R agonists as antinociceptive, anti-inflammatory, and anticancer drugs.13–15 Identification of CB1R functional residues and characterization of ligand-directed information transduction and intracellular trafficking are sought-after to inform the rational, structure-guided design of therapeutically useful CB1R agonists.16–18 Such information gains particular significance from the proposition that CB1R agonists with specific ligand-binding domains/pharmacological properties might preferentially activate therapeutic signaling cascades over those inviting adverse events, thereby reducing the risk of adverse psychobehavioral responses that may accompany high levels of CB1R activation in the CNS.19
Although X-ray crystallography can provide atomic-level insight into the three-dimensional structure of proteins, diffraction-quality CB1R crystals have proven to be elusive and cannot afford direct characterization of a receptor’s signaling output or trafficking consequent to ligand engagement.20 Attempts have thus been made by us21,22 and others23 to delineate the structural features of ligand-dependent CB1R activation using computational prediction methods, albeit with underlying assumptions regarding the states of apo- and holo-CB1R in situ. This laboratory has also utilized nuclear magnetic resonance spectroscopy (NMR) to characterize experimentally the structure and dynamics of discrete CB1R transmembrane helix (TMH) domains.24–26 Nonetheless, the physiological and therapeutic importance of CB1R warrants direct experimental analysis of CB1R activation across a range of small molecule agonists.
We have developed a ligand-assisted protein structure (LAPS) approach that affords insight into critical features of ligand binding to therapeutically relevant proteins (enzymes, receptors).27,28 This experimental paradigm employs purpose-designed, pharmacologically active covalent affinity probes in tandem with mutational analyses to identify amino acids at (or within the immediate environment of) the protein’s ligand-binding domains critical to ligand recognition/engagement. In conjunction with biological assays, LAPS allows delineation of structure-function correlates in biologically active drug targets. Given the importance of cysteine residues to protein structure and activity,29 we have exploited the spontaneous, preferential reactivity at physiological pH between isothiocyanate (NCS)-functionalized electrophilic ligands and protein cysteine nucleophiles.30 Using one such ligand, the Δ9-THC derivative AM841 [(-)-7′-isothiocyanato-11-hydroxy-1′,1′-dimethylhep-tylhexahydrocannabinol], we have implicated a cysteine residue in transmembrane helix 6 (TMH6) of human CB1R (hCB1R), C6.47(355), as a critical structural feature of hCB1R activation by this classical phytocannabinoid analogue.31 Yet, CB1R agonists are structurally quite diverse and encompass not only Δ9-THC-like classical cannabinoids such as AM841 but also nonclassical cannabinoids, aminoalkylindoles, and the eicosanoid endocannabinoids 2-AG and AEA.1,6,12 The appreciable molecular and chemotype variation among CB1R agonists, along with the potential for different CB1R agonists to activate distinct intracellular signaling pathways in a ligand-directed fashion (“biased agonism” or “functional selectivity”)17 and the corollary therefrom that CB1R may adopt several distinct, ligand-dependent active conformations, raises questions regarding the importance of C6.47(355) to the receptor’s orthosteric ligand-interaction landscape with endocannabinoids and the functional effects of the engagement of eicosanoid-type ligands on CB1R signal output and trafficking.
To address these issues experimentally, we report the profiling of a designer covalent cannabinergic ligand, 20-isothiocyanato-eciosa-5,8,11,14-tetraenoic acid cyclopropyla-mide (AM3677). Featuring an electrophilic NCS functionality, AM3677 is a direct analogue of the AEA derivative and CB1R full agonist arachidonoylcyclopropylamide [(5Z,8Z,11Z,14Z)-eicosa-5,8,11,14-tetraenoic acid cyclopropylamide] (ACPA) (Figure 1).32–34 Our data implicate C6.47(355) as the exclusive TMH cysteine involved in hCB1R activation by AM3677 and the hCB1R residue to which AM3677 covalently binds. We further demonstrate that AM3677 engagement elicits a pattern of CB1R signaling encompassing adenylyl cyclase inhibition, G-protein activation, and irreversible CB1R internalization from the plasma membrane. In silico modeling suggests that discrete hydrogen-bond and aromatic interactions within the receptor’s hydrophobic ligand-binding pocket act as critical determinants of AM3677’s disposition in active-state hCB1R, which is somewhat distinct from that of naturally occurring AEA. These data constitute the initial report and pharmacological profiling of a novel, covalent hCB1R agonist with respect to its binding motif and the functional consequences of hCB1R activation by such a ligand.
Figure 1.
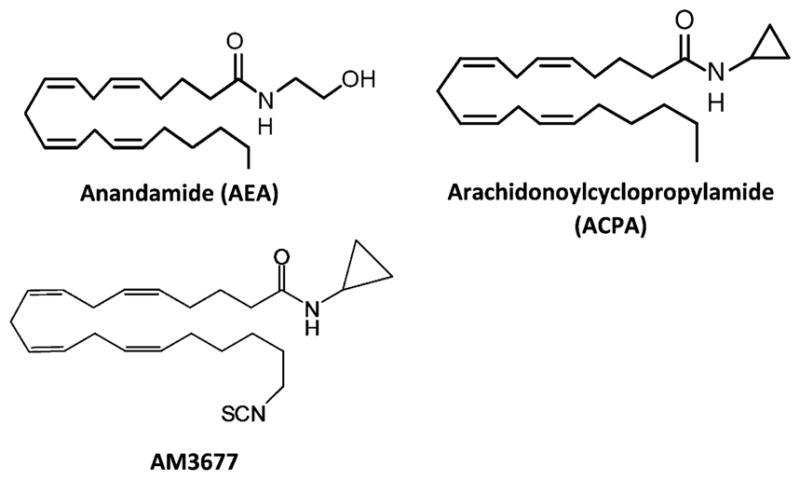
Chemical structures of the endocannabinoid agonist anandamide (AEA) and the AEA derivatives arachidonoylcyclopropylamide (ACPA) and AM3677.
RESULTS AND DISCUSSION
Wild-Type (WT) and Mutant hCB1Rs Are Expressed in Flp-In-293 Cells
Mutation of extracellular loop (ECL) C257 or C254 abrogates high-level hCB1R expression and receptor function, and N-terminal C98 and C107 are critical to hCB1R orthosteric ligand affinity,35,36 TMHs form the hCB1R binding pocket into which small molecule lipid ligands enter, likely from the membrane bilayer through an entry portal delineated by the TMH bundle itself.22 Accordingly, WT hCB1R and hCB1R variants with conservative Cys-to-Ser mutations at each of the receptor’s five TMH Cys residues (out of 13 total) were expressed in Flp-In-293 cells. Receptor functional competency was evaluated in saturation-binding assays on membrane preparations from each Flp-In-293 cell line with [3H]CP55,940, a nonclassical, high-affinity cannabinoid radioligand universally utilized for profiling CB1R/CB2R orthosteric ligand binding.31 The WT and Cys mutant hCB1R variants bound [3H]-CP55,940 with defined, saturable kinetics. Analysis of the saturation-binding data demonstrated that the five Cys mutant hCB1Rs displayed [3H]CP55,940 binding affinities (as Kd) and expression densities (as Bmax) at least comparable to those of WT hCB1R generated in the same Flp-In-293 expression system: the Bmax values across all six hCB1R variants averaged 0.96 ± 0.12 pmol/mg, and the Kd values averaged 5.4 ± 0.8 nM (means ± SEM; n = 6). These results are congruent with prior demonstration that mutation of C6.47(355), C7.38(382), or C7.42(386) to Ala does not appreciably affect hCB1R ligand binding.31,35
AM3677 Can Serve as a Chemical Probe at hCB1R
This laboratory has designed and synthesized various cannabinergic molecules as novel probes for identifying key amino acid residues involved in CB1R/CB2R ligand engagement.37,38 One of our design strategies involves incorporation into known, high-affinity CB1R/CB2R ligands functional groups potentially reactive with amino acids within (or very near) the target receptor’s ligand-binding domain followed by conservative structural modifications (if warranted) to maintain the pharmacological profile of the parent compound. From the resulting library of electrophilic or photoactivatable molecules belonging to various cannabinoid chemical classes, we selected AM3677 for the present study. The selection was predicated upon AM3677 being a direct analogue of ACPA, the latter of which is itself an AEA derivative that binds to rat CB1R (rCB1R) with high affinity (apparent Ki = 2.2–2.7 nM; [3H]CP55,940 as competitive radioligand) and selectivity (~60 to 300-fold vs CB2R) and potently activates recombinant hCB1R as full agonist [EC50 = 2 nM, inhibition of forskolin- stimulated cyclic AMP (cAMP) production] (Figure 1).32,33 The electrophilic NCS functionality was incorporated into AM3677 to allow its preferential and covalent reaction with nucleophilic hCB1R cysteine residues at physiological pH.30,31 Indeed, AM3677 has been demonstrated to bind to rCB1R with high affinity (apparent Ki = 1.3 nM; [3H]CP55,940 as competitive radioligand) and selectivity (~40-fold vs CB2R) in an irreversible manner.33
Since the foregoing precedent for AM3677 as a high-affinity, irreversible CB1R ligand was established with rCB1R,33 we determined the binding affinity of AM3677 for the WT and Cys mutant hCB1Rs that we had expressed in Flp-In-293 cells. No specific AM3677 or [3H]CP55,940 binding to membranes from nontransfected Flp-In-293 cells was observed (data not shown). As summarized in Table 1 from competition-binding assays with [3H]CP55,940, the mean apparent AM3677 Ki value for WT hCB1R (1.7 nM) was comparable to that for each of the Cys mutant hCB1R variants studied (1.6–2.3 nM) and to the apparent AM3677 Ki for rCB1R reported (2.7 nM).33 The high affinity of hCB1R for AM3677 contrasts with this receptor’s moderate affinity for AEA [(Ki = 61 nM (rCB1R); 240 nM (hCB1R)].39 These aggregate chemical and affinity binding data validate AM3677 as a suitable probe for interrogating the molecular features and functional consequences of endocannabinoid(-like) ligand engagement by hCB1R.
Table 1.
Apparent AM3677 Binding Affinities of WT hCB1R and Cys Mutants
| hCB1R variant | apparent Ki (95% CI; nM)a |
|---|---|
| WT | 1.7 (0.7–3.8) |
| C1.55(139)S | 2.3 (1.0–5.3) |
| C4.47(238)S | 2.4 (0.8–7.2) |
| C6.47(355)S | 2.2 (0.8–5.8) |
| C7.38(382)S | 1.9 (0.4–8.1) |
| C7.42(386)S | 1.6 (0.5–5.0) |
Apparent binding affinities for AM3677 (as Ki values) were derived from competitive binding assays with [3H]CP55,940 and membrane preparations from stably transfected Flp-In cells. Data are the mean of at least three independent experiments carried out in triplicate, with 95% confidence intervals (CIs) shown in parentheses.
AM3677 Binds Covalently to hCB1R at C6.47(355)
After a 1 h preincubation of membranes from Flp-In-293 cells overexpressing hCB1R with excess AM3677 (i.e., a final concentration some 10-fold the apparent hCB1R Ki for AM3677) at 30 °C followed by extensive washing to remove unbound ligand, WT hCB1R displayed a significantly reduced (by 43%) mean Bmax for [3H]CP55,940 relative to that of membranes processed in parallel but not exposed to AM3677 prior to incubation with [3H]CP55,940 radioligand (Figure 2A). This result indicates that, under our experimental conditions, AM3677 displayed a 43% level of irreversible (i.e., covalent) binding to hCB1R at a 20 nM concentration such that [3H]CP55,940 was subsequently unable to engage the AM3677-occupied ligand-binding pocket. The C1.55(139)S, C4.47(238)S, C7.38(382)S, and C7.42(386)S hCB1R mutants exhibited a degree of irreversible labeling by AM3677 comparable to that displayed by WT hCB1R (Figure 2A,B). These data allow the conclusion to be made that the respective cysteine residues in hCB1R TMH1, TMH4, and TMH7 are not critical determinants of AM3677 engagement by hCB1R. For the CB1Rs whose orthosteric ligand-binding site remained unoccupied by AM3677, the affinity (as Kd) of the non-covalently modified CB1Rs for CP55,940 was 6.1 nM (2.3–12.4, 95% confidence interval) (n = 3), a low-nanomolar value comparable to the CP55,940 affinity of CB1R in membrane preparations from various tissue and cultured-cell sources, as reported by us31,40,41 and others.39
Figure 2.

AM3677 engages hCB1R covalently at TMH6 cysteine residue C6.47(355). (A) Preincubation of Flp-In-293 membranes from cells expressing hCB1R with excess (10-fold its apparent Ki) AM3677 reduces subsequent [3H]CP55,940 specific binding (i.e., Bmax) by at least 43% to hCB1Rs containing C6.47(355) [as illustrated for WT hCB1R and the C4.47(238)S receptor mutant]. In contrast, the saturation-binding profile of [3H]CP55,940 is unaffected in the hCB1R C6.47(355)S mutant. (B) Comparison of the extent of covalent AM3677 labeling of WT hCB1R and mutants, designated as the difference in the respective [3H]CP55,940 Bmax values of each hCB1R membrane preparation with or without preincubation with AM3677. Data shown represent the mean ± SEM of three independent experiments performed in duplicate.
In marked contrast, preincubation of the hCB1R C6.47(355)S mutant with AM3677 did not affect the receptor’s Bmax for [3H]CP55,940 in a subsequent saturation-binding assay relative to control receptor that was treated in parallel but without [3H]CP55,940 preincubation (Figure 2A). Along with the data in Figure 2B, the lack of AM3677 irreversible binding to the hCB1R C6.47(355)S mutant identifies this TMH6 cysteine as the critical amino acid residue involved in the covalent attachment of the AM3677 NCS group within the hCB1R binding pocket. Although not sharing an endocanna-binoid-like eicosanoid structure with AM3677, the classical cannabinoid affinity label and isothiocyanate Δ9-THC analogue, AM841, also bound covalently to hCB1R C6.47(355),31 suggesting that this TMH6 Cys residue plays an essential role in hCB1R agonist engagement across at least two predominant chemical classes of cannabinergic ligands.
AM3677 Acts as a CB1R Agonist
Adenylyl cyclase and CB1R are negatively coupled, primarily through Gi/o. Activation of CB1R inhibits cellular cAMP formation, a response used to index CB1R G protein-dependent activation.9,17 AM3677 acted as an agonist of adenylyl cyclase-mediated signaling at hCB1R, inhibiting forskolin-activated cAMP formation in Flp-In-293 cells overexpressing hCB1R with an IC50 value of 27.1 ± 2.2 nM (mean ± SEM, n = 3) and maximal efficacy of ~60% at micromolar concentrations (Figure 3). AM3677 had no effect on Flp-In cells that were not transfected with CB1Rs, as per our routine control in this assay (data not shown).42 The molecular pharmacology of AM3677 as an agonist of adenylyl cyclase-mediated signaling is reminiscent of AEA’s partial and ACPA’s full agonist activity at hCB1R.32,34 The comparable, low-nanomolar potencies of both AM3677 and ACPA as inhibitors of forskolin-stimulated cellular cAMP production are much (~200-fold) greater than that reported for AEA in this assay.32
Figure 3.
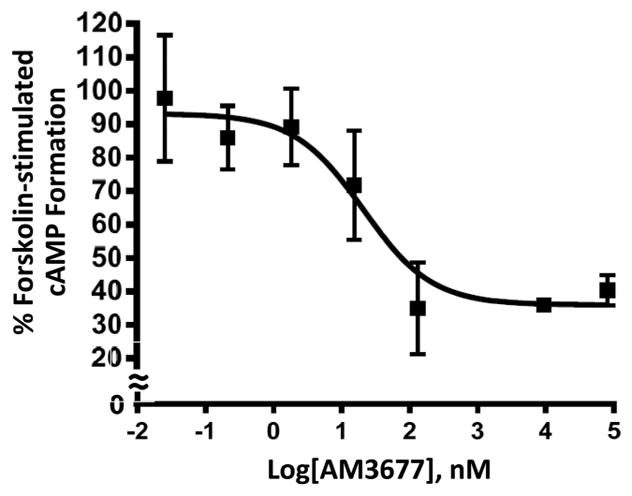
AM3677 acts as an agonist to inhibit in a concentration-dependent manner forskolin-stimulated cAMP accumulation in Flp-In-293 cells expressing WT hCB1R. Data shown represent the mean ± SEM of three independent experiments performed in duplicate.
The guanosine 5′-O-(3-[35S]thio)triphosphate ([35S]-GTPγS) binding assay reflects the functional response of GPCR ligands at the level of GDP/GTP exchange by the ternary, agonist-activated GPCR-G protein complex, an event that can modulate the activity of downstream effector proteins. The assay measures the degree of G protein activation following GPCR agonist engagement, an event more proximal to the GPCR in the biosignaling cascade than cAMP formation. While detection of [35S]GTPγS binding is, in theory, independent of the G protein subtype to which the GPCR couples, in the typical cellular environment the most prominent signal detected is that reflecting Gαi/o subtype activation, as this is a very highly expressed Gα protein.43 Accordingly, we assessed [35S]GTPγS binding to confirm AM3677’s agonist properties by utilizing endogenous CB1R in membranes from mouse hippocampus, which are naturally CB1R-rich.2,3 AM3677, the endocannabinoid AEA, and CP55,940 all stimulated GTPγS binding. However, both AM3677 and AEA elicited considerably less stimulation than did CP55,940: the maximum response produced by AM3677 was 32 ± 2.1%, and that produced by AEA was 34 ± 1.5% (mean ± SEM; n = 3) that of CP55,940 (Figure 4A). Furthermore, AM3677 (EC50 = 0.05 ± 0.02 μM) was nearly 100-fold more potent than AEA (EC50 = 4.2 ± 1.0 μM) and ~23-fold more potent than CP55,940 (EC50 = 1.1 ± 0.2 μM) (mean ± SEM, n = 3–4) as a partial agonist in this test system (Figure 4A). The rank order of potency of these three agonists in our [35S]GTPγS binding assay qualitatively parallels their relative apparent affinities for hCB1R, i.e., AM3677 (1.7 nM, Table 1) > CP55,940 (5.4 ± 0.8 nM, vide supra)31 ≫ AEA (240 nM).39 To support conclusion that the agonists’ responses reflected CB1R-mediated signaling, we employed the cannabinoid-specific antagonist, SR141716A,1,2 which dose-dependently inhibited [35S]GTPγS binding elicited by CP55,904 (IC50 = 126 ± 2.2 nM), AM3677 (IC50 = 79.4 ± 1.5 nM), and AEA (IC50 = 3.6 ± 1.7 nM) (mean ± SEM; n = 3) (Figure 4B). These collective data indicate that AM3677 stimulates G protein coupling by acting as a low-efficacy CB1R partial agonist. Similar to AM3677, both AEA and its stable chiral AEA analogue, R-(+)-methanandamide, have also been characterized as weak partial agonists of CB1R coupling to G proteins.44,45
Figure 4.
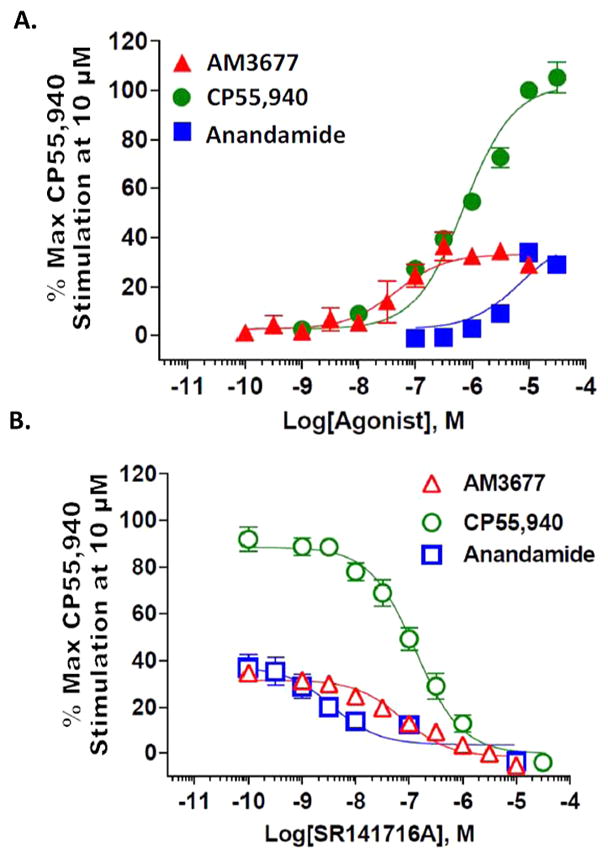
AM3677 elicits CB1R-mediated partial agonist activity when measured as [35S]GTPγS binding to CB1R in mouse hippocampus. In these studies, the maximum response is defined as the response produced by 10 μM CP55,940. (A) AM3677 elicited partial G protein coupling in this measure of response and was more potent than its endocannabinoid analogue, AEA. (B) The response produced by either 3 μM AM 3677, 10 μM CP55,940, or 10 μM anandamide could be completely inhibited by the CB1R competitive antagonist SR14716A in a concentration-dependent manner. Each data point is the mean ± SEM of three to four independent experiments performed in duplicate.
AM3677 Induces CB1R Trafficking and Irreversible Internalization
Intracellular receptor trafficking to and from the plasma membrane is a decisive contributor to the spatial and temporal control of GPCR46 and CB1R47 cell-surface density and signal output. Using a cell-based system developed and validated in this laboratory and employing HEK293 cells stably expressing hemagglutinin (HA)-tagged rCB1R,47 we next analyzed whether AM3677 induces CB1R trafficking. AM3677 was found to elicit CB1R internalization in a concentration-dependent fashion with an EC50 of 3.4 nM and a maximum internalization of ~42% (from the best-fit sigmoidal curve in Figure 5A). Under conditions whereby synthesis of new hCB1Rs was prevented by ribosomal inactivation with emetine, recycling of CB1Rs (indexed as recovery of cell-surface CB1R immunoreactivity) whose internalization had been induced by AM3677 was not observed, indicating that AM3677-induced CB1R internalization is irreversible, whereas hCB1R internalization induced by the full agonist CP55,940 was fully reversible (Figure 5B).
Figure 5.

(A) AM3677 internalizes CB1R in a concentration-dependent manner. HEK293 cells expressing hemagglutinin-tagged rCB1R were incubated with each indicated concentration of AM3677 for 30 min at 37 °C. Loss of cell-surface receptors was quantified as detailed in the text, and a curve of best fit was plotted by nonlinear regression. Values shown are from a single experiment performed in triplicate. (B) AM3677-induced CB1R internalization is irreversible. Treatment for 2 h with AM3677 (300 nM) or CP55,940 (10 nM) induced rCB1R internalization, designated as the relative percent vehicle control cell-surface expression level. Subsequent exposure to CP55,940/SR141716A (SR) (100 nM final SR141716A concentration) resulted in recycling of internalized CB1Rs to the plasma membrane, as demonstrated by the recovery of cell-surface CB1R immunoreactivity after 30 and 60 min. In contrast, CB1R recycling was not observed following AM3677/SR141716A treatment. Values shown are from three independent experiments done in quadruplicate (** p < 0.01; * p < 0.05; ns, not significant. One-way ANOVA with Dunnett’s post-test, as compared to respective vehicle-treated control).
Computational Modeling of the Interaction Profile between AM3677 and Active-State hCB1R (CB1R*)
We modeled in silico the AM3677-hCB1R* complex by docking AM3677 after its covalent attachment to C6.47(355) into our previously detailed hCB1R* homology model.48 AM3677 was found to bind within the TMH3-4-5-6 region, which is rich in aromatic residues (Figure 6).49,50 On the basis of CB1R mutation studies that identified K3.28(192) as an essential point of interaction for AEA at CB1R,51 K3.28(192) was considered to be the primary interaction site for AM3677 at CB1R*. In the resulting hydrogen bond, the AM3677 amide oxygen/CB1R K3.28(192) distance (C = O···H–N) was 2.8 Å, and the hydrogen-bond angle (O···H-N) was 154°. The position of the AM3677 amide oxygen beneath K3.28(192) allows AM3677 to assume a compact, J-shaped conformation in the hCB1R* binding pocket that facilitates an aromatic interaction between F3.25(189) and the C5–C6 double bond within the AM3677 acyl chain. This interaction is consistent with studies demonstrating that F3.25(189) mutation affects AEA binding49,50 and that F3.25(189) is part of the CB1R binding pocket.52 The modeled complex is also consistent with a previously published AEA-hCB1R* complex49 in which interactions between AEA and hCB1R* also occur at residues F3.25(189) and K3.28(192). However, the AEA fatty-acyl chain adopted a curved, U-shaped conformation in that complex,49 whereas AM3677 adopts another low free-energy shape, a J-shaped conformation, in the complex reported here.53,54 This difference likely reflects prevention of AM3677 from adopting the more compact U-shape by its covalent attachment at C6.47. In the AM3677/hCB1R* complex, the saturated tail of AM3677, covalently bound to C6.47(355), lies between toggle-switch residues F3.36(200) and W6.48(356), establishing them in active-state conformations within hCB1R*.50
Figure 6.
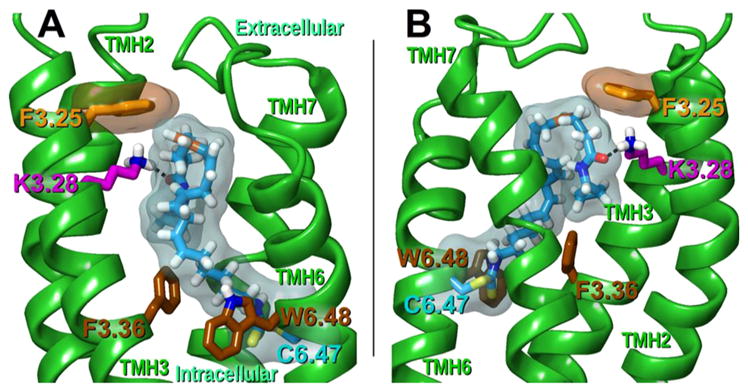
Illustration of modeled AM3677 binding pose in hCB1R*. AM3677 (cyan) is covalently linked to hCB1R* C6.47(355) (cyan). The amide oxygen of AM3677 is hydrogen-bonded to the amino group of K3.28(192) (magenta). With the AM3677 amide oxygen positioned beneath K3.28(192), the headgroup orientation allows AM3677 to assume a more compact, J-shaped conformation within the binding pocket, allowing F3.25(189) (orange) to establish an aromatic interaction with the C5–C6 double bond (orange) in the AM3677 acyl chain. Transparent molecular surfaces of AM3677/C6.47 (cyan) and F3.25 (orange) reveal these interactions. Notably, the saturated tail of AM3677 is disposed between the toggle-switch residues F3.36(200) and W6.48(356). (A) Depiction from a lipid viewpoint through TMHs 4 and 5. (B) Rotated 180° and depicted from a lipid viewpoint through TMH1. (For clarity, TMHs 1, 4, and 5 are not shown.)
In class-A GPCRs, C6.47 is part of the highly conserved CWXP hinge motif that participates in receptor conformational changes accompanying ligand-induced activation.55 Studies using the substituted cysteine accessibility method have shown that C6.47 is not ligand-accessible in the inactive β-2-adrenergic receptor but becomes accessible to the binding pocket in the receptor’s activated state.56 Consistent with these data for the β-2-adrenergic receptor, in the hCB1R* model employed here, C6.47(355) faces lipid, a disposition that enables its reaction with the isothiocyanate-derivatized cannabinoid, AM3677, likely accessing the hCB1R binding pocket via the membrane lipid bilayer.57 Since AM3677 is a CB1R agonist, our modeling results suggest that ligand motility within the binding pocket permits significant AM3677 interactions with K3.28(189) and F3.25(192). We have previously reported that AM841, an isothiocyanate-derivatized classical cannabinoid, also forms a covalent attachment with C6.47(355) and acts as a long-acting CB1R agonist of adenylyl cyclase-mediated signaling,31 but AM3677 appears to be less restrained within the binding pocket than is AM841 due to the pronounced flexibility of its arachidonic acid side chain.50,51 Although it is covalently linked to hCB1R at C6.47(355), because of its flexibility AM3677 may more easily move out of the binding pocket and, in so doing, return C6.47(355) to a lipid-facing position and the receptor to an inactive state, thereby attenuating signaling.
CONCLUSIONS
The present study constitutes the initial functional profiling of a covalent, high-affinity agonist of the eicosanoid structural class selective for CB1R, a high-abundance GPCR in the CNS and target focus of drug discovery efforts. Since AM3677 and the endocannabinoid AEA are chemically similar arachidonoylester derivatives exhibiting the molecular pharmacology of a CB1R agonist, AM3677 may be considered a novel chemical probe with which to explore the structural features of CB1R activation by endocannabinoid(-like) native ligands (AEA in particular) and define the requirements for hCB1R-mediated modulation of various signaling pathways and the distinct biological consequences that a covalent hCB1R agonist of this ligand structural class may elicit.
The interaction profile of AM3677 with CB1R involves covalent binding at C6.47, an amino acid that is a common feature of nonolfactory class-A GPCRs such as CB1R. This Cys residue is part of the highly conserved, TMH6 CWXP motif, an important microswitch modulating conformational rearrangement of the TMH6/TMH7 interface accompanying class-A GPCR inactive-active state transitions.55 The critical participation of C6.47(355) in modulating CB1R activation-state structure may underlie mechanistically why reactive isothiocyanate cannabinergic agonists of two distinct chemical classes, the Δ9-THC structural analogue and classical cannabinoid AM84131 and the endocannabinoid-like eicosanoid AM3677, both evidence as a feature of their interaction profile with hCB1R covalent binding to this TMH6 Cys residue. Given that GPCR-targeted covalent drugs enjoy clinical success across various indications,58,59 it remains to be determined whether the interaction of AM3677 with hCB1R C6.47(355) endows this ligand with pharmacological properties differentiable in vivo from typical, noncovalent hCB1R agonists that may be exploited to therapeutic advantage. In this regard, it is noteworthy that AM841 covalent binding to hCB2R C6.47(257) appeared to contribute to that ligand’s exceptional hCB2R agonist potency, which differentiates it from the relatively lower agonist potencies of both the direct AM841 analogue without the reactive NCS moiety at hCB2R28,60 and AM3677 at hCB1R, a distinction suggesting that agonist covalent reactivity at C6.47 in hCB1R and CB2R per se need not result in an exceptionally high level of receptor activation. Aside from its disposition and reactivity within the CB1R ligand-binding pocket and the formation of the resulting, covalently modified CB1R*, AM3677 may have potential effects on processes such as CB1R post-translational modification61 and homo- or heterodimerization62 that could contribute to the overall pharmacological profile of this agonist. It is also conceivable that the covalent nature of the interaction between AM3677 with CB1R might elicit cellular signaling patterns with unique temporal characteristics. The availability of AM3677 as a potent, covalent, CB1R-selective probe makes it feasible to address experimentally these and other questions for the first time.
METHODS
Materials
Standard laboratory chemicals and reagents were obtained pure or at the highest available grade from commercial sources.24–28,31 Gen Elute plasmid mini-prep kit, Dulbecco’s modified Eagle’s medium (DMEM), and cAMP kit were from Sigma (St. Louis, MO). AM3677 (Figure 1) was synthesized at the Center for Drug Discovery, Northeastern University (Boston, MA). CP55,940 and [3H]CP55,940 were obtained from the National Institute on Drug Abuse (Bethesda, MD). pRC/CMV-hCB1R was a generous gift from Dr. T.I. Bonner (National Institute of Mental Health, Bethesda, MD). Oligonucleotide primers were synthesized by Integrated DNA Technologies (Coralville, IA). The My Cycler PCR system was purchased from BioRad Laboratories (Hercules, CA). Pfu Turbo DNA polymerase was purchased as part of the QuikChange site-directed mutagenesis kit from Stratagene (La Jolla, CA). One Shot Top10 Escherichia coli cells, Flp-In-293 cells, pcDNA/FRT, hygromycin, lipofectamine 2000, and pure link hipure plasmid filter midi-prep kit were purchased from Invitrogen (Carlsbad, CA). MinElute gel-extraction kits and the QIA Prep spin mini-prep kits were from Qiagen (Valencia, CA). Restriction endonucleases were purchased from New England Biolabs (Beverly, MA). Fetal bovine serum (FBS) and penicillin-streptomycin solution were purchased from GIBCO (Rockville, MD).
Amino Acid Descriptor
The Ballesteros-Weinstein numbering scheme is used herein to designate loci of specific CB1R amino acids.63 Accordingly, the most highly conserved amino acid in a particular receptor TMH is assigned a locant of 0.5. This number is preceded by the helix number followed in parentheses by the sequence number. All other amino acids in a given TMH are assigned a locant relative to that most conserved residue, which in TMH6 of hCB1R is Pro358, i.e., P6.50(358).
Cloning of hCB1R Gene
The full-length hCB1R gene (1.4 kbp) was amplified from pRC/CMV CB1 construct as template, using Pfu DNA polymerase (Stratagene) under the following thermocycling conditions: 95 °C for 1 min, followed by 30 cycles of 95 °C for 30 s and 68 °C for 2 min, followed by an extension time of 5 min at 68 °C in an Eppendorf Mastercycler (Westbury, NY). NheI and BamHI restriction sites were incorporated into forward 5′-CGCTAGCATG-AAGTCGATCCTAGATGGCCT-3′ and reverse 5′-TATGGATCC-TCACAGAGCCTCGGCAGACGTG-3′ primers, respectively. The PCR product was purified using a MinElute PCR kit (Qiagen) and was digested using BamHI and NheI restriction enzymes. The same restriction enzymes were used for digestion of pcDNA5/FRT expression vector (Invitrogen). The vector and PCR fragment were purified using a MinElute kit and ligated at room temperature for 1 h. The ligated products were then transformed into One Shot Top10 competent E. coli cells following the vendor’s protocol (Invitrogen). Plasmid preparations were cultured in Luria broth containing 0.1 mg/mL ampicillin. Recombinant plasmid DNA was isolated using a pure link kit (Invitrogen), and DNA insertion was confirmed by sequencing (University of Connecticut Biotechnology Center, Storrs, CT).
Site-Directed Mutagenesis, Transfections, and Cell Culture
Site-directed mutagenesis of pcDNA5/FRT/hCB1R was performed as outlined in the QuikChange kit (Stratagene). Complementary oligonucleotide primers for Cys-to-Ser mutants were designed following vendor’s recommendations for cysteine residues in hCB1R TMHs 1, 4, 6, and 7 individually. Primers were annealed and extended, employing 18 cycles and utilizing the BioRad My Cycler system and Pfu Turbo DNA polymerase. The PCR parameters were as described above. Primers used to make mutations C1.55(139)S, C4.47(238)S, C6.47(355)S, C7.38(382)S, and C7.42(386)S were, respectively, as follows: forward 5′-CTC CTG GTG CTG TCC GTC ATC CTC CAC-3′ and reverse 5′-GTG GAG GAT GAC GGA CAG CAC CAG GAG-3′; forward 5′-CGT GGT GGC GTT TTC CCT GAT GTG GAC CA-3′ and reverse 5′-TGG TCC ACA TCA GGG AAA ACG CCA CCA CG-3′; forward 5′-GTG GTG TTG ATC ATC TCC TGG GGC CCT CTG-3′ and reverse 5′-CAG AGG GCC CCA GGA GAT GAT CAA CAC CAC-3′; forward 5′-CGG TGT TTG CAT TCT CCA GTA TGC TCT GCC-3′ and reverse 5′-GGC AGA GCA TAC TGG AGA ATG CAA ACA CCG-3′; forward 5′-CTG CAG TAT GCT CTC CCT GCT GAA CTC CA-3′ and reverse 5′-TGG AGT TCA GCA GGG AGA GCA TAC TGC AG-3′. The PCR products were treated with DpnI restriction enzyme to digest methylated parental nonmutated DNA. One microliter of the DpnI-treated PCR product was transformed into One Shot Top10-competent E. coli cells following the vendor’s protocol (Invitrogen), and plasmid preparations were cultured in Luria broth containing 0.1 mg/mL ampicillin. To ensure that only the desired mutations had been produced, plasmid DNA was first subjected to restriction enzyme digestion and the correct clones were then sequenced (University of Connecticut Biotechnology Center). The sequence-verified mutagenic plasmid DNA was used to transfect Flp-In-293 cells.
Flp-In-293 cells were maintained in DMEM containing 1× penicillin-streptomycin, 100 μg/mL zeocin, and 10% FBS at 37 °C under 5% CO2-95% air. Stably transfected cell lines were generated utilizing lipofectamine 2000 with an appropriate amount of plasmid DNA harboring the transgene cassette and POG44 following the vendor’s procedures (Invitrogen). Typically, three to five independent transfections were performed in parallel and duplicated over a 3 day period to maximize cell line integrity. Transformants were selected using hygromycin (100 μg/mL) over a 10 day period and passed to adherent cell culture flasks. hCB1R-transfected Flp-In-293 cell lines were maintained in DMEM supplemented with 1× penicillin-streptomycin, 10% FBS, and 100 μg/mL hygromycin, and each cell line was propagated to obtain sufficient protein for radioligand binding and cAMP assays. The maximum passage number was 20. Cells were harvested by centrifugation in PBS containing 1 mM EDTA and repeatedly washed with the same solution. The harvested cells were cryopreserved in liquid nitrogen.
Flp-In-293 Cell Membrane Preparation
Confluent Flp-In-293 cell monolayers were harvested over ice into PBS containing 1 mM EDTA. Cells were disrupted by cavitation, and the membrane fraction was obtained by ultracentrifugation, essentially as described.41 Membranes were resuspended in 25 mM Tris HCl/5 mM MgCl2/1 mM ethylenediaminetetraacetic acid (EDTA), pH 7.4 (TME), and protein concentration was determined using the DC protein assay system (Bio-Rad, Hercules, CA).
Radioligand Binding Assays
Saturation and competitive binding assays were performed in a 96-well format, as detailed previously.31,60 Briefly, membrane resuspended in TME containing 0.1% BSA (w/v) (TME-BSA) and equivalent to 25 μg of membrane protein was added to each assay well. [3H]CP55,940 was diluted in TME-BSA to yield final assay concentrations from an order of magnitude below to an order of magnitude above the ligand’s Kd. Nonspecific binding was assessed in the presence of 5 μM unlabeled CP55,940 for the saturation binding experiments. For competition binding experiments, the final concentration of [3H]CP55,940 was 0.75 nM, with increasing concentrations of competitive ligand. All binding assays were performed at 30 °C for 1 h with gentle agitation. After incubation, the samples were transferred to Unifilter GF/B-96-well filter plates, and unbound ligand was removed using a Packard Filtermate-196 cell harvester (PerkinElmer Packard, Shelton, CT). Filter plates were washed four times with ice-cold wash buffer (50 mM Tris-HCl and 5 mM MgCl2 containing 0.5% BSA, pH 7.4), and bound radioactivity was quantified by liquid scintillation counting. Nonspecific binding was subtracted from total bound radioactivity to calculate specific radioligand binding (as pmol/mg membrane protein). Bmax and Kd values were determined from the saturation binding assays using the one-site binding analysis equation: Y = Bmax × X/(Kd + X). IC50 and Ki values were determined from the competition binding assays by nonlinear regression. GraphPad Prism 3.03 (GraphPad Software, San Diego, CA) on a Windows platform was used for the calculations. Each data point represents the mean ± SEM from three independent experiments performed in triplicate. For irreversible ligand AM3677, the Ki values designate apparent receptor affinity.64
Covalent Labeling Assays
Flp-In-293-hCB1R membranes were resuspended at a concentration of 0.8 mg/mL in TME-BSA and incubated with a concentration of AM3677 some 10-fold its apparent Ki (i.e., 20 nM) at 30 °C for 1 h under gentle agitation. Control membranes were incubated under the same conditions without AM3677. The incubation was terminated by centrifugation at 27 000g for 5 min. Noncovalently bound probes were removed by washing and centrifugation, and pellets were resuspended in TME buffer containing 1% BSA and incubated at 30 °C for 20 min, followed by centrifugation at 27 000g for 5 min. The pellets were washed twice with TME buffer containing 1% BSA. Fifteen minutes was allowed between resuspension of pellets and washing to permit equilibration between buffer and membranes. Three final washes were conducted with TME buffer alone. The occupancy of the receptor by the electrophilic affinity probe was evaluated by a saturation binding assay with [3H]CP55,940 radioligand, as described above.
Cellular cAMP Assay
Flp-In-293 cells expressing hCB1R and grown to 70% confluence were collected by centrifugation (500g, 5 min) and resuspended in DMEM containing phosphodiesterase inhibitors (0.1 mM RO-20-1724 [Calbiochem, La Jolla, CA] and 1 mM IBMX [Sigma]), 20 mM HEPES (pH 7.3), (MediaTech, Inc., Herndon, VA), and 0.1% BSA. Cells (106 cells per assay) were then incubated at 30 °C for 30 min with 5 μM forskolin (Fisher Scientific, Pittsburgh, PA) in the same buffer, followed by addition of test hCB1R ligand at concentrations ranging from 0.01 to 10 000 nM and a further 5 min incubation. Basal cAMP levels were determined from cells incubated in the absence of forskolin and ligand. cAMP levels were also determined in cells incubated with forskolin alone. All incubations were terminated by boiling for 5 min and immediate cell lysis by freeze-thaw. Lysates were centrifuged at 12 000g for 5 min, and the cAMP in the supernatant was determined with an immunoassay kit (Sigma). Each cAMP determination was made in three independent experiments in duplicate and normalized to cell protein. IC50 values for inhibition of net forskolin-stimulated AMP production (above basal) were determined by nonlinear regression (GraphPad Prism).
[35S]GTPγS Binding to CB1R in Mouse Hippocampus Membranes
To assess G protein coupling in mouse brain, hippocampi from Barr2-WT mice (4-7 months old) were collected, minced, and disrupted in a glass homogenizer in homogenization buffer (10 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM DTT). The homogenate was passed through a 26-gauge needle eight times, centrifuged twice at 20 000g for 30 min at 4 °C, and resuspended in assay buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 20 μM GDP, 1 mM DTT). For each reaction, 2.5 μg of membrane protein was incubated in assay buffer containing ~0.1 nM [35S]GTPγS and increasing concentrations of test compound in a total volume of 200 μL for 2 h at room temperature. Test compounds (CP55,940, AEA, and AM3677) were first diluted through serial dilutions in DMSO and then with assay buffer to a final DMSO concentration of less than 1%. Reactions were terminated by separating membrane-bound and free [35S]GTPγS through filtration with GF/B filters using a 96-well plate harvester (Brandel Inc., Gaithersburg, MD). Filters were dried overnight, and radioactivity was determined with a microplate scintillation counter. For competition curves, both agonists (each at the concentration indicated in the legend to Figure 4) and the antagonist SR141716A (at multiple concentrations) were incubated with the membranes simultaneously. Data were fit by nonlinear regression using GraphPad Prism, version 6.0e, and the four-parameter logistic dose-response curve: response = bottom + ((top - bottom)/(1 + 10(logEC50-logX)))
AM3677-Induced CB1R Internalization
A HEK293 cell line stably expressing hemagglutinin (HA)-tagged rCB1R was used to detect functional activation by AM3677.65 Cultures were grown to 90–100% confluence on poly-D-lysine coated 96-well plates. In preparation for the assay, growth medium (DMEM containing 10% FBS and 1× penicillin-streptomycin) was replaced with 40 μL/well 10 mM HEPES, 130 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 0.2% BSA, and 0.1% DMSO (HBD), and the plate was incubated for 15 min at 37 °C. A 2× stock of the indicated concentration of either CP55,940 or AM3677 (40 μL) in HBD was added to appropriate wells, and cells were incubated for 30 min at 37 °C. Following this incubation, the cells were fixed and stained to detect cell-surface CB1R as described below.
CB1R Recycling
On the day of the experiment, the growth medium of HEK293 cells stably expressing HA-tagged rCB1R (above) was replaced with growth medium containing 10 μM emetine to inhibit new receptor synthesis. The cells were then incubated for 2 h under 37 °C in 5% CO2-95% air. The medium was then replaced with 40 μL/well HBD containing 10 μM emetine (HBDE), and the cells were incubated under the same conditions for an additional 15 min. A 2× concentration of either CP55,940 or AM3677 (40 μL) in HBDE was added to the wells, and the cells were incubated for a further 30 min. To determine receptor recycling, 40 μL of a 3× concentrated solution of SR141716A in HBDE (100 nM, final SR141716A concentration) and containing a 1× concentration of either CP55,940 or AM3677 was added per well, and the cells were incubated at 37 °C for specified times. At the end of the experiment, each plate was placed on ice, media were removed, and each well was filled with 100 μL of ice-cold 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. The plates were then incubated at room temperature for 20 min and subsequently washed five times for 5 min each with 1× Tris-buffered saline (TBS). Cells were blocked for 90 min at room temperature with 100 μL/well Odyssey blocking buffer (Li-Cor Bioscience, Lincoln, NB). Blocking buffer was next replaced with 40 μL/well blocking buffer containing a monoclonal antibody against the HA epitope (Covance Research, Emeryville, CA) at a 1:200 dilution. Following an overnight incubation at 4 °C, the cells were washed five times for 5 min each at room temperature with TBS containing 0.05% Tween-20 (TBS-T). Cells were then incubated for 1 h at room temperature with a commercial donkey anti-mouse CB1R antibody (Rockland Immunochemicals, Gilbertsville, PA) diluted 1:800 in blocking buffer. Cells were then washed five times for 5 min each in TBS-T at room temperature, and the plate was scanned on an Odyssey near-IR scanner. The extent of internalization (i.e., residual cell-surface CB1R) was calculated as the average integrated intensities of the drug-treated wells divided by the average integrated intensities of the untreated wells and is expressed as a percentage.
AM3677 Computational Docking into CB1R*
In the absence of a reported CB1R* crystal structure, we docked AM3677 into our recently detailed CB1R* homology model developed from the dark-state rhodopsin atomic structure and elaborated with the aid of published CB1R* binding-site conformational information and ligand-anchoring interactions.48,49,53 First, the covalent attachment resulting from the AM3677 isothiocyanate group reaction with CB1R C6.47(355) was modeled. On the basis of the fact that AM3677 is an AEA analogue and K3.28(192) as an essential point of interaction between AEA and CB1R,51 interactive computer graphics were then used to model a hydrogen-bonding interaction between the CB1R K3.28(192) amino group and the AM3677 amide oxygen. The AM3677/CB1R* complex was then subjected to a postdocking quantum mechanics/molecular mechanics minimization using a 12.0 Å nonbonded residue cutoff (updated every 10 steps) (Qsite, v6.6; Schrödinger, LLC, New York, NY). The molecular mechanics portion of the minimization consisted of an OPLS2005 based Polak-Ribier conjugate gradient minimization of 500 steps using a distance-dependent dielectric function with a base constant of 2. To preserve the global fold of the CB1R* model, a 1000 kcal/mol restraint was applied to all the backbone atoms, termini, and loops, while transmembrane residue side chains were free to move. In the quantum mechanics portion of the calculation, the interaction between the covalently attached AM3677 and the CB1R* model was optimized at the ab initio Hartree-Fock 6-31G* level. Up to 100 QM optimization steps were allowed on 8 processor cores.
Statistics
For pairwise comparisons, Student’s t test was used. For comparisons among more than two groups, one-way ANOVA with Bonferroni’s or Dunnett’s posthoc tests was used. Statistical significance was set at the p < 0.05 level. Unless otherwise indicated, all values are the mean ± SEM or mean with 95% confidence interval over the number of experiments and replicates specified.
Acknowledgments
Funding
This work was supported by National Institutes of Health grants DA3801, DA9152, and DA9158 (to A.M.), DA3801 (to L.M.B.), and DA021358 (to P.H.R.)
A portion of this work was submitted by S. Yaddanapudi in partial fulfillment of Ph.D. thesis requirements from the University of Connecticut, Storrs, CT, USA. We thank Alexandra Peterson for expert bibliographic assistance.
ABBREVIATIONS
- CB1R
cannabinoid 1 receptor
- CB2R
cannabinoid 2 receptor
- GPCR
G-protein coupled receptor
- 2-AG
2-arachidonoylgly-cerol
- AEA
anandamide
- CNS
central nervous system
- Δ9-THC
(-)-Δ9-tetrahydrocannabinol
- NMR
nuclear magnetic resonance spectroscopy
- TMH
transmembrane helix
- LAPS
ligand-assisted protein structure
- NCS
isothiocyanate
- AM841
(-)-7′-isothiocyanato-11-hydroxy-1′,1′-dimethylheptylhexahy-drocannabinol
- hCB1R
human cannabinoid 1 receptor
- AM3677
20-isothiocyanato-eciosa-5,8,11,14-tetraenoic acid cyclopropylamide
- ACPA
arachidonoylcyclopropylamide, (5Z,8Z,11Z,14Z)-eicosa-5,8,11,14-tetraenoic acid cyclopropylamide
- WT
wild type
- ECL
extracellular loop
- rCB1R
rat cannabinoid 1 receptor
- cAMP
cyclic AMP
- hCB1R*
human cannabinoid receptor 1 active state
- IC
intracellular
- EC
extracellular
- [35S]GTPγS
guanosine 5′-O-(3-[35S]thio)-triphosphate
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- EDTA
ethylenediaminetetraacetic acid
Footnotes
Author Contributions
Designed research: S.Y., N.Z., K.M., L.M.B., P.H.R., and A.M. Synthesized AM3677: K.V.S. and V.G.S. Conducted experiments: S.Y., N.Z., E.S., L.Z., D.H., and J.W.-M. Performed data analysis: D.R.J., S.Y., N.Z., K.M., L.M.B., P.H.R., and A.M. Wrote the manuscript: D.R.J., L.M.B., P.H.R., K.M., and A.M.
Notes
The authors declare no competing financial interest.
References
- 1.Di Marzo V. The endocannabinoid system: its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploration. Pharmacol Res. 2009;60:77–84. doi: 10.1016/j.phrs.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 2.Console-Bram L, Marcu J, Abood ME. Cannabinoid receptors: nomenclature and pharmacological principles. Prog Neuropsychopharmacol Biol Psychiatry. 2012;38:4–15. doi: 10.1016/j.pnpbp.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mechoulam R, Parker LA. The endocannabinoid system and the brain. Annu Rev Pscyhol. 2013;64:21–47. doi: 10.1146/annurev-psych-113011-143739. [DOI] [PubMed] [Google Scholar]
- 4.Ohno-Shosaku T, Kano M. Endocannabinoid-mediated retrograde modulation of synaptic transmission. Curr Opin Neurobiol. 2014;29:1–8. doi: 10.1016/j.conb.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 5.Ashton CH, Moore PB. Endocannabinoid system dysfunction in mood and related disorders. Acta Psychiatr Scand. 2011;124:250–261. doi: 10.1111/j.1600-0447.2011.01687.x. [DOI] [PubMed] [Google Scholar]
- 6.Janero DR, Makriyannis A. Terpenes and lipids of the endocannabinoid and transient-receptor-potential-channel bio-signaling systems. ACS Chem Neurosci. 2014;5:1097–1106. doi: 10.1021/cn5000875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glangetas C, Girard D, Groc L, Marsciano G, Chaouloff F, Georges F. Stress switches cannabinoid type-1 (CB1) receptor-dependent plasticity from LTD to LTP in the bed nucleus of the stria terminalis. J Neurosci. 2013;33:19657–19663. doi: 10.1523/JNEUROSCI.3175-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Díaz-Alonso J, Guzman M, Galve-Roperh I. Endocannabinoids via CB1 receptors act as neurogenic niche cues during cortical development. Philos Trans R Soc, B. 2012;367:3229–3241. doi: 10.1098/rstb.2011.0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luchicci A, Pistis M. Anandamide and 2-archidonoylglycerol: pharmacological properties, functional features, and emerging specificities of the two major endocannabinoids. Mol Neurobiol. 2012;46:374–392. doi: 10.1007/s12035-012-8299-0. [DOI] [PubMed] [Google Scholar]
- 10.Janero DR, Lindsley L, Vemuri VK, Makriyannis A. Cannabinoid 1 G protein-coupled receptor (periphero-)neutral antagonists: emerging therapeutics for treating obesity-driven metabolic disease and reducing cardiovascular risk. Expert Opin Drug Discovery. 2011;6:995–1025. doi: 10.1517/17460441.2011.608063. [DOI] [PubMed] [Google Scholar]
- 11.Talwar R, Potluri VK. Cannabinoid 1 (CB1) receptor—pharmacology, role in pain and recent developments in emerging CB1 agonists. CNS Neurol Disord: Drug Targets. 2011;10:536–544. doi: 10.2174/187152711796235005. [DOI] [PubMed] [Google Scholar]
- 12.Pertwee RG. Targeting the endocannabinoid system with cannabinoid receptor agonists: pharmacological strategies and therapeutic possibilities. Philos Trans R Soc, B. 2012;367:3353–3363. doi: 10.1098/rstb.2011.0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cumella J, Hernández-Folgado L, Girón R, Sanchez E, Morales P, Hurst DP, Gómez-Cañas M, Gómez-Ruiz M, Pinto DC, Goya P, Reggio PH, Martin MI, Fernández-Ruiz J, Silva AM, Jagerovic N. Chromenopyrazoles: non-psycho-active and selective CB1 cannabinoid agonists with peripheral antinociceptive properties. ChemMedChem. 2012;7:452–463. doi: 10.1002/cmdc.201100568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fichna J, Bawa M, Thakur GA, Tichkule R, Makriyannis A, McCafferty DM, Sharkey KA, Storr M. Cannabinoids alleviate experimentally induced intestinal inflammation by acting at central and peripheral receptors. PLoS One. 2014;9:e109115. doi: 10.1371/journal.pone.0109115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kenessey I, Bánki B, Márk A, Varga N, Tóvári J, Landányi A, Rásó E, Tímár J. Revisiting CB1 receptor as drug target in human melanoma. Pathol Oncol Res. 2012;18:857–866. doi: 10.1007/s12253-012-9515-y. [DOI] [PubMed] [Google Scholar]
- 16.Shim J, Padgett L. Functional residues essential for the activation of the CB1 cannabinoid receptor. Methods Enzymol. 2013;520:340–345. doi: 10.1016/B978-0-12-391861-1.00016-2. [DOI] [PubMed] [Google Scholar]
- 17.Turu G, Hunyady L. Signal transduction of the CB1 cannabinoid receptor. J Mol Endocrinol. 2010;44:75–85. doi: 10.1677/JME-08-0190. [DOI] [PubMed] [Google Scholar]
- 18.Rozenfeld R. Type I cannabinoid receptor trafficking: all roads lead to lysosome. Traffic. 2011;12:12–8. doi: 10.1111/j.1600-0854.2010.01130.x. [DOI] [PubMed] [Google Scholar]
- 19.Bosier B, Muccioli GG, Herman E, Lambert DM. Functionally selective cannabinoid receptor signaling: therapeutic implications and opportunities. Biochem Pharmacol. 2010;80:1–12. doi: 10.1016/j.bcp.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 20.Reynolds CH. Protein-ligand cocrystal structures: we can do better. ACS Med Chem Lett. 2014;5:727–729. doi: 10.1021/ml500220a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nikas SP, Grzybowska J, Papahatjis DP, Charalambous A, Banijamali AR, Chari R, Fan P, Kourouli T, Lin S, Notowski AJ, Marciniak G, Guo Y, Li, Xiuyan L, Wang CJ, Makriyannis A. The role of halogen substitution in classical cannabinoids: a CB1 pharmacophore model. AAPS J. 2004;6:1–13. doi: 10.1208/aapsj060430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hurst DP, Schmeisser M, Reggio PH. Endogenous lipid activated G protein-coupled receptors: emerging structural features from crystallography and molecular dynamics simulations. Chem Phys Lipids. 2013;169:46–56. doi: 10.1016/j.chemphyslip.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ai R, Chang CA. Ligand-specific homology modeling of human cannabinoid (CB1) receptor. J Mol Graphics Modell. 2012;38:155–164. doi: 10.1016/j.jmgm.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 24.Tiburu EK, Bowman AL, Struppe JO, Janero DR, Avraham HK, Makriyannis A. Solid-state NMR and molecular dynamics characterization of cannabinoid receptor-1 (CB1) helix 7 conformational plasticity in model membranes. Biochim Biophys Acta. 2009;1788:1159–1167. doi: 10.1016/j.bbamem.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tyukhtenko S, Tiburu EK, Deshmukh L, Vinogradova O, Janero DR, Makriyannis A. NMR solution structure of human cannabinoid receptor-1 helix 7/8 peptide: candidate electrostatic interactions and microdomain formation. Biochem Biophys Res Commun. 2009;390:441–446. doi: 10.1016/j.bbrc.2009.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiburu EK, Gulla SV, Tiburu M, Janero DR, Budil DE, Makriyannis A. Dynamic conformational responses of a human cannabinoid receptor-1 helix domain to its membrane environment. Biochemistry. 2009;48:4895–4904. doi: 10.1021/bi802235w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zvonok N, Pandarinathan L, Williams J, Johnston M, Karageorgos I, Janero DR, Krishnan SC, Makriyannis A. Covalent inhibitors of human monoacylglycerol lipase: ligand-assisted characterization of the catalytic site by mass spectrometry and mutational analysis. Chem Biol. 2008;15:854–862. doi: 10.1016/j.chembiol.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szymanski DW, Papanastasiou M, Melchior K, Zvonok N, Mercier RW, Janero DR, Thakur GA, Cha S, Wu B, Karger B, Makriyannis A. Mass spectrometry-based proteomics of human cannabinoid receptor 2: covalent cysteine 6.47(257)-ligand interaction affording megagonist receptor activation. J Proteome Res. 2011;10:4789–4798. doi: 10.1021/pr2005583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pace NJ, Weerapana E. Diverse functional roles of reactive cysteines. ACS Chem Biol. 2013;8:283–296. doi: 10.1021/cb3005269. [DOI] [PubMed] [Google Scholar]
- 30.Tahtaoui C, Balestre MN, Klotz P, Rognan D, Barberis C, Mouillac B, Hilbert M. Identification of the binding sites of the SR49059 nonpeptide antagonist into the V1a vasopressin receptor using sulfydryl-reactive ligands and cysteine mutants as chemical sensors. J Biol Chem. 2003;278:40010–40019. doi: 10.1074/jbc.M301128200. [DOI] [PubMed] [Google Scholar]
- 31.Picone RP, Khanolkar AD, Xu W, Ayotte AL, Thakur GA, Hurst DP, Abood ME, Reggio PH, Fournier DJ, Makriyannis A. (-)-7′-Isothiocyanato-11-hydroxy-1′, 1′-dimethylheptylhexahydrocannabinol (AM841), a high-affinity electro-philic ligand, interacts covalently with a cysteine in helix six and activates the CB1 cannabinoid receptor. Mol Pharmacol. 2005;68:1623–1635. doi: 10.1124/mol.105.014407. [DOI] [PubMed] [Google Scholar]
- 32.Hillard CJ, Manna S, Greenberg MJ, Dicamelli R, Ross RA, Stevenson LA, Murphy V, Pertwee RG, Campbell WB. Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1) J Pharmacol Exp Ther. 1999;289:1427–1433. [PubMed] [Google Scholar]
- 33.Li C, Xu W, Vadivel SK, Pusheng F, Makriyannis A. High affinity electrophilic and photoactivatable covalent endocannabinoid probes for the CB1 receptor. J Med Chem. 2005;48:6423–6429. doi: 10.1021/jm050272i. [DOI] [PubMed] [Google Scholar]
- 34.McHugh D, Page J, Dunn E, Bradshaw HB. Δ9-Tetrahydrocannabinol and N-arachidonyl glycine are full agonists at GPR18 receptors and induce migration in human endometrial HEC-1B cells. Br J Pharmacol. 2012;165:2414–2424. doi: 10.1111/j.1476-5381.2011.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fay JF, Dunham TD, Farrens DL. Cysteine residues in the human cannabinoid receptor: only C257 and C264 are required for a functional receptor, and steric bulk at C386 impairs antagonist SR141716A binding. Biochemistry. 2005;44:8757–8769. doi: 10.1021/bi0472651. [DOI] [PubMed] [Google Scholar]
- 36.Fay JF, Farrens DL. The membrane proximal region of the cannabinoid receptor CB1 N-terminus can allosterically modulate ligand affinity. Biochemistry. 2013;52:8286–8294. doi: 10.1021/bi400842k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khanolkar AD, Palmer SL, Makriyannis A. Molecular probes for the cannabinoid receptors. Chem Phys Lipids. 2000;108:37–52. doi: 10.1016/s0009-3084(00)00186-9. [DOI] [PubMed] [Google Scholar]
- 38.Thakur GA, Bajaj S, Paronis C, Peng Y, Bowman AL, Barak LS, Caron MG, Parrish D, Deschamps JR, Makriyannis A. Novel adamantyl cannabinoids as CB1 receptor probes. J Med Chem. 2013;56:3904–3921. doi: 10.1021/jm4000775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McPartland JM, Glass M, Pertwee RG. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: interspecies differences. Br J Pharmacol. 2007;152:583–593. doi: 10.1038/sj.bjp.0707399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kapur A, Hurst DP, Fleischer D, Whitnell R, Thakur GA, Makriyannis A, Reggio PH, Abood ME. Mutation studies of Ser3.79 and Ser2.60 in the human CB1 cannabinoid receptor: evidence for a serine-induced bend in CB1 transmembrane helix 7. Mol Pharmacol. 2007;71:1512–1524. doi: 10.1124/mol.107.034645. [DOI] [PubMed] [Google Scholar]
- 41.Xu W, Filppula SA, Mercier R, Yadanapudi S, Pavlopoulos S, Cai J, Pierce WM, Makriyannis A. Purification and mass spectroscopic analysis of human CB1 cannabinoid receptor functionally expressed using the baculovirus system. J Pept Res. 2005;66:138–150. doi: 10.1111/j.1399-3011.2005.00283.x. [DOI] [PubMed] [Google Scholar]
- 42.Mercier RW, Pei Y, Pandarinathan L, Janero DR, Zhang J, Makriyannis A. hCB2 ligand-interaction landscape: cysteine residues critical to biarylpyrazole antagonist binding motif and receptor modulation. Chem Biol. 2010;17:1132–1142. doi: 10.1016/j.chembiol.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strange PG. Use of the GTPγS ([35S]GTPγS and Eu-GTPγS) binding assay for analysis of ligand potency and efficacy at G protein-coupled receptors. Br J Pharmacol. 2010;161:1238–1249. doi: 10.1111/j.1476-5381.2010.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Breivogel CS, Childers SR. Cannabinoid agonist signal transduction in rat brain: comparison of cannabinoid agonists in receptor binding, G-protein activation, and adenylyl cyclase inhibition. J Pharmacol Exp Ther. 2000;295:328–336. [PubMed] [Google Scholar]
- 45.Childers SR. Activation of G-proteins in brain by endogenous and exogenous cannabinoids. AAPS J. 2006;8:E112–E117. doi: 10.1208/aapsj080113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Magalhaes AC, Dunn H, Fergurson SS. Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. Br J Pharmacol. 2012;165:1717–1736. doi: 10.1111/j.1476-5381.2011.01552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flores-Otero J, Ahn KH, Delgado-Peraza F, Mackie K, Kendall DA, Yudowski GA. Ligand-specific endocytic dwell times control functional electivity of the cannabinoid receptor 1. Nat Commun. 2014;5:4589. doi: 10.1038/ncomms5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nikas SP, Sharma R, Paronis CA, Kulkarni S, Thakur GA, Hurst D, Wood JT, Gifford RS, Rajarshi G, Liu Y, Raghav JG, Guo JJ, Järbe TU, Reggio PH, Bergman J, Makriyannis A. Probing the carboxyester side chain in controlled deactivation (-)-delta(8)-tetrahydrocannabinols. J Med Chem. 2015;58:665–681. doi: 10.1021/jm501165d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McAllister SD, Rizvi C, Anavi-offer S, Hurst DP, Barnett-Norris J, Lynch DL, Reggio PH, Abood ME. An aromatic microdomain at the cannabinoid CB1 receptor constitutes an agonist/inverse agonist binding region. J Med Chem. 2003;46:5139–5152. doi: 10.1021/jm0302647. [DOI] [PubMed] [Google Scholar]
- 50.McAllister SD, Hurst DP, Barnett-Norris J, Lynch D, Reggio PH, Abood ME. Structural mimicry in class A G protein-coupled receptor rotamer toggle switches: the importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation. J Biol Chem. 2004;279:48024–48037. doi: 10.1074/jbc.M406648200. [DOI] [PubMed] [Google Scholar]
- 51.Song ZH, Bonner TI. A lysine residue of the cannabinoid receptor is critical for receptor recognition by several agonists but not WIN55212-2. Mol Pharmacol. 1996;49:891–896. [PubMed] [Google Scholar]
- 52.Tuccinardi T, Ferrarini PL, Manera C, Ortore G, Saccomanni G, Martinelli A. Cannabinoid CB2/CB1 selectivity. Receptor modeling and automated docking analysis. J Med Chem. 2006;49:984–994. doi: 10.1021/jm050875u. [DOI] [PubMed] [Google Scholar]
- 53.Barnett-Norris J, Hurst DP, Lynch DL, Guarnieri F, Makriyannis A, Reggio PH. Conformational memories and the endocannabinoid binding site at the cannabinoid CB1 receptor. J Med Chem. 2002;45:3649–3659. doi: 10.1021/jm0200761. [DOI] [PubMed] [Google Scholar]
- 54.Barnett-Norris J, Guarnieri F, Hurst DP, Reggio PH. Exploration of biologically relevant conformations of anandamide, 2-arachidonylglycerol, and their analogues using conformational memories. J Med Chem. 1998;41:4861–4872. doi: 10.1021/jm9803471. [DOI] [PubMed] [Google Scholar]
- 55.Olivella M, Caltabiano G, Cordomi A. The role of cysteine 6.47 in class A GPCRs. BMC Struct Biol. 2013;13:3. doi: 10.1186/1472-6807-13-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. β2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- 57.Hurst DP, Grossfield A, Lynch DL, Feller S, Romo TD, Gawrisch K, Pitman MC, Reggio PH. A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J Biol Chem. 2010;285:17954–17964. doi: 10.1074/jbc.M109.041590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discovery. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 59.Kalgutkar AS, Dalvie DK. Drug discovery for a new generation of covalent drugs. Expert Opin Drug Discovery. 2012;7:561–581. doi: 10.1517/17460441.2012.688744. [DOI] [PubMed] [Google Scholar]
- 60.Pei Y, Mercier RW, Anday JK, Thakur GA, Zvonok AM, Hurst D, Reggio PH, Janero DR, Makriyannis A. Ligand-binding architecture of human CB2 cannabinoid receptor: evidence for receptor subtype-specific binding motif and modeling GPCR activation. Chem Biol. 2008;15:1207–1219. doi: 10.1016/j.chembiol.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oddi S, Dainese E, Feza F, Lanuti M, Chiurchiŭ V, Totaro A, Catanzaro G, Barcaroli D, De Laurenzi V, Centonze D, Mukkopadhyay S, Selent J, Howlett AC, Maccarrone M. Effects of palmitoylation of Cys(415) in helix 8 of the CB1 cannabinoid receptor on membrane localization and signaling. Br J Pharmacol. 2012;165:2635–2651. doi: 10.1111/j.1476-5381.2011.01658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hudson B, Hébert TE, Kelly MEM. Ligand- and heterodimer-directed signaling of the CB1 cannabinoid receptor. Mol Pharmacol. 2010;77:1–9. doi: 10.1124/mol.109.060251. [DOI] [PubMed] [Google Scholar]
- 63.Ballesteros JA, Weinstein H. Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 64.Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 65.Diagle TL, Kwok ML, Mackie K. Regulation of CB1 cannabinoid receptor internalization by a promiscuous phosphorylation-dependent mechanism. J Neurochem. 2008;106:70–82. doi: 10.1111/j.1471-4159.2008.05336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]