

Abstract
Investigations on the chemistry and mechanism of action of 2-methyl-1,4-naphthoquinone (or menadione) derivatives, revealed 3-phenoxymethyl menadiones as a novel antischistosomal series. These newly synthesized compounds 1–7 and their difluoromethylmenadione counterparts 8–9 were found to be potent and specific inhibitors of Schistosoma mansoni thioredoxin-glutathione reductase (SmTGR) identified as a potential target. The compounds were also tested in enzymic assays using both human flavoenzymes, i.e. the glutathione reductase (hGR) and the selenium-dependent human thioredoxin reductase (hTrxR) to evaluate the specificity of the inhibition. Structure-activity relationships as well as physico- and electro-chemical studies showed a high potential for the 3-phenoxymethyl menadiones to inhibit SmTGR selectively versus hGR and hTrxR enzymes, in particular those bearing α-fluorophenol methyl ether moieties to improve antischistosomal action. In particular, the (substituted phenoxy)methyl menadione derivative 7 displayed time-dependent SmTGR inactivation, correlating with unproductive NADPH-dependent redox-cycling of SmTGR, and potent antischistosomal action in ex vivo worms. In contrast, the difluoromethylmenadione analogue 9, which inactivates SmTGR through an irreversible non-consuming NADPH-dependent process, has little killing effect in cultured ex vivo worms. Because none of the compounds tested in vivo was active, a limited bioavailability might compromise compound activity and future studies will be directed toward improving pharmacokinetics properties.
Keywords: flavoenzyme inhibitor; glutathione disulfide reductase; 1,4-naphthoquinone; Schistosoma mansoni; thioredoxin disulfide reductase
Introduction
Schistosomiasis, also called bilharzia after its discovery by Theodor Bilharz [1], is a widespread tropical disease caused by the helminth parasites of the genus Schistosoma, including S. mansoni.[2] It is a devastating tropical disease ranking second after malaria in terms of social and economic impact and public health importance, affecting more than 200 million people worldwide.[3,4] No vaccine is available and the only available drug, praziquantel, is extensively used, currently administered to more than 10 million people annually.[5,6] No new drugs have been introduced since praziquantel, and prior drugs are not produced anymore or are not effective against all species of Schistosoma parasites.[7] As it is likely that praziquantel-resistant/tolerant parasites will develop,[8] there is an urgent need to find new antischistosomal drugs.
Adult S. mansoni worms live in the mesenteric veins of their human hosts, where they can survive for up to 30 years.[9] Residing in an aerobic environment, they must have effective mechanisms to maintain their cellular redox balance. In addition, worms must be able to reduce reactive oxygen species generated by the host’s immune response. In most eukaryotes there are two major systems based on NADPH-dependent flavoenzymes to regenerate thiols from disulfide substrates and to detoxify reactive oxygen species: one is based on the tripeptide glutathione (GSH) and glutathione reductase (GR, E.C. 1.8.1.7) and the other, on the protein thioredoxin (Trx) and thioredoxin reductase (TrxR, E.C. 1.8.1.9). In S. mansoni these two enzymes are absent and replaced by a unique bi-functional seleno-enzyme, thioredoxin-glutathione reductase (SmTGR, E.C. 1.8.1.9).[10] This enzyme plays a crucial role in S. mansoni thiol redox metabolism and has been identified as a key drug target.[11] The aim of this study was to identify new lead chemical series and to design novel inhibitors of this essential enzyme.
A preliminary inhibitor screen led to the identification of the (substituted phenoxy)methyl menadione derivative 1 (Fig. 1) with a carboxylic acid function, as a potent SmTGR inhibitor, with inhibitory activity in the nanomolar range. However, compound 1 was able to kill cultured adult worms at only relatively high concentrations (50 μM). Because of this limited activity, likely due to the known limited membrane permeability of carboxylic acids, a series of new menadione derivatives bearing lipophilic moieties or fluorinated groups on the oxyphenylmethylene arm were synthesized to improve the pharmacological profile. These compounds were tested both in enzyme and biological assays to identify potential new antischistosomal agents.
Fig. 1.

Structures of the 3-phenoxymethyl menadione series.
Results and Discussion
Chemistry
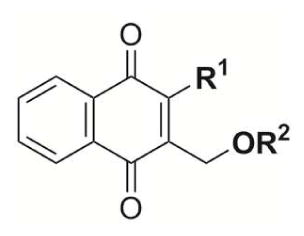
A new series of (substituted phenoxy)methyl menadione derivatives was synthesized based on the structure of 1 (Fig. 1). The 2,6-difluorophenol group is a known bioisostere of the carboxylic acid function.[12] The introduction of bioisosteric moieties of the carboxylic acid could improve the pharmacokinetic profiles of the inhibitors.[ 13 ] In addition, compounds were generated to allow the introduction of fluorine groups in order to enhance lipophilicity[ 14 ] and cell permeability of the compounds. A synthetic route for the preparation of the chemical series was investigated and allowed the preparation of new bioisosteric analogues and prodrugs based on 1 structure as potent SmTGR inhibitor scaffold with optimized inhibition and pharmacokinetic profiles, i.e. the preparation of bioisosteres/prodrugs of the -COOH moiety of 1 by replacing the benzoic acid group by a nitrile (2), or a difluoromethoxyphenol (3), which are known to enhance the cellular permeability of the parent carboxylic acid, or analogues to introduce chemical diversity, with halogens e.g. chloro (4), bromo (5), or CF3 (6, 7) groups (Fig. 1).
From the synthetic point of view, the (substituted phenoxy)methyl menadione derivative 2 bearing a cyano group instead of the benzoic acid function found in 1 was obtained with a 13% global yield from commercially available 4-cyanophenol (Fig. 2, route A). Compound 2 can be considered as a prodrug of 1. The side chain of 4-cyanophenol was elongated by the reaction with ethylchloroacetate under basic conditions to afford 2c as white crystals with 97% yield. The ethyl ester was then saponified to give the acid 2d with 65% yield. Compound 2d was reacted with menadione in the Kochi-Anderson radical decarboxylation [15] to obtain the final (p-cyanophenoxy)methyl menadione derivative 2 with 21% yield. Starting from the commercially available difluorophenol, (difluoro phenoxy)methyl menadione derivative 3 was obtained with an overall yield of 5% (Fig. 2, route A). The hydroquinone 3a was obtained through Elbs oxidation [16] with a yield of 44%, and then was subjected to selective methylation with dimethylsulfate under mild basic conditions to give the 4-methoxy-3,5-difluorophenol 3b with 50% yield. The side chain of the phenol 3b was elongated by reaction with ethylchloroacetate under basic conditions to afford the ester 3c with 71% yield. Saponification of 3c led to the carboxylic acid 3d with a yield of 77%. The acid 3d was introduced in the Kochi-Anderson radical decarboxylation to obtain the final difluorophenol methoxy ether derivative 3 with 41% yield. Then, to introduce more structural diversity in the (substituted phenoxy)methyl menadione series, other analogues were synthesized, such as molecules bearing different halogens. The addition of halogen increases the lipophilicity of the compounds, changes their redox potential value, and improves their metabolic stability in the host. Commercially available 2-(4-chlorophenoxy)acetic acid and 2-(4-bromophenoxy)acetic acid were allowed to react with menadione in the Kochi-Anderson radical decarboxylation to afford the corresponding 3-phenoxymenadione derivatives, 4 and 5, with 35% and 24% yield, respectively.
Fig. 2.

Synthesis of 3-phenoxymethylmenadione derivatives (Route A) and its 2-difluoromethyl analogs (Route B).
Finally, another series of compounds was investigated by introducing fluorine directly on the methyl group of the menadione core (Fig. 2, route B). Commercially available 1,4-naphthoquinone was reduced using SnCl2/HCl and the resulting dihydronaphthoquinone was methylated by dimethylsulfate under mild basic conditions. The dimethoxynaphthalene intermediate was then successively formylated (98% yield) and treated with 2.0 equiv. of diethylaminosulfur trifluoride (DAST) to obtain the 2-(difluoromethyl)-1,4-dimethoxynaphthalene with a yield of 92%, according described procedures.[ 17 ] Subsequent oxidation with cerium ammonium nitrate (CAN) led to the difluorinated menadione with 93% yield. The difluoromethylmenadione derivative and the p-cyanophenylacetic acid were subjected to the Kochi-Anderson radical decarboxylation to afford the final difluoromethylmenadione derivatives bearing an oxyphenylmethylene arm, 8 (p-cyano-, with 35% yield), or 9 (3-trifluoromethyl-4-methoxy-, with 57% yield).
Electrochemistry
The redox potentials of the different (substituted phenoxy)methyl menadione derivatives were determined by cyclic voltammetry in DMSO containing 0.1 M NBu4PF6 (tetrabutylammonium hexafluorophosphate) as the electrolyte system. The results obtained with the 1,4-naphthoquinones 2, 8, 5, 1, 7 and 9, are compiled in Table 1. For all the compounds a 1e− quasi-reversible redox wave affording the monoradical-anion can be observed (ΔEp ~ 96–170). The quasi-reversibility of this electron transfer process can be assessed by the large ΔEp separation (a theoretical ΔEp of 60 mV is expected for an ideal 1e− transfer) as well as by the Ipc1/Ipa1 ratio significantly higher than 1. A second quasi-reversible redox wave can be observed for 2 (Figure in Table 1) and 7, also it is not fully reversible with ΔEp < 70 mV. However, we estimated the half-wave potential E2½ as well as the ΔE½ separation (E1½-E2½), which were found to be comparable. For compounds 1 and 5, the second electron transfer affording the quinone dianion appears to be an irreversible process thus precluding half-wave potential determination. This electrochemical behavior might result from either comproportionation reactions or by fast and irreversible dimerization process between the quinone dianion (NQ2−) and the naphthoquinone (NQ) to afford an electro-inactive dimeric species NQ2 2−. When the methyl of the menadione core was replaced by CHF2 in 2 and 7 there was no second redox wave, and the 1e− reduction potential experienced a significant anodic shift of 200 mV with respect to CH3-substituted analogue (see 2 versus 8 in the figure of Table 1). This feature clearly reflects the most oxidant character for 8 (Figure in Table 1) and 9, as previously observed for fluoromethyl menadione analogues,[17,18,19] and subsequently the most reactive properties.
Table 1. Electrochemical data measured using cyclic voltammetry (CV) for 3-phenoxymethylmenadione derivatives examined in this work.
Cyclic voltammograms of 3-phenoxymethylmenadione representatives, the menadione 2 (black bold line) and its difluoromethyl analogue 8 (red dashed line) were recorded at platinum electrode in CHCl2 with 0.1 M Tetra-n-butylammonium tetrafluoroborate (Bu4NBF4) as supporting electrolyte at 200 mV s−1 scan rate.
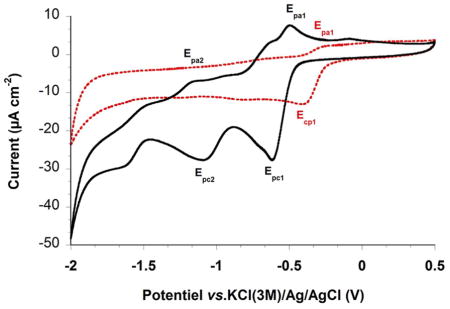
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Epc1 (V) | Epa1 (V) | Ipc1/Ipa1 | E1½ (V) | ΔEp (mV) | Epc2 (V) | Epa2 (V) | Ipc2/Ipa2 | E2½ (V) | ΔEp (mV) | ΔE½ (mV) | |
| 1 | −0.628 | −0.532 | 1.27 | −0.580 | 96 | −1.265 | irr | irr | irr | irr | NA |
| 2 | −0.620 | −0.492 | 2.29 | −0.556 | 128 | −1.154 | −1.100 | 2 | −1.127 | 54 | 571 |
| 5 | −0.627 | −0.508 | 1.89 | −0.508 | 119 | −1.158 | irr | irr | irr | irr | NA |
| 7 | −0.620 | −0.506 | 2.92 | −0.563 | 114 | −1.132 | −1.120 | 1.75 | −1.126 | 12 | 563 |
| 8 | −0.414 | −0.244 | 3.31 | −0.329 | 170 | ND | ND | ND | ND | ND | ND |
| 9 | −0.326 | −0.215 | 1.67 | −0.271 | 111 | ND | ND | ND | ND | ND | ND |
DMSO;I = 0.1 M n-Bu4NPF6, E½ = (Epc+Epa)/2 (V), ΔE = Epa-Epc (mV), ΔE½ = E1½-E2½ (V)). v = 200 mV s−1; reference electrode = KCl(3 M)/Ag/AgCl; working electrode = glassy carbon disk of 0.07 cm2 area; auxiliary electrode = Pt wire. ND: no second wave observed, irr: irreversible reduction, NA: not applicable.
Physicobiochemistry of glutathionylation
Absorption spectrophotometry was first used to estimate the pKa value of the difluoromenadione derivative 9. Significant spectral variation and color change of compound 9 were observed when raising the pH from acidic (below pH 7, colorless solution) to basic conditions (pale yellow solution pH ~ 11.5–12). These observations clearly indicate that thanks to the fluorine atoms, deprotonation of the difluoromenadione species might occur in a biocompatible pH window. The statistical processing of the data allowed evaluating a single pKa value, pKa ~ = 7.1 ± 0.5 (Fig. 3A).
Fig. 3.






Determination of the pKa value (Panel A) and changes of the absorption spectra (Panels B-E) for the difluoromethyl derivative 9 (BJ735) upon glutathione addition. (B) Absorption spectrophotometric variation and (C) Absorbance at 350 nm measured in the time course of the glutathionylation reaction with the difluoromethyl derivative 9. Solvent: 200μL MeCN + 265 μL NH4OH buffer at pH 7.5; [9] = 2 × 10−4 M (10 μL stock at 10 mM); [GSH] = 10−3 M (25 μL stock at 20 mM); T = 25°C. (1) t = 0 s; (2) t = 1800 s. (D) Electronic absorption spectra measured for the difluoromethyl derivative 9 and its glutathione adducts and (E) Distribution diagram of the difluoromethyl derivative 9 and its products as a function of time upon the glutathionylation reaction. Solvent: 200μL MeCN + 265 μL NH4OH buffer at pH 7.5; [9] = 2 × 10−4 M (10 μL stock at 10 mM); [GSH] = 10−3 M (25 μL stock at 20 mM); T = 25°C.
We then investigated using absorption spectrophotometry the glutathionylation reaction kinetics of three difluoromenadione species, the difluoromenadione itself, and both difluoromethylmenadione derivatives bearing an oxyphenylmethylene arm, 8 and 9 (see supplementary material in Fig. S1–S2 for difluoromenadione and compound 8, and Fig. 3B–E for compound 9). Taking into account the estimated acido-basic properties, it can be anticipated that a mixture of neutral protonated difluoromenadione and deprotonated derivatives dominates under our experimental conditions. Figure S1 displays the absorption changes of a 0.2 mM of difluoromethylmenadione solution in the course of glutathionylation following addition of 5 equiv of GSH (1mM final). The spectral window used (300–500 nm) allowed monitoring the weak n-π* transitions of the quinone moiety. Interestingly, the glutathionylation induces the formation of intense absorption band in the visible region (~ 440 nm) which allowed easily monitoring the reaction. A biphasic kinetic behavior can be observed (Fig. S1, panels A–B). Statistical tools were used to process the spectral and kinetic data and allowed calculating the pseudo-first order rate constants of the two steps process as well as the electronic spectra of the kinetic intermediate (P1), and the final product P2 (Scheme in Table 2). These spectral observations can be rationalized by the formation of the quinone methide (kinetic intermediate P1) prior to a Michael addition of glutathione and concomitant fluorine elimination (concerted mechanism, product P2). However, as previously reported, both the structure and reactivity of difluoroquinone methides can be compared to those of related β-fluorocarbanions known to generate further additions with nucleophiles and cross or self-polymerization (see Scheme in Table 2) [19]. The same kinetic approach was used for the compounds 8 and 9, which share a common spectral behavior upon addition of GSH at pH 7.5 (Fig. S2 in the Supplementary material and Fig. 3, respectively). The n-π* transitions apparently experience a significant hyperchromic shift upon addition of GSH and allowed monitoring the glutathionylation kinetics of these two phenoxymethyl-substituted difluoromenadiones. Similarly to difluoromenadione, a biphasic kinetic process was also observed. Interestingly, the three compounds examined in this work are characterized by a common fast kinetic step as deduced from their comparable k1 values (Table 2) and electronic spectra (Fig. 3E, Fig. S1C, Fig. S2C). This would suggest that the three compounds are experiencing the same reaction (i.e. deprotonation and addition-elimination) leading to the reactive quinone methide intermediate P1, and then the GSH-conjugates P2 and other products. As previously discussed,[17,19] the difluoromenadione core through the quinone methide is highly reactive towards nucleophiles; it acts as a difluoroacrolein prone to polymerization (Scheme in Table 2). The kinetics of addition of GSH is, however, greatly influenced by the substitution pattern of the difluoromenadione core as observed by the almost 10-fold decrease of the k2 values measured for both compounds 8 and 9 compared to the difluoromenadione itself. On the other hand, no significant distinct behaviour can be deduced between the two phenoxymethyl-substituted 8 and 9.
Table 2.
Proposed chemical reactivity of difluoromethylmenadiones and rates constants for difluoro-based quinone methide (P1) and glutathione adducts (P2) formation in glutathionylation kinetics.
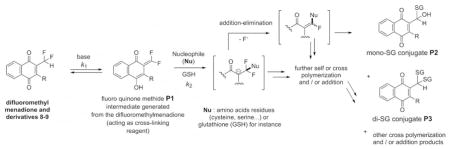
| ||
|---|---|---|
| 2-difluoromethyl-3-H or 3-phenoxymethyl menadiones | K1 (s−1) | K2 (s−1) |
| difluoromethylmenadione | 2.13 10−2 | 1.57 10−2 |
| Compound 8 | 3.22 10−2 | 3.52 10−3 |
| Compound 9 | 2.04 10−2 | 2.76 10−3 |
Glutathionylation reactions and LC–MS analyses of the reaction mixtures containing the difluoromethylmenadione–GSH conjugates
The spontaneous reaction of difluoromethylmenadione with free reduced GSH gave rise to a complex mixture due to the various reactions occurring at different sites of the electrophile molecule (see the UV-Vis chromatograms and LC-MS traces in Fig. S3, Supplementary material). Our first step for quantifying the potential glutathione adducts was to develop a chromatographic method to resolve the conjugates, which could be detected using positive ESI-MS techniques. As shown in Fig. S3, the LC–MS chromatogram from the difluoromenadione–GSH conjugates contains quite a few peaks due to the presence of several expected diastereo- and regioisomers upon reaction with GSH at the difluoroquinone methide or at C-3 of the quinone core. All difluoromethylmenadione–GSH conjugates showed the same pattern in the LC–MS traces (Fig. S3 in the Supplementary material), and the mono– and di–GSH conjugates were all detected (Table S1 in the Supplementary material).
Biological activities
SmTGR inhibitory activity
The library of representative (substituted phenoxy)methyl menadione derivatives was tested for inhibitory activity using the SmTGR DTNB assay (Table 3). All compounds inhibited SmTGR activity with IC50 values in the nanomolar range (1, 482 nM; 2, 462 nM; 3, 407 nM; 4, 457 nM; 5, 784 nM) indicating excellent recognition of the menadione-anchored oxyphenylmethylene arm by SmTGR. Interestingly, the most potent SmTGR inhibitors were the difluoromenadione derivatives 8 and 9, whose difluoromenadione core [17] has been shown to act as a suicide substrate of the related flavoenzymes, namely GRs from human and Plasmodium falciparum.[17] Similar results were found with mono- [18] or trifluoromenadione core [19], as a template to undergo fast, irreversible mechanism-based inhibition of both flavoenzymes. This result validates the difluoromenadione core as a suicide substrate of the parasitic SmTGR.
Table 3.
In vitro inhibition of SmTGR and both human related disulfide reductases hGR and hTrxR.
| Compound |
|
IC50 (nM) SmTGR assay[a] |
IC50 (nM) hGR assay[b] |
IC50 (nM) hTrxR assays | |||
|---|---|---|---|---|---|---|---|
| Placenta wild type hTrxR | Sec498Cys TrxR | ||||||
| R1 | R2 | Trx assay[c] | DTNB assay[d] | DTNB assay[d] | |||
| 1 | – Me |
|
482 | 2 300 | 2 000 | 3 000 | 40 000 |
| 2 | – Me |
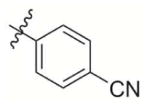
|
462 | 8 500 | 1 500 | 1 200 | 6 000 |
| 3 | – Me |
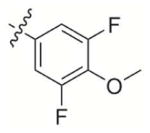
|
407 | > 25 000 | 2 000 | 2 000 | > 100 000 |
| 4 | – Me |
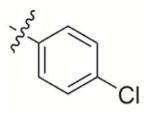
|
457 | 5 500 | 700 | 700 | > 100 000 |
| 5 | – Me |
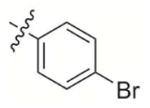
|
784 | > 5 000 | 800 | 800 | > 100 000 |
| 6 | – Me |
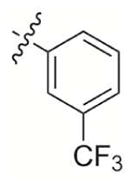
|
166 | 600 | 3 000 | 1 500 | > 50 000 |
| 7 | – Me |
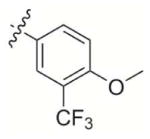
|
200 | 12 000 | 800 | 1 500 | > 50 000 |
| 8 | – CHF2 |
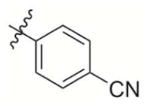
|
144 | 2 100 | 13 | 14 | 370 |
| 9 | – CHF2 |
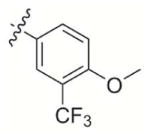
|
46 | - | - | - | - |
| PAT[a] |
|
173 | - | - | - | - | |
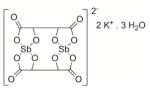
The values were determined at pH 7 and 25°C in the presence of 100 μM NADPH, 3 mM DTNB. PAT (potassium antimonyl tartrate) is used as drug reference.
The values were determined at pH 6.9 and 25°C in the presence of 100 μM NADPH, 1 mM GSSG
The values were determined at pH 7.4 and 25°C in the presence of 100 μM NADPH and 20 μM hTrxC72S after 10 min preincubation.[30]
The values were determined at pH 7.4 and 25°C in the presence of 200 μM NADPH and 3 mM DTNB after 10 min preincubation.[30]
Time-dependent inactivation of Schistosoma mansoni TGR activity
Time-dependent inactivation of SmTGR was studied using the DTNB reduction assay to evaluate the residual activity of the reacted enzyme (Table 3). The pair of menadione/difluoromethylmenadione derivatives, compound 7 versus compound 9, was selected on the basis of inhibition potency. Upon preincubation with NADPH and the inhibitor for 0, 2.5, 5 and 10 min, inactivation of SmTGR by 7 at low concentration (≤ 3 μM) followed pseudo-first order reaction kinetics (Fig. 4A). The experimental data allowed the application of the derivation by Kitz and Wilson [20] for irreversible inactivation. A semi-logarithmic plot of the fraction of non-inhibited enzyme activity ln(vi/v0) versus incubation time yielded straight lines with increasing slopes at short time periods, equivalent to the apparent rate constant of irreversible inhibition (kobs). The secondary plot expressing kobs as a function of inhibitor concentration followed equation (1): kobs = (ki x [I])/(KI + [I]) (1), where KI represents the dissociation constant of the inhibitor, and ki is the first order rate constant for irreversible inactivation, respectively. An hyperbolic curve at low inhibitor concentration allowed estimation of ki as 0.57 ± 0.02 min−1, and KI = 0.8 ± 0.08 μM, and second order rate constant ki/KI as 1.18 × 103 M−1 s−1 (Fig. 4B). The resulting half-time value (t1/2) of inactivation of the SmTGR was determined as 1.29 min. The results are consistent with our concept of mechanism-based inhibition by menadione derivatives acting as NADPH-consuming redox-cyclers (Fig. 5, mechanism A), ending with dead-end enzymes, as previously discussed. [13,17,18,19,21,22]
Fig. 4.



Time-dependent inactivation of Schistosoma mansoni thioredoxin glutathione reductase by compounds 7 (panels A and B) or 9 (panel C). Panels A-B: SmTGR (40 nM in 100 μL) was incubated in the presence of 100 μM NADPH and various inhibitor concentrations at 25°C for different times of incubation periods (0, 2.5, 5, and 10 min). Compound 7 concentrations used were 0 (●), 0.1(□), 0.3 (◆), 1.0 (◇), and 3.0 (▼) μM (panel A). The kobs data versus [I] were fitted to equation 5 (see text) which resulted in the hyperbolic curve (dashed line). At low inhibitor 7 concentration the dissociation constant and the first order rate constant for irreversible inactivation were determined as KI = 0.8 ± 0.08 μM and ki as 0.57 ± 0.02 min−1, respectively. Panel C: SmTGR (40 nM in 100 μL) was incubated in the presence of 100 μM NADPH and inhibitor at varying concentrations (0 – 0.03 – 0.1 – 0.3 – 1.0 – 3.0 – 10.0 – 30.0 μM) for 0 (●) and (■) 5 min-incubation periods. Reactions were also carried out in the absence of NADPH with the pre-reacted enzyme for 5 min (□). Then, 5 μL aliquots were removed and the remaining activity was measured in a standard DTNB reduction assay as described under Experimental Procedures. 3.4% final DMSO was present in all incubation mixtures.
Fig. 5.

Cascade of redox reactions responsible for the antischistosomal activity of 3-phenoxymethyl menadione as 2 or 7 (pathway A) and abolishment of the toxicity by a suicide-substrate, the difluoro analogue 8 or 9.
In the case of the difluoromethyl analogue 9, inactivation of SmTGR was evaluated with preincubation times of 0 and 5 min in the presence or in the absence of NADPH. At low inhibitor concentrations, an NADPH-dependent and time-dependent inhibition was observed at very low inhibitor concentration (data not shown) as it was observed with a monofluoromethyl naphthoquinone derivative inactivating hGR[18] and the estimation of the dissociation and first order rate constants for irreversible inactivation were respectively determined as KI = 0.26 ± 0.17 μM and ki = 0.64 ± 0.12 min−1. In this mechanism, NADPH-dependent reduction of the napthoquinone is faster than proton subtraction of the difluoromethyl group. At high inhibitor concentrations, in contrast to the NADPH-dependent TGR-catalyzed activation observed earlier at low concentration, incubation of the enzyme with compound 9 in the absence of NADPH resulted in inactivation (Fig. 4C), suggesting that the difluoromethylmenadione derivative 9 followed two reaction mechanism kinetics. This observation could be rationalized as followed: the low pKa value of the difluoromethyl group, determined around 7.1, implies that proton subtraction quickly occurred at physiological pH and dominates (Fig. 5, mechanism B), mainly before enzymic reduction, and the highly reactive difluoroquinone methide intermediate (P1) is likely to act as a cross-linking reagent, by analogy to difluoro acrolein analogue in polyacroleins formation, or to the gem-difluoroalkenes, which are known to be exclusively attacked by nucleophiles at the gem-difluoromethylene carbon atoms to form β-fluorocarbanions.[17, 23] Further additions to the carbanions might lead to insoluble cross-linked protein (polymers). However, the elucidation of the structure of the cross-linked protein is extremely difficult due to their lack of stability and solubility. In attempts to co-crystallize SmTGR with the difluoromethylmenadione analog 8 a fast precipitation of an inactivated insoluble 8–reacted SmTGR enzyme was observed upon polyalkylation or crosslinking reactions (data not shown). Noteworthy is to mention that the inhibitor concentration-dependent flavoenzyme inactivation was previously observed with the mono- and di-fluoromethyl menadione derivatives in hGR and PfGR inactivation studies.[18,19] Consequently, NADPH-dependent reduction of difluoromethylmenadione derivatives was not a prerequisite for this mechanism-based SmTGR inactivation and the precipitated cross-linked inactive SmTGR could not enter a continuous NADPH-dependent redox-cycling.
hGR inhibitory and hTrxR inhibitory activity
Examination of these novel small molecules against human disulfide reductase “orthologues” demonstrated an impressive degree of selectivity for SmTGR inhibition over other human flavoenzymes. To assess the specificity of those molecules inhibiting SmTGR, the library was also tested for inhibitory activity against hGR. With the exception of 6 (600 nM), and in contrast to the results with SmTGR, none of the tested compounds were inhibitors of hGR in the nanomolar range. Their inhibitory activity is at least 5 fold less based on IC50 values. The library was also tested against hTrxR. Two assays were performed: the Trx assay and the DTNB assay (see experimental procedures). The results were comparable between the two methods (Table 3). For the majority of compounds the IC50 values were in the μM or high nanomolar range (1, 2–3 μM; 2, 1.2–1.5 μM; 3, 2 μM; 4, 0.7 μM; 5, 0.8 μM; 6, 1.5–3 μM; 7, 0.8–1.5 μM). In particular, the IC50 for 6 was around 10 fold higher for hTrxR than for SmTGR and the one for 7 was around 5 fold higher. A notable exception was 8, the most potent inhibitor of SmTGR, which is also a very potent inhibitor of hTrxR (IC50 = 13–14 nM), albeit a weak inhibitor of hGR (IC50 = 2.1 μM).
In vitro cytotoxicity against S. mansoni worms and human cells
All compounds were tested at 50 μM alone (Fig. 6) for a period of 4 days. Besides the starting hit 1, the most potent antischistosomal 3-phenoxymethyl menadiones were the compounds bearing the α-fluorophenol methyl ether in the oxyphenylmethylene arm, i.e. 3 and 7, but not the difluoromethyl menadiones 8 and 9. This can be explained according to distinct mechanisms (Fig. 5). First, as predicted the difluorophenol is known to mimic a carboxylic function,[12] like in 1, because the fluorine atoms are known to influence the pKa value of the phenol to lower values. The methyl ether of the phenol might increase the bioavailability in the worm and might release the phenol in situ in the worms.[24] Also, with several fluorine atoms, the compounds are thought to effectively cross the lipophilic tegument of the worms, in addition to conferring better metabolic stability in vivo. The bioisosteric effect of the difluorophenol (transiently protected under the methyl ether form) was validated by the pair of compounds 7 (active) versus 6 (inactive), suggesting the importance of the prodrug effect to release a “negative” charge inside the parasite. In a second assay, all compounds were tested at 50 μM with hemoglobin (Hb) at 10 μM. After 7h incubation with the lead compounds a majority of the worms were dead. After 22h all worms were dead whatever the compound tested. Interestingly compound 2 displayed a specific effect depending on the gender of the parasites. Indeed, after 22h incubation time, this compound killed 100% of the female worms and only 50% of the male worms. These results suggest that compound 2 may affect a worm target that is particularly sensitive/more expressed in female than in male worms. The mechanism of action of 2 is still unclear, but it might involve hemoglobin digestion, as female Schistosoma worms are known to have a higher hemoglobin intake and a more intense metabolism than males.[25] We also tested the activity of select compounds against cultured ex vivo parasites in the presence of human red blood cells (10 μl/well). The purpose of the addition of human red blood cells is to allow worms to undergo drug bioactivation following hemoglobin digestion in the worm intestine. The final concentrations of compounds were 50 μM. While compound 7-based survival rates after 24 hours were identical in the presence and absence of red blood cells (RBCs), more parasites were dead in the absence of RBCs than in the presence of RBCs after 48 hours. RBCs may increase the oxidative metabolism of compound 7 and the reactive metabolites react quickly with electrophilic sites with nonessential components in the culture media external to the worm. The oxidative metabolism of 7 may lead to the generation of the phenol upon cleavage of the methoxy group or of phenoxymethyl bridge.[24] In that case, exposure to a Michael acceptor site (quinone methide) might be followed by thiol alkylation in the parasite.[26]
Fig. 6.

Survival curves of S. mansoni worms cultured in vitro and treated with the representatives of the 3-phenoxymethylmenadione series. Worms were cultured in the presence of 1 (
 ), 2 (
), 2 (
 ), 3 (
), 3 (
 ), 4 (
), 4 (
 ), 5 (
), 5 (
 ), 6 (
), 6 (
 ), 7 (
), 7 (
 ), 8 (
), 8 (
 ), 9 (
), 9 (
 ) or control with DMSO alone (
) or control with DMSO alone (
 ). PAT (potassium antimonyl tartrate) is used as drug reference; it led to 100 % dead worms after two days.
). PAT (potassium antimonyl tartrate) is used as drug reference; it led to 100 % dead worms after two days.
Finally, as a proof of concept to validate the essential TGR-catalyzed bioactivation step for the observed antischistosomal activity of the 3-phenoxymethyl menadione series, the synthesis of a difluoromethyl analogue (8, Fig. 1) of the lead 3-phenoxymethyl menadione 2 was designed to alkylate SmTGR in situ (i.e. in the parasite) by preventing the formation of the corresponding reduced 3-phenoxymethyl menadione metabolite (dihydronaphthoquinone). This compound, the 2-difluoromethyl-3-phenoxymethyl naphthoquinone 8 (Fig. 1), was shown to act as a potent suicide-substrate of isolated TGR and hTrxRs (Table 3). The introduction of 2 fluorines at the methyl group of menadione led to abolishment of the antiparasitic activity of the compound, i. e. (moderately) active 2 versus inactive 8 (phenoxymethyl series). This result has been confirmed with another pair of compounds: the active compound 7 (without RBC) versus the inactive difluoromethyl analogue, 9. As shown in Fig. 6, both compounds 8 and 9 displayed no effect on killing the worms, no matter whether RBCs were present or not. Hence, the TGR inactivation by the bioreductible fluorinated alkylating (substituted phenoxy)methyl menadione derivatives in situ was directly correlated to the abolishment of the antischistosomal activity of the parent menadione analogue 2. Our data strongly support the essential requirement of an active TGR, within a cascade of redox reactions to bioactivate potent antischistosomal 1,4-naphthoquinones. More studies are required to identify the cellular oxidant (Fig. 5) that is reduced in schistosomes, leading to the death of the worms.
Cytotoxic Activities Against Human Cells in Vitro
When tested for cytotoxicity against the human lung MRC-5 fibroblasts, by using the Alamar blue assay (Table S2 in the Supplementary material), most of the 1,4-naphthoquinone derivatives were only mildly toxic (IC50 from 21.7 μM to 31.07 μM), with the exception of the antischistosomal hit 1 (IC50 = 6.55 μM) and both difluoromethylmenadione derivatives 8 (IC50 = 6.70 μM) and 9 (IC50 = 11.20 μM). This cytotoxicity likely stems from the higher lipophilicity due to the presence of fluorine atoms and/or from their high oxidant character [18], respectively.
In vivo antischistosomal activities of lead (substituted phenoxy)methyl menadiones in S. mansoni-infected mice
Selected compounds were tested in a laboratory model of S. mansoni. All injections were ip, the first six weeks post-infection. Perfusions were 7 days after the last injection. Doses were: 4 - 1 × 50 mg/kg in 100% DMSO; 1 - 4 daily ip injections at 20 mg/kg in 10% DMSO; 5 - 4 daily ip injections at 20 mg/kg in 100% DMSO. Control animals received an equal volume of 10% DMSO. None of the compounds resulted in significant decreases in worm burdens. Furthermore, 1 was quite toxic and three of five mice treated died. The remaining 1-treated mice were severely affected.
Conclusions
We have designed and synthesized new 3-benzyloxo menadione and difluoromethylmenadione derivatives, displaying specific inhibitory activities in vitro against SmTGR versus hGR and hTrxR. Most of the compounds displayed sub-micromolar inhibitory capabilities toward SmTGR. Structure-activity relationship studies showed a high potential for (substituted phenoxy)methyl menadione derivatives bearing a substituted phenyloxo moiety to inhibit TGR versus GR/TrxR enzymes. Interestingly, one representative of these, the redox-active 3-benzyloxo menadione 7 displayed a time-dependent and NADPH-dependent SmTGR inactivation, correlating with potent antischistosomal action in vitro. The enzyme is expected to turn over in an unproductive redox-cycling, which ends by the formation of an inactive TGR dimer as a reversible dead-end product. Therefore, the redox-active 3-benzyloxo menadiones, like 7, might be considered as inhibitors of possible half-site reactivity of SmTGR because they might prevent the transfer of the reactivity toward NADPH binding of one subunit to the second subunit to allow disulfide reduction.[21] The formation of reversible dead-end flavoenzyme species as the cause for the time- and NADPH-dependent SmTGR inactivation by naphthoquinones could be supported by the distinct time-dependent SmTGR inactivation mechanism displayed by the difluoromethyl analog of inhibitor 7, the difluoromethylmenadione derivative 9. In preincubation SmTGR assays, in the presence of the difluoromethylmenadione 9, irreversible inactivation was found extremely rapid and resulted in precipitation of a cross-linked and polymerized inactivated enzyme. However, the fact that the difluoromethylmenadione 9, which inactivates SmTGR through a non-consuming NADPH-dependent redox-active cycle, has little killing effect in cultured ex vivo worms, is a nice illustration that the effect of the other active compounds against S. mansoni worms, like 7, is due to a continuous NADPH-consuming redox cycling of SmTGR. Several analogs of compound 7 showed significant antischistosomal action against cultured parasites but none of the compound tested in vivo were active. Limited bioavailability may compromise compound activity and future studies will be directed toward improving this.
Experimental Procedures
Chemistry
General methods
Melting points were determined with a Büchi Melting Point B-540 apparatus. 1H NMR and 13C NMR spectra were recorded on Brucker ARX 250, Brucker DRX 300 or Brucker DRX 500 spectrometers by using CDCl3, if not otherwise indicated. Chemical shifts (δ) are expressed in ppm relative to TMS. Multiplicity is indicated as s (singlet), d (doublet), t (triplet) and m (multiplet). Har stands for an aromatic proton in 1H NMR. CIV indicates a quaternary carbon in the 13C NMR assignation. J values are given in Hz. 19F NMR was performed using 1,2-difluorobenzene as external standard (δ= −139.0 ppm). Electron Impact (EI) mass spectra were recorded at 70 eV on Jeol JMS-700 and Finnigan TSQ 700 spectrometers at the Institut für Organische Chemie der Ruprecht-Karls Universität Heidelberg. Elemental analyses were carried out in the Mikroanalytisches Laboratorium der Chemischen Fakultät der Ruprecht-Karls Universität Heidelberg. Analytical thin layer chromatography (TLC) was performed on pre-coated silica gel plates Polygram® SIL G/UV254 and silica gel G60 from Macherey-Nagel (230–400 mesh) was used for flash column chromatography.
Side chain elongation (general procedure 1)
The phenol (1.0 equiv.), ethylchloroacetate (1.15 equiv.), sodium iodide (1.0 equiv) and anhydrous potassium carbonate (2.0 equiv.) were refluxed in dry acetone for 4h. The mixture was cooled and filtered. The solid was washed with CH2Cl2. The washings were combined with the filtrate and evaporated in vacuo.
Kochi-Anderson reaction (general procedure 2)
menadione (1.0 equiv.) and the phenoxyacetic acid (2.0 equiv.) were dissolved in a 3:1 solution of MeCN:H2O and heated to 85°C. Silver nitrate (0.1 equiv.) was added and ammonium peroxodisulfate (1.3 equiv.) in solution in the previous solvent mixture was added dropwise. The reaction mixture was stirred under reflux for 3h. It was then cooled and MeCN was removed in vacuo. The residue was extracted with CH2Cl2, dried over MgSO4 and evaporated in vacuo.
4-((3-Methyl-1,4-dioxo-1,4-dihydronaphthalen-2-yl)methoxy)benzoic acid (1)
Commercially available [4-(carboxy)phenoxy]acetic acid (2.27 g, 11.6 mmol) was used as the starting material and treated according to general procedure 2. A recrystallization in pure acetone gave yellow crystals of the final derivative 1 (563 mg, 30%); mp: 222–223 °C; 1H NMR (300 MHz, [D6]DMSO): δ= 8.04–8.08 (m, 2H, Har), 7.87–7.94 (m, 4H, Har), 7.12 (d, 2H, Har), 5.16 (s, 2H), 2.23 (s, 3H); 13C NMR (75 MHz, [D6]DMSO): δ= 184.6 (C=O), 183.2 (C=O), 167.0, 161.7, 147.8 (CIV), 138.8 (CIV), 134.2, 131.5, 131.4, 126.0, 123.5, 121.3, 115.0, 61.0, 12.8; HRMS-FAB m/z: 323.1 [M+H]+; Anal. calcd for C19H14O5 · 0.25 H2O: C 69.83, H 4.47, found: C 69.94, H 4.55.
Ethyl 2-(4-cyanophenoxy)acetate (2c)
Commercially available 4-cyanophenol (1 g, 8.40 mmol) was used as the starting material and treated according to general procedure 1. The crude product was obtained as a yellow oil. After flash chromatography purification (CH2Cl2) white crystals of 2c were obtained (1.475 g, 86%); mp: 50–53°C; 1H NMR (300 MHz, CDCl3): δ= 7.58 (d, 2H, Har), 6.94 (d, 2H, Har), 4.66 (s, 2H), 4.26 (q, 2H), 1.28 (t, 3H); 13C NMR (75 MHz, CDCl3): δ= 167.8, 160.9, 134.0, 118.8, 115.3, 105.0, 65.1, 61.6, 14.0; EI MS (70 eV, m/z (%)): 205.1 ([M]+·, 100), 132.1 (95), 102.1 (41); Anal. calcd for C11H11NO3: C 64.38 H 5.40, found: C 64.37, H 5.44.
2-(4-Cyanophenoxy)acetic acid (2d)
Ethyl 2-(4-cyanophenoxy)acetate 2c (500 mg, 2.44 mmol) was added to 50mL of a 10% solution of KOH in MeOH and the reaction mixture was refluxed for 5h. The resulting solution was cooled and the solvent evaporated in vacuo. The residue was diluted with water and washed with Et2O. The aqueous layer was then acidified and extracted with Et2O. The organic layer was dried over MgSO4 and concentrated in vacuo to give 2d as a white solid (275 mg, 65%); mp 169–172°C; 1H NMR (300 MHz, [D6]DMSO): δ= 7.77 (d, 2H, Har), 7.09 (d, 2H, Har), 4.82 (s, 2H); 13C NMR (75 MHz, [D6]DMSO): δ= 169.5, 161.2, 134.1, 119.0, 115.6, 103.3, 64.6; EI MS (70 eV, m/z (%)): 177.07 ([M]+·, 86), 132.06 (100), 119.05 (34), 102.04 (63); Anal. calcd for C9H7NO3: C 61.02 H 3.98, N 7.91, found: C 60.75, H 4.01, N 7.76.
4-((3-Methyl-1,4-dioxo-1,4-dihydronaphthalen-2-yl)methoxy)benzonitrile (2)
Compound 2d (200 mg, 1.12 mmol) was used as the starting material and treated according to general procedure 2. A flash chromatography purification Hexane:EtOAc 4:1 gave final derivative 2 as a yellow solid (40 mg, 21%); mp: 181–183°C; 1H NMR (300 MHz, CDCl3): δ= 8.10–8.55 (m, 2H, Har), 7.72–7.76 (m, 2H, Har), 7.39 (d, 2H, Har), 6.87 (d, 2H, Har), 5.10 (s, 2H), 2.31 (s, 3H); 13C NMR (75 MHz, CDCl3): δ= 184.9 (C=O), 183.5 (C=O), 161.6, 148.7 (CIV), 138.9 (CIV), 134.1, 134.0, 132.1, 131.7, 126.6, 119.0, 115.4, 104.8, 61.0, 13.3; EI MS (70 eV, m/z (%)): 303.1 ([M]+·, 100), 185.1 (94), 157.1 (96), 128.1 (40); Anal. calcd for C19H13NO3: C 75.24 H 4.32, N 4.62, found: C 75.40, H 4.22, N 4.51.
2,6-Difluorobenzene-1,4-diol (3a)
The 2,6-difluorophenol (200 mg, 1.54 mmol) was dissolved in 10mL of NaOH 6%. Solid potassium persulfate (425 mg, 1.55 mmol) was added to this stirred solution in several portions over 10min. The orange mixture was stirred overnight at RT. It was then concentrated to 1/3 its original volume on the rotary evaporator. The solution was neutralized with concentrated HCl and extracted with water. The aqueous solution was acidified with 3mL of concentrated HCl and boiled for 1h. It was then concentrated to 1/4 on the rotary evaporator. Addition of 10mL acetone precipitated the inorganic salts, which were removed by filtration. The filtrate was evaporated in vacuo. The brown residue was dissolved in acetone and was added to silica gel. The solvent was removed and the material added to a column of silica gel. A flash chromatography purification (Hexane:EtOAc 2:1) gave compound 3b as a white solid (100 mg, 44%); mp: 149–151°C; 1H NMR (300 MHz, [D6]DMSO): δ= 9.55 (s, 1H, Ph-OH), 9.14 (s, 1H, Ph-OH), 6.36–6.43 (m, 2H, Har); 13C (75 MHz, [D6]DMSO): δ= 152.8 (dd, 1JC-F=239.9 Hz, 3JC-F=9.4 Hz, 2C), 149.6 (t, 3JC-F =13.2 Hz), 125.7 (t, 2JC-F =16.7 Hz), 99.2 (dd, 2JC-F =25.1 Hz, 2C); EI MS (70 eV, m/z (%)): 146.06 ([M]+·, 100), 98.04 (18), 70.03 (23); Anal. calcd for C6H4F2O2: C 49.33 H 2.76, found: C 49.08, H 2.87.
Ethyl 2-(3,5-difluoro-4-methoxyphenoxy)acetate (3c)
3,5-difluoro-4-methoxyphenol 3b (500mg, 3.12 mmol) was used as the starting material and treated according to general procedure 1. The crude product was obtained as a yellow oil. A flash chromatography purification (CH2Cl2) led to white crystals of 3c (720 mg, 71%); mp: 29–30°C; 1H NMR (300 MHz, CDCl3): δ= 6.40–6.51 (m, 2H, Har), 4.52 (s, 2H), 4.24 (q, 2H), 3.87 (s, 3H), 1.27 (t, 3H); 13C NMR (75 MHz, CDCl3): δ= 168.1, 156.2 (dd, 1JC-F =247.5 Hz, 3JC-F =8.1 Hz, 2C), 153.1 (t, 3JC-F =12.8 Hz), 131.2 (t, 2JC-F =14.8 Hz), 99.4 (dd, 2JC-F=26.6 Hz, 2C), 65.7, 62.0 (t, 4JC-F =2.8 Hz), 61.5, 14.0; EI MS (70 eV, m/z (%)): 246.1 ([M]+·, 100), 173.08 (26), 159.06 (34), 145.05 (14); Anal. calcd for C11H12F2O4: C 53.66 H 4.91, found: C 53.51, H 4.88.
2-(3,5-Diflluoro-4-methoxyphenoxy)acetic acid (3d)
The ethyl ester 3c (500 mg, 2.03 mmol) was hydrolysed in 25mL of 10% aqueous NaOH. After the mixture was refluxed for 2h, it was cooled and then acidified with concentrated HCl whereupon the acid crystallised as a white solid. It was filtered and dried to obtain 3d as a white solid (340 mg, 77%); mp 115–117°C; 1H NMR (300 MHz, [D6]DMSO): δ= 6.80 (d, 2H, Har), 4.68 (s, 2H), 3.82 (s, 3H); 13C NMR (75 MHz, [D6]DMSO): δ= 169.7, 155.6 (dd, 1JC-F=244.8Hz, 3JC-F=8.4 Hz, 2C), 153.5 (t, 3JC-F=13.3 Hz), 129.9 (t, 2JC-F=15.1 Hz), 99.6 (dd, 2JC-F=26.1 Hz, 2C), 65.2, 62.1 (t, 4JC-F=2.4 Hz); EI MS (70 eV, m/z (%)): 218.09 ([M]+·, 100), 203.06 (19), 159.06 (54), 145.04 (60), 125.03 (26); Anal. calcd for C9H8F2O4: C 49.55 H 3.70, found: C 49.29, H 3.67.
2-((3,5-Difluoro-4-methoxyphenoxy)methyl)-3-methylnaphthalene-1,4-dione (3)
Compound 3d was used as the starting material and treated according to general procedure 2. Flash chromatography purification Hexane:EtOAc 10:1 gave final derivative 3 as a light yellow powder (55 mg, 41%); mp: 181–183°C; 1H NMR (300 MHz, CDCl3): δ= 8.13 (m, 2H, Har), 7.75 (m, 2H, Har), 8.56 (d, 2H, Har), 5.04 (s, 2H), 3.91 (s, 3H), 2.31 (s, 3H), 13C NMR (75 MHz, CDCl3): δ= 185.0 (C=O), 183.5 (C=O), 156,3 (dd, 1JC-F =247.4 Hz, 3JC-F =8.2 Hz, 2C), 153.8 (t, 3JC-F =12.7 Hz), 148.5 (CIV), 139.1 (CIV), 133.9, 132.1, 131.7, 131.1 (t, 2JC-F =14.8 Hz), 126.6, 99.4 (dd, 2JC-F=26.4 Hz, 2C), 62.2 (t, 4JC-F =2.7 Hz), 61.4, 13.2; EI MS (70 eV, m/z (%)): 344.1 ([M]+·, 61), 160.1 (100); Anal. calcd for C19H14F2O4: C 66.28 H 4.10, found: C 75.77, H 4.31.
2-((4-Chlorophenoxy)methyl)-3-methylnaphthalene-1,4-dione (4)
Commercially available [4-(chloro)-phenoxy]acetic acid (2.16 g, 11.6 mmol) was used as the starting material and treated according to general procedure 2. A flash chromatography purification petroleum ether:Et2O 4:1 gave 4 (633 mg, 35%); mp: 106–108°C; 1H NMR (300 MHz, CDCl3) : δ= 8.10–8.15 (m, 2H, Har), 7.73–7.76 (m, 2H, Har), 7.25 (d, 2H, Har), 6.92 (d, 2H, Har), 5.10 (s, 2H), 2.32 (s, 3H); 13C NMR (75 MHz, CDCl3): δ= 185.1 (C=O), 183.6 (C=O), 157.0, 148.4 (CIV), 139.6 (CIV), 133.8, 132.0, 131.8, 129.4, 126.5, 126.3, 116.1, 61.1, 13.2; HRMS-FAB m/z: 313.1 [M+H]+; Anal. calcd for C18H13ClO3: C 69.13 H 4.19, Cl 11.34 found: C 69.35, H 4.20.
2-((4-Bromophenoxy)methyl)-3-methylnaphthalene-1,4-dione (5)
Commercially available [4-(bromo)-phenoxy]acetic acid (2.68 g, 11.6 mmol) was used as the starting material and treated according to general procedure 2. A flash chromatography purification petroleum ether:Et2O 4:1 gave the final derivative 5 (500 mg, 24%); mp: 108–110 °C; 1H NMR (300 MHz, CDCl3): δ= 8.10–8.15 (m, 2H, Har), 7.73–7.76 (m, 2H, Har), 7.39 (d, 2H, Har), 6.87 (d, 2H, Har), 5.10 (s, 2H), 2.32 (s, 3H); 13C NMR (75 MHz, CDCl3): δ= 185.1 (C=O), 183.6 (C=O), 157.5, 148.4 (CIV), 139.6 (CIV), 133.8, 132.4, 126.5, 116.6, 113.7, 61.0, 13.2; HRMS-FAB m/z: 357.1 [M+H]+; Anal. calcd for C18H13BrO3: C 60.52 H 3.67, Br 22.37, found: C 60.73, H 3.82.
2-Methyl-3-((3-(trifluoromethyl)phenoxy)methyl)naphthalene-1,4-dione (6)
Commercially available [3-(trifluoromethyl)phenoxy]acetic acid (500 mg, 2.28 mmol) was used as the starting material and treated according to general procedure 2. A flash chromatography purification Hexane:EtOAc 10:1 gave yellow crystals of final derivative 6 (80 mg, 20%); mp: 106–108°C; 1H NMR (300 MHz, CDCl3): δ= 8.14 (m, 2H, Har), 7.75 (m, 2H, Har), 7.41 (m, 1H, Har), 7.14–7.26 (m, 3H, Har), 5.16 (s, 2H), 2.34 (s, 3H); 13C NMR (75 MHz, CDCl3): δ= 184.0 (C=O), 181.5 (C=O), 139.4, 133.9, 133.8, 130.1, 126.6, 126.5, 118.1, 112.1 (q, 3JC-F=3.9 Hz), 111.7, 111.6, 61.0, 13.2; 19F NMR (282MHz, CDCl3): δ = −63.10; EI MS (70 eV, m/z (%)): 346.1 ([M]+·, 100), 331.1 (21), 201.1 (21), 185.1 (61), 157.1 (83), 128.1 (39); Anal. calcd for C19H13F3O3: C 65.90 H 3.78, found: C 65.87, H 4.03.
Ethyl 2-(4-methoxy-3-(trifluoromethyl)phenoxy)acetate (7c)
Commercially available 4-methoxy-3-trifluoromethyl-phenol (550 mg, 2.86 mmol) was used as the starting material and treated according to general procedure 1. The pure product 7c was obtained as a white solid (760 mg, 2.73 mmol, 95%); mp: 63–64°C; 1H NMR (300 MHz, CDCl3): δ= 7.16 (d, 1H, Har), 7.05 (dd, 1H, Har), 6.93 (d, 1H, Har), 4.59 (s, 2H), 4.27 (q, 2H), 3.86 (s, 3H), 1.30 (t, 3H); 13C NMR (75 MHz, CDCl3): δ= 168.7 (C=O), 152.4 (CIV), 151.2 (CIV), 123.2 (q, 1JC-F = 272.5 Hz, CF3), 119.5 (q, 2JC-F = 31.2 Hz), 118.9, 114.3, 113.4, 66.3, 61.5, 56.5, 14.1; 19F NMR (282MHz, CDCl3): δ = − 62.97; EI MS (70 eV, m/z (%)): 278.1 ([M]+, 100), 205.08 (33), 191.05 (54); Anal. calcd for C12H13F3O4: C 51.80, H 4.71; found: C 51.48, H 4.66.
2-(4-Methoxy-3-(trifluoromethyl)phenoxy)acetic acid (7d)
The ethyl ester (500 mg, 1.80 mmol) was hydrolysed in 25mL of 10% aqueous NaOH. After the mixture was refluxed for 2h, it was cooled and then acidified with concentrated HCl whereupon the acid crystallised as a white solid. It was filtered and dried to obtain 7d as a white solid (415 mg, 92%); mp: 133–135°C; 1H NMR (300 MHz, [D6]DMSO): δ= 7.20 (m, 2H, Har), 7.13 (m, 1H, Har), 4.71 (s, 2H), 3.82 (s, 3H); 13C NMR (75 MHz, [d6]DMSO): δ= 170.1 (C=O), 151.3 (CIV), 151.0 (CIV), 119.7, 114.2, 113.1, 65.2, 56.4; 19F NMR (282MHz, [D6]DMSO): δ= − 61.17; EI MS (70 eV, m/z (%)): 250.1 ([M]+·, 90), 191.1 (94), 177.0 (21), 129.0 (19); Anal. calcd for C10H9F3O4: C 48.01 H 3.63; found: C, 47.75 H, 3.62.
2-((4-Methoxy-3-(trifluoromethyl)phenoxy)methyl)-3-methylnaphthalene-1,4-dione (7)
2-(4-methoxy-3-(trifluoromethyl)phenoxy)acetic acid 7d (400 mg, 1.60 mmol) was used as the starting material and treated according to general procedure 2. A flash chromatography purification Hexane:EtOAc 10:1 gave the final derivative 7 as yellow crystals (59 mg, 20%); mp 146–148°C; 1H NMR (300 MHz, CDCl3): δ= 8.10–8.15 (m, 2H, Har), 7.72–7.76 (m, 2H, Har), 7.22 (d, 1H, Har), 7.12 (dd, 2H, Har), 6.95 (d, 1H, Har), 5.09 (s, 2H), 3.86 (s, 3H), 2.32 (s, 3H); 13C NMR (75 MHz, CDCl3): δ= 185.2 (C=O), 183.6 (C=O), 152.1 (CIV), 151.7 (CIV), 148.3 (CIV), 139.6 (CIV), 133.8, 132.1 (CIV), 131.8 (CIV), 126.6, 123.3 (q, 1JC-F = 272.7 Hz, CF3), 119.5 (q, 2JC-F = 31.2 Hz), 118.9, 114.2 (q, 3JC-F= 5.4 Hz), 113.5, 61.7, 56.6, 13.2; 19F NMR (282MHz, CDCl3): δ = − 63,09; EI MS (70 eV, m/z (%)): 376.2 ([M]+·, 66), 192.1 (100), 191.1 (80), 157.1 (25), 128.1 (19), 127.1 (7), Anal. calcd for C20H15F3O4: C 63.83 H 4.02, found: C 63.56, H 4.19.
1,4-Dimethoxy-naphthalene, 1,4-dimethoxy-naphthalene-2-carbaldehyde, 2-(difluoromethyl)-1,4-dimethoxynaphthalene, 2-difluoromenadione were synthesized as previously described.[17]
4-((3-(Difluoromethyl)-1,4-dioxo-1,4-dihydronaphthalen-2-yl)methoxy)benzonitrile (8)
Compound 2d (1 g, 5.64 mmol) and difluoromenadione (600 mg, 2.8 mmol) were used as the starting materials and treated according to general procedure 2. A flash chromatography purification Hexane:EtOAc 2:1 gave the final derivative 8 as a yellow solid (344 mg, 35%); mp: 143–145°C; 1H NMR (300 MHz, CDCl3): δ= 8,17 (m, 2H, Har); 7,84 (m, 2H, Har); 7,62 (d, 2H, Har); 7,17 (t, 2JH-F=53,5Hz, CHF2); 7,04 (d, 2H, Har); 5,29 (s, 2H);13C NMR (75 MHz, CDCl3) : δ= 175.9 (C=O), 174.5 (C=O); 161.5 (CIV), 142.5 (CIV), 138.2 (t, 2JC-F=21 Hz), 134.9, 134.8, 134.10 (2C), 131.5 (CIV), 131.1 (CIV), 127.1, 126.8, 119.0 (CIV), 115.5 (2C), 110.1(t, 1JC-F=241 Hz), 105.0 (CIV), 60.0; 19F NMR (282 MHz, CDCl3): δ = −116.36; EI MS (70 eV, m/z (%)): 340.3 ([M]+·, 100), Anal. calcd for C19H11F2NO3 · 0.5 H2O: C 67.26 H 3.27, N 4.13; found: C 67.33, H 3.43, N 4.12.
2-(difluoromethyl)-3-((4-methoxy-3-(trifluoromethyl)phenoxy)methyl)naphthalene-1,4-dione (9)
2-(4-methoxy-3-(trifluoromethyl)phenoxy)acetic acid 7d (2.40 g, 9.6 mmol), and difluoromenadione (1 g, 4.8 mmol) were used as the starting materials and treated according to general procedure 2. A flash chromatography purification Hexane:EtOAc 10:1 gave 9 as an orange solid (1.116 g, 2.71 mmol, 57%); m.p.: 84–86°C; 1H NMR (300 MHz, CDCl3): = 8.15–8.18 (m, 2H, Har), 7.82–7.85 (m, 2H, Har), 7.23 (d, 4JH-H = 3 Hz, 1H, Har), 7.17 (t, 2JH-F = 54 Hz, 1H, CHF2), 7.14 (dd, 3JH-H = 9 Hz, 4JH-H = 3 Hz, 1H, Har), 6.96 (d, 3JH-H = 9 Hz, 1H, Har), 5.21 (s, 2H), 3.87 (s, 3H); 13C NMR (75 MHz, CDCl3) : δ= 183.4 (C=O), 182.6 (C=O), 152.4 (CIV), 151.7 (CIV), 143.3 (CIV), 137.8 (t, 2JC-F = 21 Hz), 134.8, 134.7, 131.6 (CIV), 131.2 (CIV), 127.1, 126.8, 126.2 (q, 1JC-F = 272.5 Hz, CF3), 119.5, 119.4 (q, 2JC-F = 31.2 Hz), 114.7 (q, 3JC-F = 5 Hz), 113.5, 110.2 (t, 1JC-F = 241 Hz), 61.1, 56.6; 19F NMR (282 MHz, CDCl3): δ = −116.98 (d, JH-F = 54 Hz, CHF2), δ = −62.48 (CF3); −116.98; HRMS-FAB m/z: 412.3 ([M]+); Anal. calcd for C20H13F5O4: C 58.26 H 3.18, found: C 57.96, H 3.35.
Cyclic Voltammetry
Cyclic voltammetry of the menadione derivatives (~ 10−3 M) was performed using a Voltalab 50 potentiostat/galvanostat (Radiometer Analytical MDE15 polarographic stand, PST050 analytical voltammetry and CTV101 speed control unit) controlled by the Voltamaster 4 electrochemical software. A conventional three-electrodes cell (10 mL) was employed in our experiments with a glassy carbon disk (GC, s = 0.07 cm2) set into a Teflon rotating tube as a working electrode, a Pt wire as a counter electrode, and KCl(3 M)/Ag/AgCl reference electrode (+210 mV vs NHE).[27] Prior to each measurement, the surface of the GC electrode was carefully polished with 0.3 μm aluminium oxide suspension (Escil) on a silicon carbide abrasive sheet of grit 800/2400. Thereafter, the GC electrode was copiously washed with water and dried with paper towel and argon. The electrode was installed into the voltammetry cell along with a platinum wire counter electrode and the reference. The solutions containing ca. 10−3 M of the menadione derivatives were vigorously stirred and purged with O2-free (Sigma Oxiclear cartridge) argon for 15 minutes before the voltammetry experiment was initiated, and maintained under an argon atmosphere during the measurement procedure. The voltammograms were recorded at room temperature (23°C) in DMSO with 100 mM tetra-n-butylammonium hexafluorophosphate as supporting and inert electrolyte. The voltage sweep rate was varied from 50 to 300 mV s−1 and several cyclic voltammograms were recorded from +0.5 V to −2.2 V. Peak potentials were measured at a scan rate of 200 mV s−1. Redox potentials were determined from oxidation and reduction potentials.
Glutathionylation reactions
The GSH conjugates were prepared by incubating the starting difluoromethylmenadione and its derivatives 8 and 9 with 5 equivalents of reduced glutathione. Reduced glutathione was purchased from Sigma–Aldrich and a fresh 100 mM stock solution was daily prepared by dissolution in pure Milli-Q water. Stock solutions (10 mM) of difluoromethylmenadione and its derivatives 8 and 9 were prepared in acetonitrile (MeCN) and kept at 4 °C. Glutathionylation of the difluoromethylmenadione and its derivatives 8 and 9 was first investigated by LC-MS/UV-Vis over a 1 h-period at 25 °C. According to the results in LC-MS/UV, an investigation in UV spectrophotometry was applied and based on the experimental protocol of GSH reaction for LC-MS/UV. All reaction mixtures (final volume of 500 μL) used in the UV-vis and in LC-MS/UV analyses contained: 200 μL acetonitrile, 1 mM GSH (25 μL of a 20 mM stock solution in pure water), 200 μM naphthoquinone (10 μL of the 10 mM stock solution in MeCN), and 265 μL of an aqueous buffer adjusted at pH 7.5 with aqueous NH4OH. For the LC-MS analysis, formic acid was used in a solution of 50% with 10 μl addition in the vial to stop the GSH-reaction. In contrast, UV-Vis spectra were recorded as soon as the buffer was added. In parallel, an additional control without MeCN was prepared for compound 8, in an aqueous buffer adjusted at pH 7.5 with aqueous NH4OH. Slight modifications of the protocols for the glutathionylation reactions were applied for the LC-MS analyses, in particular in order to optimize the m/z signals by increasing (5-fold) the starting naphthoquinone and GSH concentrations. All reaction mixtures (final volume of 500 μL) used in the LC-MS analysis contained: 160 μL acetonitrile, 5 mM GSH (25 μL of the 100 mM stock solution), 1 mM naphthoquinone (50 μL of the 10 mM stock solution), 265 μL of an aqueous buffer adjusted at pH 7.5 with aqueous NH4OH, and 10 μl of 50% formic acid solution. At different time points (0 – 1h – 2h – 4h – 24h), 5 μL-aliquots of each reaction mixture were removed and analyzed by LC–MS/MS and UV-Vis spectrophotometry for GSH-conjugate formation (see Fig. S3 in the Supplementary material for the LC-MS chromatograms, the equipment and details of the LC-MS analyses).
Enzymes
Recombinant human glutathione reductase was purified according to reported procedures [ 28 ] and had a specific activity of 182 U/mg (9500 units/μmol subunit). Recombinant SmTGR was expressed in Escherichia coli and purified by affinity chromatography as described [11].
SmTGR inhibitory activity
The DTNB (5,5′-dithiobis-(2-nitrobenzoic acid)) activity of SmTGR was used to determine the potencies of all compounds. SmTGR was preincubated with 100 μM NADPH and 50 μM compounds at room temperature for 10 min. Then, 3 mM DNTB and 100 μM NADPH were added to initiate the reactions. The rate of formation of 2-nitro-5-thiobenzoic acid from DTNB was determined spectrally using an ε412nm of 13.6 mM−1 cm−1. The absorbance of each reaction was followed with a Thermo Scientific Multiskan Spectrum Microplate Spectrophotometer using an experimentally determined pathlength correction factor. To determine the IC50 values against TGR in enzymic assays, compounds were tested at multiple concentrations between 0.01 and 30 mM. The DTNB reduction by S. mansoni TGR treated with different compounds was compared to that of untreated enzyme preincubated with 100 μM NADPH but without drug.
Time-dependent inactivation of SmTGR by Compound 7 or Compound 9
For determining the rate constants of S. mansoni TGR inactivation (A-B panels in Fig. 4) by compound 7, the residual DTNB reduction activity was monitored over time by following an incubation protocol.[29] Stock solutions (10 mM) of (A) compound 7 and (B) compound 9 were prepared in DMSO and kept at 25 °C. All pre-reaction mixtures (final volume of 100 μL) contained 100 μM NADPH, varying inhibitor concentrations (0 – 10 μM), SmTGR (40 nM), 1.8% DMSO in TGR buffer at 25 °C. At different time points (0 – 2.5 – 5.0 – 10.0 min), 100 μL of reaction buffer (3 mM DTNB and 100 μM NADPH in TGR reaction buffer) was added to each pre-reaction and residual activity was measured in the standard DTNB reduction assay at 25 °C. In the case of the inhibitor 9, SmTGR inactivation at varying inhibitor concentrations (0 – 10 μM), was tested with preincubation times of 0 and 5 min, with and without NADPH in TGR buffer at 25 °C (panel C in Fig. 4). The final DMSO concentration in all assays was 3.4%.
hGR inhibitory activity
The standard assay was conducted at 25 °C as described previously.[17] Briefly hGR was added to the assay mixture which contained 100 μM NADPH, 1 mM GSSG and the inhibitor (0 to 100 μM) in buffer (100 mM potassium phosphate buffer, 200 mM KCl, 1 mM EDTA, pH 6.9. The reaction was started by adding the enzyme hGR and initial rates of NADPH oxidation were monitored at 340 nm (ε340 nm = 6.22 mM−1 cm−1) and IC50 calculated.
hTrxR inhibitory activity
Wildtype human placenta hTrxR, the recombinant hTrxR mutant Sec498Cys, and hTrxC72S were purified/recombinantly produced and assayed on a Hitachi U-2100 spectrophotometer as previously described.[30] Inhibition studies on the wildtype enzyme were conducted in the Trx assay as well as in the DTNB assay. Due to loss of Trx-reducing activity, the Sec498Cys mutant was assayed in the DTNB assay only. Half-maximal inhibition of the enzymes was determined by incubating wild-type hTrxR (5–25 nM FAD-containing subunits), or mutant hTrxR (2 μM FAD-containing subunits) for 10 min with NADPH (100 μM or 200 μM) and inhibitor at 25°C. The enzyme activity was then measured at 340 nm or 412 nm by adding hTrxC72S (20 μM) or DTNB (3 mM) to the mixtures.
Evaluation of the cytotoxicity of the different drugs against human MRC-5 cells
Human MRC-5SV2 cells were cultured in Earl’s MEM + 5 % FCSi. Assays were performed in 96-well microtiter plates, each well containing about 104 cell/well. After 3 days incubation, cell viability was assessed fluorimetrically after addition of resazurin and fluorescence was measured (λex 550 nm, λem 590 nm).[31] The results are expressed as % reduction in cell growth/viability compared to untreated control wells and IC50 was determined. Compounds were tested at 5 concentrations (64–14– 4–1–0.25 μM or mg/ml). When the IC50 is lower than 4 μg/mL or μM, the compound is classified as toxic.
Antischistosomal activity against adult, ex vivo worms
Adult worms were harvested by perfusion of mice with DMEM 6–7 weeks after infection with S. mansoni.[32] Harvested worms were washed thoroughly with DMEM medium. The parasites were then cultured in RPMI medium with 2 mM glutamine, 25 mM HEPES, 10% heat-inactivated fetal bovine serum, 125 μg/ml streptomycin, and 150 units/ml penicillin G in a CO2 incubator at 37°C overnight. The overnight culture allows the worms to recover from the Nembutal anesthesia and to adapt to the culture media. All compounds were dissolved in DMSO and their activities were tested against adult worms at a concentration of 50 μM. The mobility of the parasites was observed under a stereomicroscope over 48 hours. The parasites were considered dead after they had completely lost mobility. Human red blood cells were isolated by centrifugation of whole blood at 400 x g without break for 10 min. The pelleted cells were washed three times with RPMI + 10% fetal calf serum. Finally the wash supernatant was removed and the cells were resuspended in the same volume of RPMI + 10% fetal calf serum as the initial blood sample. Worms were cultured in the presence of 10 μl human red blood cells/well. When present in the cultures, hemoglobin (human, Sigma) was used at 10 μM.
In vivo drug treatments
Compounds were dissolved in DMSO and administrated by intraperitoneal (ip) injection as indicated in the text. Control S. mansoni–infected mice were administrated a corresponding amount of DMSO. Worm burdens were determined by hepato-portal perfusion seven days after the last drug administration. Animal Welfare: This study was approved by the Institutional Animal Care and Use Committee at Rush University Medical Center (IACUC number 11-064; DHHS animal welfare assurance number A3120-01). Rush University Medical Center’s Comparative Research Center (CRC) is operated in accordance with the Animal Welfare Act {(Public Law (P.L.) 89–544) as amended by P.L.91–579 (1970); P.L.94–279 (1976); P.L. 99–198(1985); and P.L 101–624 (1990)}, the Public Health Service’s Policy on Humane Care and Use of Laboratory Animals (revised, 2002), the Guide for the Care and Use of Laboratory Animals (revised, 1996) and the U.S. Government Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research and Training. The CRC is registered with the Animal and Plant Health Inspection Service (APHIS) arm of the United States Department of Agriculture (USDA). The Institution has an Animal Welfare Assurance on file with the National Institutes of Health, Office of Laboratory Animal Welfare (OLAW), A-3120-01. The facilities are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International). The CRC is directed by the Senior Director of the CRC, a Doctor of Veterinary Medicine (D.V.M.) and a Diplomate of the American College of Laboratory Animal Medicine (ACLAM), who reports to the Associate Provost and Vice President for Research, who is also the Institutional Official for Animal Care and Use.
Supplementary Material
Acknowledgments
The authors thank Mourad Elhabiri and Mouhamad Jida for interpretation assistance with the electro- and physico-chemistry and mass spectrometry analysis of the GSH conjugates. This work was partly supported by the ANRémergence program (grant SCHISMAL [E.D.-C.]), the Laboratoire d’Excellence (LabEx) ParaFrap (grant LabEx ParaFrap ANR-11-LABX-0024 [E.D.-C.]), the NIH/National Institute of Allergy and Infectious Disease (grant R01AI065622 to D.L.W.), and the Deutsche Forschungsgemeinschaft (BE1540/11-2 to K.B.). Schistosome-infected mice were provided by the NIAID Schistosomiasis Resource Center at the Biomedical Research Institute (Rockville, MD) through NIH-NIAID Contract HHSN272201000005I for distribution through BEI Resources.
Abbreviations
- DAST
diethylaminosulfur trifluoride
- DMEM
Dulbecco’s Modified Eagle’s Medium
- DTNB
(5,5′-dithiobis-(2-nitrobenzoic acid)
- CAN
cerium ammonium nitrate
- GSH
glutathione
- GSSG
glutathione disulfide
- (h)GR
(human) glutathione reductase
- (h)TrxR
(human) thioredoxin reductase
- MeCN
acetonitrile
- NQ2−
naphthoquinone dianion
- NQ
1,4-naphthoquinone
- NBu4PF6
tetrabutylammonium hexafluorophosphate
- PAT
potassium antimonyl tartrate
- RBCs
red blood cells
- RPMI
Roswell Park Memorial Institute medium
- (Sm)TGR
(Schistosoma mansoni) thioredoxin-glutathione reductase
- TLC
thin layer chromatography
- Trx
thioredoxin
Footnotes
Author Contribution Statement
E.D.-C. conceived the medicinal chemistry approach, and L.J. contributed to the synthesis of the naphthoquinones. E.D.-C. and D.B. co-wrote the manuscript. L.J. and H.-H.H. respectively, implemented the protocols in chemistry and in parasitology, respectively. D.B. and M.C. performed the electrochemical, UV-vis spectrophotometry and LC-MS measurements. K.B. and D.L.W. supervised the enzymic studies and wrote the corresponding parts. D.L.W. analyzed the parasitological data and revised the manuscript. All authors produced the figures and commented on the manuscript.
Supporting Information Additional supporting information may be found in the online version of this article at the publisher’s web site:
Fig. S1. Fig. S1. Changes of the Absorption Spectra of the difluoromethylmenadione upon glutathione addition (Panels A–D).
Fig. S2. Changes of the Absorption Spectra of the difluoromethyl derivative 8 upon glutathione addition (Panels A–D). Methods about LC–MS analysis of the difluoromethylmenadiones–glutathione conjugates
Fig. S3. UV-Vis chromatograms and LC–MS traces After analysis of the glutathionylation reaction mixtures for difluoromethylmenadiones–GSH conjugate formation.
Table S1. Proposed chemical structures of difluoromethylmenadiones–glutathione adducts analyzed from the glutathionylation reaction mixtures by LC-MS analysis.
Table S2. IC50 values for M5 derivatives IC50 Values of 3-phenoxymethyl menadiones and their difluoromethymenadione derivatives as cytotoxic agents against human lung fibroblasts MRC-5 in vitro.
References
- 1.Jordan P. From katayama to the Dakhla Oasis: the beginning of epidemiology and control of bilharzia. Acta Trop. 2000;77:9–40. doi: 10.1016/s0001-706x(00)00121-2. [DOI] [PubMed] [Google Scholar]
- 2.Gryseels B, Polman K, Clerinx J, Kestens L. Human schistosomiasis. Lancet. 2006;368:1106–1118. doi: 10.1016/S0140-6736(06)69440-3. [DOI] [PubMed] [Google Scholar]
- 3.Steinmann P, Keiser J, Bos RR, Tanner MM, Utzinger J. Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect Dis. 2006;6:411–425. doi: 10.1016/S1473-3099(06)70521-7. [DOI] [PubMed] [Google Scholar]
- 4.Chitsulo L, Engels D, Montresor A, Savioli L. The global status of schistosomiasis and its control. Acta Trop. 2000;77:41–51. doi: 10.1016/s0001-706x(00)00122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stothard JR, Chitsulo L, Kristensen TK, Utzinger J. Control of schistosomiasis in sub-Saharan Africa: progress made, new opportunities and remaining challenges. Parasitology. 2009;136:1665–1675. doi: 10.1017/S0031182009991272. [DOI] [PubMed] [Google Scholar]
- 6.Fenwick A, Webster JP, Bosque-Oliva E, Blair L, Fleming FM, Zhang Y, Garba A, Stothard JR, Gabrielli AF, Clements AC, Kabatereine NB, Toure S, Dembele R, Nyandindi U, Mwansa J, Koukounari J. The Schistosomiasis Control Initiative (SCI): rationale, development and implementation from 2002–2008. Parasitology. 2009;136:1719–1730. doi: 10.1017/S0031182009990400. [DOI] [PubMed] [Google Scholar]
- 7.Fenwick A, Savioli L, Engels D, Bergquist RN, Todd MH. Drugs for the control of parasitic diseases: current status and development in schistosomiasis. Trends Parasitol. 2003;19:509–515. doi: 10.1016/j.pt.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 8.Ismail M, Botros S, Metwally A, William S, Farghally A, Tao LF, Day TA, Bennett JL. Resistance to praziquantel: direct evidence from Schistosoma mansoni isolated from Egyptian villagers. Am J Trop Med Hyg. 1999;60:932–935. doi: 10.4269/ajtmh.1999.60.932. [DOI] [PubMed] [Google Scholar]
- 9.Vermund SH, Bradley DJ, Ruiz-Tiben E. Survival of Schistosoma mansoni in the human host: estimates from a community-based prospective study in Puerto Rico. Am J Trop Med Hyg. 1983;32:1040–1048. doi: 10.4269/ajtmh.1983.32.1040. [DOI] [PubMed] [Google Scholar]
- 10.Alger HM, Williams DL. The disulfide redox system of Schistosoma mansoni and the importance of a multifunctional enzyme, thioredoxin glutathione reductase. Mol Biochem Parasitol. 2002;121:129–139. doi: 10.1016/s0166-6851(02)00031-2. [DOI] [PubMed] [Google Scholar]
- 11.Kuntz AN, Davioud-Charvet E, Sayed AA, Califf LL, Dessolin J, Arnér ES, Williams DL. Thioredoxin glutathione reductase from Schistosoma mansoni: an essential parasite enzyme and a key drug target. PloS Med. 2007;4:e206. doi: 10.1371/journal.pmed.0040206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qiu J, Stevenson SH, O’Beirne MJ, Silverman RB. 2,6-Difluorophenol as a bioisostere of a carboxylic acid: bioisosteric analogues of gamma-aminobutyric acid. J Med Chem. 1999;42:329–332. doi: 10.1021/jm980435l. [DOI] [PubMed] [Google Scholar]
- 13.Biot C, Bauer H, Schirmer RH, Davioud-Charvet E. 5-Substituted tetrazoles as bioisosters of carboxylic acids. Bioisosterism and mechanistic studies on glutathione reductase inhibitors as antimalarials. J Med Chem. 2004;47:5972–5983. doi: 10.1021/jm0497545. [DOI] [PubMed] [Google Scholar]
- 14.Smart BE. Fluorine substituent effects (on bioactivity) J Fluorine Chem. 2001;109:3–11. [Google Scholar]
- 15.Anderson JM, Kochi JK. Silver(I)-catalyzed oxidative decarboxylation of acids by peroxydisulfate. Role of silver(II) J Am Chem Soc. 1970;92:1651–1659. [Google Scholar]
- 16.Feiring AE, Sheppard WA. Fluorinated hydroquinones. J Org Chem. 1975;40:2543–2545. [Google Scholar]
- 17.Müller T, Johann L, Jannack B, Brückner M, Lanfranchi DA, Bauer H, Sanchez C, Yardley V, Deregnaucourt C, Schrével J, Lanzer M, Schirmer RH, Davioud-Charvet E. Glutathione reductase-catalyzed cascade of redox reactions to bioactivate potent antimalarial 1,4-naphthoquinones - a new strategy to combat malarial parasites. J Am Chem Soc. 2011;133:11557–11571. doi: 10.1021/ja201729z. [DOI] [PubMed] [Google Scholar]
- 18.Bauer B, Fritz-Wolf K, Winzer A, Kühner S, Little S, Yardley V, Vezin H, Palfey B, Schirmer RH, Davioud-Charvet E. A fluoro analogue of the menadione derivative 6-[2′-(3′-methyl)-1′,4′-naphthoquinolyl]hexanoic acid is a suicide substrate of glutathione reductase. Crystal structure of the alkylated human enzyme. J Am Chem Soc. 2006;128:10784–10794. doi: 10.1021/ja061155v. [DOI] [PubMed] [Google Scholar]
- 19.Lanfranchi DA, Belorgey D, Müller T, Vezin H, Lanzer M, Davioud-Charvet E. Exploring the trifluoromenadione core as template to design antimalarial redox-active agents interacting with glutathione reductase. Org Biomol Chem. 2012;10:4795–4806. doi: 10.1039/c2ob25229e. [DOI] [PubMed] [Google Scholar]
- 20.Kitz R, Wilson IB. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J Biol Chem. 1962;237:3245–3249. [PubMed] [Google Scholar]
- 21.Belorgey D, Lanfranchi DA, Davioud-Charvet E. 1,4-Naphthoquinones and others NADPH-dependent glutathione reductase-catalyzed redox cyclers as antimalarial agents. Curr Pharm Des. 2013;19:2512–2528. doi: 10.2174/1381612811319140003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morin C, Besset T, Moutet JC, Fayolle M, Brückner M, Limosin D, Becker K, Davioud-Charvet E. The aza-analogues of 1,4-naphthoquinones are potent substrates and inhibitors of plasmodial thioredoxin and glutathione reductases and of human erythrocyte glutathione reductase. Org Biomol Chem. 2008;6:2731–2742. doi: 10.1039/b802649c. [DOI] [PubMed] [Google Scholar]
- 23.Amii H, Uneyama K. C-F bond activation in organic synthesis. Chem Rev. 2009;109:2119–2183. doi: 10.1021/cr800388c. [DOI] [PubMed] [Google Scholar]
- 24.Mansuy D, Dansette PM, Pecquet F, Chottard JC. Iron-porphyrin catalysis of the oxidative dealkylation of para-nitro-anisole and 7-ethoxycoumarin by cumylhydroperoxide: a possible model for the corresponding cytochrome P 450-dependent reactions. Biochem Biophys Res Commun. 1980;96:433–439. doi: 10.1016/0006-291x(80)91233-4. [DOI] [PubMed] [Google Scholar]
- 25.Oliveira MF, d’Avila JC, Torres CR, Oliveira PL, Tempone AJ, Rumjanek FD, Braga CM, Silva JR, Dansa-Petretski M, Oliveira MA, de Souza W, Ferreira ST. Haemozoin in Schistosoma mansoni. Mol Biochem Parasitol. 2000;11:217–221. doi: 10.1016/s0166-6851(00)00299-1. [DOI] [PubMed] [Google Scholar]
- 26.Thompson DC, Perera K, London R. Spontaneous hydrolysis of 4-trifluoromethylphenol to a quinone methide and subsequent protein alkylation. Chem Biol Interact. 2000;126:1–14. doi: 10.1016/s0009-2797(99)00162-3. [DOI] [PubMed] [Google Scholar]
- 27.Sawyer DT, Sobkowiak A, Roberts JL. Electrochemistry for Chemists. 2. John Wiley & Sons; New York, NY: 1995. [Google Scholar]
- 28.Nordhoff A, Bücheler US, Werner D, Schirmer RH. Folding of the four domains and dimerization are impaired by the Gly446-->Glu exchange in human glutathione reductase. Implications for the design of antiparasitic drugs. Biochemistry. 1993;32:4060–4066. doi: 10.1021/bi00066a029. [DOI] [PubMed] [Google Scholar]
- 29.Gromer S, Merkle H, Schirmer RH, Becker K. Human placenta thioredoxin reductase: preparation and inhibitor studies. Methods Enzymol. 2002;347:382–394. doi: 10.1016/s0076-6879(02)47038-3. [DOI] [PubMed] [Google Scholar]
- 30.Urig S, Fritz-Wolf K, Réau R, Herold-Mende C, Tóth K, Davioud-Charvet E, Becker K. Undressing of phosphine gold(I) therapeutic agents as irreversible inhibitors of human disulfide reductases. Angew Chem Int Ed Engl. 2006;45:1881–1886. doi: 10.1002/anie.200502756. [DOI] [PubMed] [Google Scholar]
- 31.O’Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem. 2000;267:5421–5426. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- 32.Lewis F. Schistosomiasis. In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, editors. Current protocols in immunology. John Wiley & Sons; New York, NY: 1998. pp. 19–28. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.