

Abstract
A small molecule inhibitor (QLT0267) targeting integrin-linked kinase is able to slow breast tumor growth in vivo; however, the mechanism of action remains unknown. Understanding how targeting molecules involved in intersecting signaling pathways impact disease is challenging. To facilitate this understanding, we used tumor tissue microarrays (TMA) and digital image analysis for quantification of immunohistochemistry (IHC) in order to investigate how QLT0267 affects signaling pathways in an orthotopic model of breast cancer over time. Female NCR nude mice were inoculated with luciferase-positive human breast tumor cells (LCC6Luc) and tumor growth was assessed by bioluminescent imaging (BLI). The plasma levels of QLT0267 were determined by LC-MS/MS methods following oral dosing of QLT0267 (200 mg/kg). A TMA was constructed using tumor tissue collected at 2, 4, 6, 24, 78 and 168 hr after treatment. IHC methods were used to assess changes in ILK-related signaling. The TMA was digitized, and Aperio ScanScope and ImageScope software were used to provide semi-quantitative assessments of staining levels. Using medium-throughput IHC quantitation, we show that ILK targeting by QLT0267 in vivo influences tumor physiology through transient changes in pathways involving AKT, GSK-3 and TWIST accompanied by the translocation of the pro-apoptotic protein BAD and an increase in Caspase-3 activity.
Keywords: Aperio, apoptosis, breast cancer model, BAD, Caspase-3 pharmacokinetics, immunohistochemistry, integrin-linked kinase, QLT0267, pAKT, tissue microarray, TWIST
Introduction
Targeted cancer therapies are becoming increasingly more prevalent as the molecular basis for pathogenesis is being uncovered. Although targeted agents have had a profound impact on cancer treatment progress, investigating the mechanism of target ablation on normal cell function as well as in diseased states remains challenging. This is particularly true for targets that have multiple roles in cell physiology. As an example, integrin-linked kinase (ILK) is a pleiotropic kinase central to several signal transduction pathways involving downstream molecules such as p(ser473)-AKT, pser9/21GSK-3, HIF-1α, Wnt, E-cadherin, TWIST and VEGF (Delcommenne et al. 1998; Deng et al. 2002; Guo et al. 2007; Imanishi et al. 2007; Joshi et al. 2007; Li et al. 2003; McDonald et al. 2008; Maydan et al. 2010; Mills et al. 2003; Muranyi et al. 2010; Naska et al. 2006; Persad et al. 2001; Rosano et al. 2006; Serrano et al. 2013; White et al. 2006; Xie et al. 2004; Yamaji et al. 2001). These pathways are known regulators of several key cancer processes, such as epithelial-to-mesenchymal transition (EMT), angiogenesis, and apoptosis. In fact, ILK overexpression has been documented in many cancers, including lung (Chen et al. 2012; Takanami 2005; Watzka et al. 2010), prostate (Graff et al. 2001), pancreatic (Sawai et al. 2006), colorectal (Bravou et al. 2003, 2006), gastric (Ito et al. 2003), ovarian (Ahmed et al. 2003, 2004) and thyroid (Younes et al. 2005) cancers, as well as glioma (Obara et al. 2004), squamous cell carcinoma of the head and neck (Younes et al. 2007), Ewing’s sarcoma and PNET (Chung et al. 1998), mesothelioma (Watzka et al. 2008), retinoblastoma (Chen et al. 2011) and malignant melanoma (Dai et al. 2003); in some instances, the expression of ILK has also been associated with poor prognosis (Dai et al. 2003; Graff et al. 2001). Given the importance of ILK in cancer development and progression, it is anticipated that ILK inhibition and/or silencing may be an effective way of treating cancer. Preclinical studies completed to date provide support for targeting ILK in cancer (Brazdziute and Laurinavicius 2011; Edwards et al. 2008; Kalra et al. 2009, 2011; Younes et al. 2005, 2007). Many studies have shown that in vivo inhibition of ILK using antisense oligonucleotides or siRNA targeting ILK as well as small molecule inhibitors such as KP392, QLT0254, QLT0267 and, more recently, N-methyl-3-(1-(4-(piperazin-1-yl)phenyl)-5-(4’-(trifluoromethyl)-[1,1’-biphenyl]-4-yl)-1H-pyrazol-3-yl) propanamide reduces tumor growth in animal models of prostate cancer (Lee et al. 2011; Tan et al. 2004), pancreatic cancer (Yau et al. 2005), glioblastoma (Edwards et al. 2005, 2008), gastric cancer (Zhao et al. 2011), melanoma (Wong et al. 2007), lung cancer (Liu et al. 2006), thyroid cancer (Younes et al. 2005), and breast cancer (Kalra et al. 2009). However, questions concerning the mechanisms through which ILK inhibition leads to reduced tumor growth in vivo remain unanswered. Some investigators have attempted to detail the in vivo impact of ILK inhibition by following one or two downstream effectors of ILK in overexpression or knock-out studies. A study done by White et al. (2001) showed that the transgenic MMTV/ILK mouse produces spontaneous mammary gland tumors that constitutively express phosphorylated forms of AKT, GSK-3 and MAPK. Further, Assi et al. (2008) used an intestinal epithelial cell ILK knockout animal to show that ILK regulates cyclin D1 and MMP-9 in vivo. Several groups have demonstrated that silencing ILK in prostate cancer (Yuan et al. 2013), ovarian cancer (Li et al. 2013, 2012) gastric carcinoma (Zhao et al. 2011) and melanoma (Wong et al. 2007) leads to decreased growth and increased apoptosis. Others have used ILK inhibitors to show that decreased ILK activity leads to growth suppression (Becker-Santos et al. 2012; Dobreva et al. 2008; Edwards et al. 2008; Eke et al. 2009; Faralli et al. 2011; Fielding et al. 2011; Jones et al. 2014; Kalra et al. 2009, 2010, 2011; Koul et al. 2005; Li et al. 2009, 2013; Lim et al. 2013; Muranyi et al. 2010, 2009; Oloumi et al. 2006; Santos et al. 2007; Sikkema et al. 2014; Steinbrunn et al. 2012; Tabe et al. 2007; Troussard et al. 2006; Wang et al. 2011; Younes et al. 2005, 2007). For example, Edwards and colleagues have shown that suppression of ILK in vivo leads to reduced expression of VEGF and decreased angiogenesis (Edwards et al. 2008; Tan et al. 2004). It has also been demonstrated that suppression of ILK activity in vivo can influence the phosphorylation of AKT at serine 473 (Edwards et al. 2008; Yau et al. 2005), potentially leading to reduced growth through multiple mechanisms. However, the exact pathways involved in growth suppression remain unclear. For instance, dysregulation of AKT may impact both growth as well as survival, and yet downstream effectors in the AKT pathway have not been examined in preclinical studies of ILK inhibition.
The vast number of downstream effectors that may be influenced by altered ILK signaling poses a challenge for the identification of a specific mechanism for ILK-related suppression of tumor growth in vivo. It is possible to use microarrays to establish alterations in gene expression in response to ILK inhibition using agents such as QLT0267; however, these data reflect alterations in transcript levels only, and constraints on experimental design often allow for the consideration of only one time-point even though signal transduction is an evolving process. It is also likely that a targeted agent will suppress a downstream signal transduction pathway temporarily, during the time period in which the drug concentration is at its peak. Once the agent is cleared, the suppressive effects will likely wane. Whether this leads to phenotypic alterations in downstream pathways will depend on what pathways have been transiently activated or inactivated, as well as the cell line being evaluated. In vitro studies done in our laboratory exemplify this concept in the context of ILK-mediated PI3K/AKT signaling in vitro. Verreault and Bally (2009) demonstrated, that in PC3 cells, ILK silencing has only transient effects on p(ser473)AKT levels. Moreover, the effect of ILK silencing is cell line specific, as U251 cells were seemingly more resistant to ILK-mediated down-regulation of p(ser473)AKT. The latter observations suggests that some cell lines have redundant pathways involved in the regulation of the PI3K/AKT pathway (Verreault and Bally 2009) whereas other cell lines may exhibit a preference for ILK-mediated activation of AKT (Troussard et al. 2006). Preliminary data in breast cancer cell lines, including LCC6 cells, show that QLT0267 is able to decrease p(ser473)AKT through ILK inhibition at early time-points; however, p(ser473)AKT levels normalize very quickly thereafter. Using ILK siRNA, we were able to demonstrate a similar pattern whereby a transient decrease in p(ser473)AKT is observed at 24 hr. After 48 hr, despite continued silencing of ILK, p(ser473)AKT levels returned to baseline, suggesting compensatory pathways are at play (unpublished data). Collectively, these data are intriguing. The transient down-regulation of p(ser473)AKT in vitro suggests that the same transience is likely seen in vivo when ILK activity is inhibited. For this reason, we believed it was important to look at signaling kinetics in vivo after treatment with QLT0267, as it is likely that a targeted agent will suppress a downstream signal transduction pathway temporarily during the time period in which the drug concentration is at its peak. Therefore, the ideal preclinical study examining signal transduction pathways is one which examines the sum total effects of ILK ablation using agents such as QLT0267 in the context of the drugs pharmacokinetics. Studies like the one described become very large, requiring medium- to high-throughput tools and methods. To evaluate gene expression changes over a time course using microarray would be laborious and costly and still require confirmation using IHC. Further, microarray analyses do not consider alterations in subcellular localization or activation of proteins involved in some of these pathways. The studies herein were designed to consider the pharmacokinetics of the ILK inhibitor, QLT0267, first, and addresses the kinetics of signal transduction changes after treatment using tissue microarrays (TMA) and digital quantification of immunohistochemistry (IHC). The latter tools enabled the examination of multiple effector molecules over time.
We demonstrate, using bioluminescence imaging (BLI), that tumor growth suppression is observed at 7 days after treatment initiation. The pharmacokinetic profile of QLT0267 demonstrates drug clearance within the first 6 hr after treatment. Finally, tumor tissue was collected at time-points related to the pharmacokinetics of the drug, as well as within the 7-day window where growth suppression was observed. A TMA was constructed and analyzed for changes in expression, subcellular localization, and activation of downstream effectors of ILK. We were able to show decreases in p(ser473)-AKT, pGSK-3 and TWIST within 24 hr after treatment. We show for the first time that treatment of animals with QLT0267 is related to a translocation of BAD from the cytoplasm to discrete puncta, possibly at the mitochondrial membrane in tumor tissues; a change that is known to activate apoptosis.
An understanding of early changes in growth, along with small molecule inhibitor pharmacokinetics, provides parameters around which we can rationally design large, multi-endpoint studies that enable investigation into alterations in signal transduction pathways as they evolve over time. These data can be used to support a better understanding of the mechanisms of targeted agents, such as QLT0267, in vivo.
Materials & Methods
Chemicals and Reagents
QLT0267 (267) was a generous gift from QLT Inc. (Vancouver, BC, Canada) and was diluted in PTE (polyethylene glycol, Tween-80, 95% ethanol, and citric acid). QLT0267 is a second-generation ILK inhibitor, derived from KP-392. The selectivity of this small molecule for ILK relative to that of other kinases has been determined by analyzing its effects on 150 recombinant kinases. Among these kinases tested, QLT0267 is highly specific, showing 1000-fold selectivity over kinases including CK2, CSK, DNA-PK, PIM-1, PKB/Akt and PKC, and 100-fold selectivity over other kinases, such as Erk-1, GSK-3β, LCK, PKA, p70S6K, and RSK1 (Troussard et al. 2006; Younes et al. 2007). Furthermore, the in vitro kinase activity of purified ILK is sensitive to QLT0267 (Maydan et al., 2010). QLT0267 is an accepted inhibitor of ILK kinase activity (Becker-Santos et al. 2012; Dobreva et al. 2008; Edwards et al. 2008; Eke et al. 2009; Faralli et al. 2011; Fielding et al. 2011; Jones et al. 2014; Kalra et al. 2009, 2010, 2011; Koul et al. 2005; Li et al. 2009, 2013; Lim et al. 2013; Muranyi et al. 2009, 2010; Oloumi et al. 2006; Santos et al. 2007; Sikkema et al. 2014; Steinbrunn et al. 2012; Tabe et al. 2007; Troussard et al. 2006; Wang et al. 2011; Younes et al. 2005, 2007). A dose of 200 mg/kg was chosen based on previous studies (Kalra et al. 2009, 2011). All antibodies were purchased from Cell Signaling Technology (Beverly, MA). All other reagents were of reagent grade and purchased from sources as indicated in the various sections below.
Cell Lines and Culture
MDA-MB-435/LCC6 (LCC6) estrogen receptor-negative breast cancer cells (Leonessa et al. 1996) were a generous gift from Dr. Robert Clarke (Georgetown University, Washington, D.C.). There has been much debate surrounding the origin of the LCC6 parental cell line, MDA-MB-435. Ross et al. (2000) and Rae et al. (2007) suggest that the MDA-MB-435 cell line is of melanoma origin, sharing molecular features with the M14 melanoma cell line, whereas Neve et al. (2006) have demonstrated that the MDA-MB-435 cell line shares many molecular features with other breast cancer cell lines. Sellappan et al. (2004) demonstrate that MDA-MB-435 cells can be induced to express breast differentiation-specific proteins and secrete milk lipids. The fact that MDA-MB-435 cell line shows molecular similarities with the M14 cell line is not sufficient evidence to conclude that MDA-MB-435 is melanoma in origin, as it has been established that human breast cancers do indeed express melanocytic markers (Bachmeier et al. 2008; Montel et al. 2009) and, more recently, even neuronal markers (Yu et al. 2011). The most compelling evidence for the origin of MDA-MB-435 cells comes from recent karyotype evidence showing that both M14 and MDA-MB-435 cell lines were derived from a female patient consistent with the origin of MDA-MB-435 and not M14 cells (Chambers 2009). In light of these data, we continue to use MDA-MB-435 as a model breast cancer cell line.
Cells lines were maintained in the absence of penicillin and streptomycin and screened for mycoplasma prior to preparing the stock of cells that were frozen for future use in animal experiments. Cells were re-suspended in freezing media (10% DMSO in FBS) and slowly frozen in Nalgene 1°C freezing containers (Rochester, NY) containing 100% isopropanol at -80°C for 24 hr before storage in liquid nitrogen. Frozen cells were quickly thawed at 37°C, centrifuged to remove freezing media, plated, and passaged twice before use in experiments. LCC6 and LCC6WT-luc cells were maintained in DMEM/high glucose supplemented with L-glutamine (2 mM; DMEM and L-glutamine from Stem Cell Technologies; Vancouver, BC, Canada), 5 mM penicillin/streptomycin (Stem Cell Technologies), and 10% FBS (Hyclone; Logan, UT). All cells were maintained at 37°C and 5% CO2 in a humidified atmosphere and allowed to undergo no more than 20 passages.
Lentivirus Transfections
The transfection procedure used to create the LCC6WT-Luc cell line has been previously described (Kalra et al. 2011). Briefly, constructs for the lentivirus (LV) vector containing the luciferase (Luc) and Green Fluorescent Protein (GFP) genes were obtained from Dr. Alice Mui (Jack Bell Research Center, Vancouver General Hospital). To generate luciferase-expressing lentivirus (Lenti-Luc), the vector was co-transfected using calcium phosphate with packaging constructs pRSVREV, pMDLg/pRRE, and the VSV-G expression plasmid pHCMVG into the packaging cell line HEK-293T. Conditioned medium was collected daily for 4 days and cleared of debris by low-speed centrifugation, filtered, and stored at -70°C. The LCC6 cells were then infected with Lenti-Luc virus (25 µl concentrated viral supernatant/mL medium). Subsequently, cells were re-plated in low concentrations into soft agar in the wells of a 6-well plate. Luciferin was added to each well and plates were imaged using IVIS (see below) to identify luciferase-positive colonies. Positive colonies were selected, expanded, and used for the in vitro and in vivo studies described below.
Western Blot
Total protein lysates were prepared from cells incubated in the presence of QLT0267 or PTE vehicle. Briefly, cells were rinsed with PBS, harvested from plates with trypsin and centrifuged at 1500 ×g for 5 min. Cell pellets were then re-suspended in lysis buffer [150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 2.5 mM EDTA, 0.1% SDS, mini-protease inhibitor cocktail tablets (Roche Diagnostics; Mannheim, Germany)], sheared using a 25-gauge needle, incubated on ice for 30 min, and finally centrifuged at 10,000 ×g for 10 min to remove insoluble material. Protein concentrations were determined from the supernatants using the Bradford Method and approximately 50 μg of total protein from each sample were denatured in loading buffer (Invitrogen) by boiling for 10 min and then loaded onto 10% SDS-polyacrylamide gels. Proteins separated by electrophoresis were transferred to nitrocellulose membranes (Millipore; Bedford, MA) and blocked for 1 hr at room temperature in Odyssey blocking buffer (Licor Biosciences; Lincoln, NBR). Membranes were incubated at 4°C overnight in Odyssey blocking buffer containing polyclonal anti-ILK, anti-AKT, anti-P-AKT, anti-pBAD, or anti-β-actin antibodies (1:1000 dilution; Cell Signaling Technology). Membranes were then washed three times for five minutes 5 min with PBS-Tween (1% v/v) and incubated with either anti-rabbit IRDYE (Rockland; Gilbertsville, PA) or anti-rabbit Alexa 680 (Invitrogen, Molecular Probes; Burlington, ON) at 1:10,000 for 1 hr at room temperature. Signals were detected using the Odyssey Infrared Detection System and associated software (Odyssey v1.2; Licor). The studies were done at least three times and representative immunoblots are shown.
Immunofluorescence
Cells grown on coverslips were rinsed with PBS (pH 7.4), fixed using 2.5% paraformaldehyde (w/v) in PBS for 20 min at room temperature and permeabilized using 0.5% Triton X-100 (v/v) in PBS for 5 min at room temperature. Coverslips were then washed three times with PBS, incubated for 1 hr in 2% bovine serum albumin (BSA) (w/v) in PBS to block non-specific binding, washed three times in PBS, and then incubated with anti-BAD and anti-BCL-xl antibodies (Santa Cruz Biotechnology Inc.; Dallas, TX) for 1 hr at room temperature. All antibodies were diluted in BSA/PBS. Coverslips were washed three times for 5 min using PBS. Primary antibody binding was detected by further incubations with anti-rabbit Alexa 546 or Alexa 488 (Molecular Probes). To ensure that there was no non-specific antibody binding, a secondary antibody control coverslip was used for each experiment, where coverslips were stained with either Alexa 546 or Alexa 488 alone. Nuclei were stained using Hoechst nuclear stain (10 mg/ml; Molecular Probes) at 1:1000 for 5 min at room temperature. Coverslips were rinsed once with double-distilled water and mounted to microscope slides using a 9:1 solution of glycerol and PBS (Air Products & Chemicals, Inc.; Allentown, PA). Images were viewed and captured using a Leica CTR-mic UV fluorescence microscope (Wetzlar, Germany) and a DC100 digital camera with Open Lab software (Improvision; Lexington, MA). The studies were done at least three times and representative immunofluorescence images are shown.
Caspase Activation Assay
Cells were subject to the Caspase-Glo 3/7 luminescent assay (Promega; Madison, WI) according to the manufacturer’s instructions. Briefly, cells grown in 96-well plates were treated with QLT0267 or PTE vehicle for 12, 24, 48 or 72 hr. At treatment endpoint, cells were incubated with 100 μl of the prepared Caspase-Glo 3/7 reagent at room temperature. The plates were covered and sealed with a plate sealer and the contents were mixed gently using a plate shaker at 300–500 RPM for 30 sec. The plates were then incubated at room temperature for 3 hr. Subsequently, the luminescence of each sample was measured using an Optima fluorescence/luminescence plate reader (BMG Labtech; Durham, NC). Luminescence data was collected at 420–540 nm. Raw data from treated cells was expressed as a percentage of normalized to vehicle-treated controls. The studies were done at least three times and luminescence data is expressed as mean values ± SD.
Animal Studies
All animal studies were conducted in accordance with and approved by the institutional (University of British Columbia) Animal Care Committee. The work was conducted in a manner that meets the Canadian Council of Animal Care guidelines for the care and use of experimental animals. Animals were maintained at 22°C in a 12-hr light/dark cycle with ad libitum access to water and food. Female NCr nude mice, weighing between 18 and 25 g, were obtained from Taconic (Oxnard, CA) and maintained in an SPF facility (Animal Resource Centre, BC Cancer Agency). Mice were terminated for humane reasons based on a health scoring method conducted under a health monitoring standard operating procedure. Reasons for euthanasia included tumor ulceration, the presence of tumors with volumes in excess of 500 mg, and/or signs of deteriorating animal health.
LCC6 cells (2×106) in a volume of 50 µl media were injected using a 28-gauge needle into the mammary fat pad of female NCr nude mice (Taconic), as previously described (Conley 1979). These mice were than randomized into two groups: vehicle and treated. After treatment initiation (see below), tumor growth was monitored using non-invasive luminescence-based imaging with the IVIS 200 imaging system from Xenogen (Caliper LS; Almeda, CA). In another study, tumor-bearing animals were given 200 mg/kg QLT0267 p.o. and killed with CO2 asphyxiation at 2, 4, 6, and 24 hr. Blood was collected immediately via cardiac puncture. Blood was rapidly transferred to EDTA-containing Microtainers and the samples placed on ice. Plasma was separated by centrifugation (15 min at 1500 × g at 4°C) and stored at -80°C until they were prepared for analytical work (see below).
Additional studies used mice randomized into three groups: control, vehicle and treatment arms. Tumors were established as described above. On day 7 post-inoculation, animals were treated p.o. with 200 mg/kg QLT0267 every day for seven days. Eight animals were sacrificed per group at 2, 4, 6, 24, and 168 hr, at which time the tumor tissue was harvested as outlined in the methods below. Figure 1 illustrates the timeline for drug dosing and harvest of tumor tissue for this study.
Figure 1.

An illustration of the treatment schedule and timing of tissue harvesting is shown for tissue samples used in the construction of the TMA. Animals were inoculated with tumor cells at seven days prior to treatment initiation. Animals were dosed daily for seven days at times 0, 24, 48, 72, 96, 120, and 144 hr (solid arrows). Tumor tissue was harvested (dashed arrows) at 2, 4, 6 and 24 hr after the first treatment, and 6 hr after treatment on days 3 and 5. The last tumor harvest was performed at 24 hr following the final treatment (168 hr).
In Vivo Imaging System (IVIS)
Imaging was performed daily to monitor tumor growth for seven days during daily treatment. LCC6WT-luc tumor-bearing mice were injected i.p. with 500 μl D-luciferin (15 mg/ml) (CaliperLS, Alameda, CA). Mice were anesthetized using isoflurane, and post-luciferin injection mice were imaged at 20 min (± 2 min). Photographic and luminescence images were taken at exposure times of 1, 2 and 5 sec, and Xenogen IVIS software was used to quantify non-saturated bioluminescence in whole-body images. Light emission between 2.0×106 to 10.0×1010 was assumed to be indicative of viable luciferase-labeled tumor cells, whereas emissions below this range were considered as background and above this range, saturation. Bioluminescence (FLUX) was quantified as photons/sec for each ROI (cm2).
Pharmacokinetics
Two stock solutions of QLT0267 were prepared in methanol. One stock solution was used to prepare the working solutions to generate calibration standard curves and the other for preparing working solutions used for generating quality control (QC) samples. Working solutions were prepared with 1:1 methanol:volume (v/v) and stored at -70°C. Calibration standards were prepared by spiking aliquots of the blank matrix with the appropriate level of anylates to create standards that spanned the calibration range of interest with a minimum of six calibration levels. QC samples were prepared by spiking aliquots of the blank matrix with QLT0267 working solutions. The QCs were prepared at four concentrations. One concentration was prepared at the intended lower limit of quantitation whereas the remaining three levels spanned the concentration range of the calibration curve. The low concentration sample was less than three times the lower limit of quantitation; the medium-level concentration near the midpoint of a semi-log plot of the calibration range; the high-level concentration at 75% of the highest calibration level. An internal standard stock solution was prepared at a concentration of 1 mg/ml in methanol in a volumetric flask, aliquoted, and stored at -70°C. A working solution was prepared at 100 ng/ml by diluting the stock solution with 1:1 methanol:water (v/v) in serial dilution and stored at -70°C. To prepare the sample, 20 µl of the plasma was aliquoted to a centrifuge tube, to which 200 µl of sodium tetraborate solution was added. Ethyl acetate (1.5 ml) was added and the tube was vortexed for 10 min. The samples were then centrifuged at 3000 RPM for 20 min at room temperature and then frozen at -70°C for 20 min. The organic phase was removed and placed into Eppendorf tubes and the samples were dried in an evaporator. Samples were reconstituted in 100 µl of 0.1% formic acid in 1:1 (v/v) methanol:water. Extracts were vortexed, and 20 µl was transferred to glass inserts of labeled amber auto-sampler vials, which were capped and placed on the auto-sampler tray of the LC-MS/MS for analysis to determine plasma concentration of QLT0267 at each time-point.
Tissue Collection, TMA Construction and Immunohistochemistry
Four or eight animals were sacrificed by CO2 asphyxiation at each of the indicated time-points and tumors in the mammary fat pad tissues were excised, fixed in 10% neutral-buffered formalin and embedded in paraffin. Representative areas of non-necrotic tumor tissue were selected by Dr. Margaret Sutcliffe, a pathologist at the BC Cancer Agency (Vancouver, BC, Canada), and marked on hematoxylin and eosin-stained slides and on their corresponding tissue blocks to indicate where the cores were to be obtained for each sample. TMAs were constructed by removing two 0.6-mm cores from selected formalin-fixed, paraffin-embedded tissue blocks and transferred to a recipient paraffin block using a manual arrayer (Pathology Devices Inc.; Westminster, MD). Four µm-thick sections of the TMA blocks were cut and stained using the DakoCytomation EnVision and System-HRP (Dako; Carpinteria, CA) in a two-step IHC technique. Sections were deparaffinized in xylene, dehydrated through three alcohol changes, and transferred to Ventana Wash solution (Ventana Medical Systems; Oro Valley, AZ). Endogenous peroxidase activity was blocked in 3% hydrogen peroxide. Antigen retrieval was performed and slides were incubated with primary antibodies against pAKT serine 473 (1:50 dilution), BAD (1:300 dilution), cleaved Caspase-3 (1:50 dilution), TWIST (1:200 dilution) or p(ser9/21)GSK3αβ serine 9/21 (1:10 dilution), all from Cell Signaling Technology. Finally, sections were incubated with the pre-diluted Ventana Universal Secondary Antibody and DAB Map detection system, counterstained with hematoxylin, dehydrated, cleared, and mounted. Optimization of the IHC protocol involved three antigen-retrieval conditions and a serial dilution of the antibody to establish the optimal staining concentration. Although freezing of the tissue samples prior to formalin fixation could have potentially affected the immunoreactivity, appropriate negative and positive controls were performed to ensure the quality and adequacy of staining. The negative control was performed by omission of the primary antibody. Human tonsil tissues were used as an internal positive control for several apoptotic markers.
Aperio Digital IHC Analysis and Quantification
TMA and tumor slides were scanned using the Aperio ScanScope System (Aperio, Technology; Vista, CA) at 40× magnification to provide a high-resolution digital image. The ImageScope software (free from Aperio.com) was used to analyze and quantify marker expression. An ROI for individual tumor cores was selected and analyzed using the positive pixel count algorithm. The ImageScope analysis tool allows the user to set the threshold for color saturation, as well as the upper and lower limits for the intensities of the weak- and strong-positive pixels. These thresholds were set using positive and negative control tissues. To ensure that settings were appropriate for the analysis of tumor tissue, randomly selected tumor cores on the TMA were tested with the parameters set using positive and negative controls tissues and adjusted accordingly. Algorithm parameters were customized to differentiate between negatively (blue), moderately (orange) and strongly (red) stained cells. Raw data were provided in terms of the number of positive pixels and intensity of positive pixels, which were normalized to the number of total pixels counted or area of ROI in µm2. For each sample, two representative cores were used to construct the TMA; thus data from two cores were averaged to give a pixel count for any given sample. In order to confirm the accuracy of the digital quantification, selected TMAs were evaluated by inspection by a pathologist, using a scoring system from 0 to 3. These data were also averaged for the two samples representing each tumor sample.
Statistical Analysis
All statistical data was collected using GraphPad Prism (San Diego, CA). Parametric analysis was done using SD or SEM and n, in an unpaired Student’s t-test. Area under the curve analysis was done using Phoenix WinNonlin software (Pharsight; St. Louis, MO).
Results
QLT0267 Is Able to Suppress p(ser473)AKT Expression In Vitro
AKT is a well-accepted downstream effector of ILK (Delcommenne et al. 1998; Guo et al. 2007; Hannigan et al. 2011; Joshi et al. 2007; Koul et al. 2005; Persad et al. 2001; Serrano et al. 2013; Xie et al. 2004). For this reason, we started our evaluation of the mechanism of action for QLT0267 by confirming the impact of ILK inhibition using QLT0267 on AKT activity. LCC6WT-luc cells were incubated with increasing doses of QLT0267 in vitro for a period of 24 hr. Protein was collected and subjected to western blot analysis for the expression of p(ser473)AKT. Figure 2A is a representative image showing that QLT0267 is able to suppress p(ser473)AKT in a dose-dependent manner in vitro without altering the expression of ILK.
Figure 2.

At 7 days post-treatment, tumor growth suppression is observed but p(ser473)AKT levels are not altered. (A) In vitro western blot analysis of LCC6WT-luc cells illustrates that treatment with QLT0267 has a dose-dependent effect on the suppression of p(ser473)AKT. (B) Representative images from bioluminescent analysis of orthotopic LCC6WT-Luc treated with vehicle and QLT0267 are shown. (C) Total light emission from tumors in animals was visualized and quantified at the time of tumor cell inoculation (IN), and 4, 24, and 168 hr after treatment initiation. At 168 hr, animals treated with QLT0267 showed lower total light flux than vehicle control (p<0.05). (D–G) Day 7 IHC of p(ser473)AKT was performed on a TMA containing tumor cores from vehicle- and QLT0267-treated animals. Positive (breast cancer tissue) (D) and negative (primary Antibody omitted) (E) controls are shown. At the 7 day time-point, no significant difference (p>0.05) in p(ser473)AKT staining is observed when comparing tumors from vehicle (F) and QLT0267 treated (G) animals (n=4 and 8, respectively) as quantified by positive pixel counting (H). Scale, 50 μm.
QLT0267 Treatment Effects Are Observed in the Orthotopic LCC6 Tumor Model Rapidly After Treatment Is Initiated
To establish how early QLT0267-mediated growth suppression occurs, luciferase-positive LCC6 (LCC6WT-Luc) tumor cells were inoculated into the mammary fat-pad of female NCr nude mice, as described in the Materials & Methods, and tumor size was evaluated using BLI at the time of inoculation (IN), and at 4, 24, and 168 hr after treatment was initiated (see Fig. 1). The results are summarized in Fig. 2B and 2C. Figure 2B shows representative images from a single animal and Fig. 2C shows the average light FLUX of six animals for each of the time-points measured. Similar to previously published results (Kalra et al. 2009), after animals have been treated with 7 daily doses of QLT0267, tumors exhibit significantly lower FLUX (p=0.038) as compared with tumors from vehicle-treated animals.
After Seven Days of QLT0267 Treatment, Levels of p(ser473)AKT Are Comparable to Vehicle-treated Tumors
As indicated above, an established downstream effector of ILK is AKT (Cabodi et al. 2010; McDonald et al. 2008). For this reason, the in vivo expression of p(ser473)-AKT was compared in vehicle- and QLT0267-treated animals using tumor tissues collected after 7 days of daily dosing (p.o.) with 200 mg/kg of QLT0267. Twenty-four hr after the last treatment (168-hr time-point; Fig. 1), animals were sacrificed, and the tumor tissue was harvested and then preserved in paraffin blocks for TMA generation. TMA sections were subjected to IHC of p(ser473)AKT and digital quantitative analysis using the Aperio ImageScope software. The resulting data have been summarized in Fig. 2D–2H. Figure 2D–2G shows representative tissue cores from positive control samples of breast tumor tissue known to express high levels of p(ser473)AKT (Fig. 2D); negative control samples, where the primary antibody was omitted (Fig. 2E); vehicle-treated tumors (Fig. 2F), and QLT0267-treated tumors (Fig. 2G). p(ser473)AKT expression was quantified using the Aperio ImageScope positive-pixel algorithim (Fig. 2H). Surprisingly, after 7 consecutive, daily treatments, there was no statistically significant difference in p(ser473)AKT expression between vehicle- and QLT0267-treated tumors. From the data summarized in Fig. 2, it was hypothesized that QLT0267 mediated the suppression of ILK activity and, therefore, suppression of p(ser473)AKT occurred at earlier time-points and was no longer observed after 7 days, possibly due to redundant pathways mediating the phosphorylation of AKT. The following studies evaluated how QLT0267 influenced ILK-associated signaling pathways in vivo over a time course of 24 hr following administration. This kinetic approach allowed us to examine the effects of an ILK-targeted agent (QLT0267) in vivo when the established tumor was first exposed to the drug.
QLT0267 Pharmacokinetic Profile Indicates Clearance of the Drug from the Plasma Compartment Occurs within 24 Hours
When considering the time course of treatment effects, it is important to first have an understanding of drug concentration versus time profiles. A pharmacokinetic analysis of QLT0267 was performed to determine the rate of drug clearance from the plasma compartment. Animals were treated with 200 mg/kg of QLT0267 and blood was collected and processed at 2, 4, 6 and 24 hr, as described in the Materials & Methods. Results summarized in Fig. 3 indicate that, following oral administration of QLT0267, peak serum concentrations of the drug were observed at 2 hr after drug administration. By 6 hr, greater than 90% of the drug had been eliminated. The calculated AUC0-24h was 37,809 ng/ml per hr, as determined using WinNonlin software. Drug levels at the 24-hr time-point were still above the limits of quantification; however, 98% of the drug had been cleared by this time.
Figure 3.
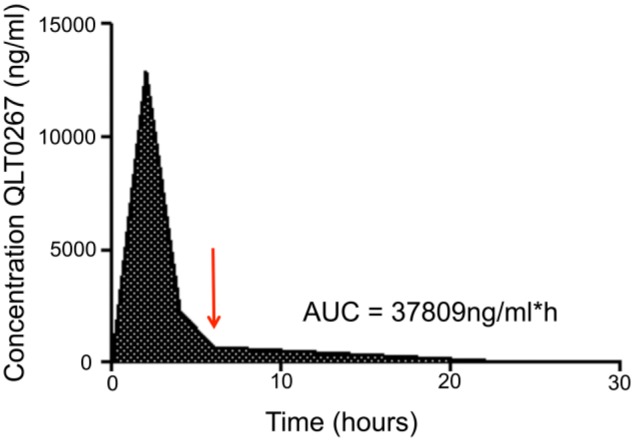
QLT0267 pharmacokinetic profile. QLT0267 treated animals (n=4 per time-point) were terminated by CO2 asphyxiation at 2, 4, 6 and 24 hr after treatment, and blood was collected by cardiac puncture. Subsequently, plasma was separated from blood cells and subject to LC-MS to determine concentrations of QLT0267 in plasma. The majority of QLT0267 was eliminated from the blood compartment within 6 hr (red arrow), where the AUC = 37,809 ng/ml per hr.
At Early Time Points, QLT0267 Suppresses Phosphorylation of AKT at Serine 473 while Altering Pro-apoptotic Markers, BAD and Caspase-3
For the following IHC studies, a TMA was constructed to evaluate changes in expression, activation, or subcellular localization of selected proteins following treatment using QLT0267 as a function of time. Animals were treated (p.o.) with 7 daily doses of QLT0267, and animals were sacrificed at 2, 4, 6, 24, 78, 126 and 168 hr (see Fig. 1). Tumor tissue was harvested and then preserved in paraffin blocks for TMA generation. Slides were subjected to IHC and digital quantitative analysis using the Aperio ImageScope software. The resulting data has been summarized in Figs. 4–8.
Figure 4.

Kinetics of p(ser473)AKT expression in vivo. (A and B) Quantification of p(ser473)AKT-positive pixels in TMA tumor cores from vehicle- and QLT0267-treated animals was performed using the Aperio ImageScope software. A decrease in positivity of p(ser473)AKT in the QLT0267-treated tumors is seen when compared to the vehicle-treated tumors at early time-points; 2–24 hr (p<0.005). After 24 hr, p(ser473) AKT levels rose beyond the expression seen in the vehicle-treated animals. p(ser473) AKT levels in vehicle-treated tumors declined with increasing tumor size. Representative p(ser473)AKT IHC images from tumors collected after 2 and 24 hr of vehicle and QLT0267 treatment are shown. (C) P(ser473)AKT expression was evaluated by visual scoring on whole tumor tissues from each time-point. Scale, 50 μm.
Figure 5.

QLT0267 triggers a change in the subcellular localization of the pro-apoptotic protein BAD. (A) In vitro examination of p(ser136)BAD expression shows that QLT0267-treated LCC6WT-luc cells have decreased expression of p(ser473)AKT and increased expression of activated BAD (p(ser136)BAD). (B) Immunofluorescence of BAD (red) and Bcl-xl (green) was examined after treatment. An alteration in the subcellular localization of BAD is observed (white arrows). (C) In vivo, the negative and positive control staining for BAD is provided using tonsil tissue. Primary antibody was omitted in the negative control. The positive control shows that BAD is localized to punctate structures in the cytoplasm (black arrow). (D) BAD expression was assessed in tumors from animals treated with vehicle or QLT0267 at 2 hr. Staining was quantified using the Aperio ImageScope software. (E) Representative tumor cores from the 2-hr time-point for vehicle-treated and QLT0267-treated tumors (n=4 and 8, respectively). In the tumors from animals treated with the vehicle control, BAD distribution appears to be diffuse and cytoplasmic, whereas, in tumor tissue from animals treated with QLT0267, BAD appears to be localized to puncta (black arrows). (F) The presence of puncta was evaluated using visual scoring, where no puncta was given a score of 0, low numbers of cells with puncta, a score of 1, and high numbers, a score of 2. Bar (B) 5 μm; (C, E) 10 μm.
Figure 6.

Caspase-3 expression is increased in tumors treated with QLT0267 at 6 hr. (A) In vitro analysis of Caspase3/7 activity was performed using a luminescence-based Caspase-3/7 assay. As shown, treatment using QLT0267 was able to promote increased Caspase activity in vitro with a peak at 12 hr after treatment. (B) Evaluation of Caspase-3 was performed on the full tissue microarray using digital quantification. A significant increase in Caspase-3 expression was observed at the 6-hr time-point (p<0.05). (C) Whole tissue sections were analyzed for Caspase-3 expression using the Aperio ImageScope Software. The whole tumor section was marked off as the ROI, except for the region outlined in green, which represents necrotic tissue plus a margin that was excluded from the analysis. The colorized image represents the mark-up after the positive pixel count algorithm was performed: blue represents negative pixels; orange, positive pixels; and red, strongly positive pixels. Note that the region selected as necrotic was not included in the mark-up image. (D) Quantification of all sections analyzed from the 6-hr time-point shows that QLT0267-treated tissues have significantly higher Caspase-3 activation than vehicle-treated tumors (n=6 and 4, respectively, p<0.05). (E) Positive control tonsil tissue exhibits single-positive cells scattered throughout negative tissue (see arrows). Vehicle-treated tumor tissues exhibit diffuse weak cytoplasmic staining, with some positive cells, whereas QLT0267-treated tumors show increased cytoplasmic staining and an increase in the number of positive cells. Scale (C) 100 μm; (E) 50 μm.
Figure 7.

TWIST expression is suppressed by QLT0267 in vivo. Quantification of TWIST staining on the full TMA was performed using Aperio ImageScope software. (A) Tonsil tissue was used as the negative control. At 24 hr, vehicle-treated tumor samples showed cytoplasmic, peripheral and nuclear staining of TWIST. Comparatively, QLT0267-treated tumors from the same time-point exhibited an overall decrease in TWIST, with little to no evidence of peripheral and nuclear staining. (B, C) Quantification of TWIST expression showed that TWIST is significantly decreased in QLT0267-treated tumors as compared to vehicle controls at 24 hr after the first treatment. (D) TWIST expression was also evaluated by visual scoring of the TMA cores from each time-point. Scale, 50 μm.
Figure 8.

GSK-3 is activated by QLT0267 in vivo. (A, B) Vehicle- and QLT0267-treated tumor cores were subject to p(ser9/21)GSK-3αβ staining and quantification using Aperio ImageScope software. Tonsil tissue exhibits very little moderate intensity staining. (C–F) Digital images of vehicle (C, D) and QLT0267 (E, F) cores indicate reduced p(ser9/21)GSK-3αβ in QLT0267-treated tumor cores as compared to vehicle-treated samples. The colorized mark-up image shows different levels of pixel intensity, where blue represents negative pixels; orange, moderately intense positive pixels; and red, strongly intense positive staining. Strong staining (red) is decreased in the tumor cores treated with QLT0267. Quantification of overall positivity (G), and strongly positive pixels based on higher threshold (H) was performed using the Aperio ImageScope software analysis. QLT0267-treated tumors show a significant decrease in positivity (p=0.0089) and strongly positive (p=0.0113) pixels as compared with vehicle-treated tumors (n=5 and 4, respectively). Scale (E, F) 10 μm.
There was a significant decrease in positivity (the number of positive pixels over total number of pixels) of p(ser473)AKT in the QLT0267-treated tumors as compared with the vehicle-treated tumors at time-points within the first 24 hr of treatment [2 hr (p=0.0095), 24 hr (p=0.0046); Fig. 4B). After 24 hr, p(ser473)AKT levels began to normalize and even exceed that seen in the vehicle treatment group. These data are consistent with in vitro data collected by our laboratory and published by Verreault and Bally (2009), which suggests that suppression of p(ser473)AKT is transient and may instigate compensatory pathways leading to the activity of AKT. Interestingly, we also show that, as tumors increase in size, both control, untreated tumors (data not shown) and vehicle-treated tumors have reduced levels of p(ser473)AKT. Figure 4B shows representative images of p(ser473)AKT staining in tumors from animals treated with vehicle or QLT0267 for 2 or 24 hr. These images illustrate that p(ser473)AKT staining is cytoplasmic and diffuse in the tumors from both vehicle- and QLT0267-treated animals. To validate the TMA and digital quantification of IHC methodology, p(ser473)AKT expression was evaluated by visual scoring of whole tumor tissues from each time-point. These data are summarized in Fig. 4C. As shown, scoring of tissues obtained from treated animals for p(ser473)AKT is low at early time-points; however, levels recover after 24 hr.
In an effort to establish the molecular mechanism through which QLT0267 causes growth suppression (see Fig. 2A and 2B), LCC6WT-luc cells were examined for alterations in pro-apoptotic marker expression that are downstream of AKT. Preliminary in vitro studies, shown in Fig. 5A and 5B, indicate that the pro-apoptotic protein BAD experiences changes in activity and subcellular localization in response to QLT0267. Figure 5A is a representative western blot image that shows when LCC6WT-luc cells have been treated with QLT0267 in vitro, expression of p(ser473)AKT decreases while the expression of activated BAD (p(ser136)BAD) increases. Figure 5B illustrates BAD (red) and Bcl-xl (green) immunofluorescence staining in LCC6WT-luc cells that have been treated with vehicle or QLT0267 in vitro. After 4 hr of treatment, BAD looks to have been translocated to subcellular structures, as indicated by the perinuclear pattern that appears in treated cells (Fig. 5B, see white arrows). TMA tumor cores were assessed for the expression of BAD and Caspase-3 in order to determine if the tumor growth suppression observed in Fig. 2 is perhaps due to an early induction of apoptosis. Figure 5C shows the negative control for BAD expression, where samples were treated with secondary antibody alone. The positive control for BAD expression was tonsil tissue, where apoptosis is known to occur regularly in the germinal centers. Note, in the positive control tonsil tissue, BAD appears punctate in the cytoplasm (Fig. 5C, arrow). BAD expression was quantified using the Aperio ImageScope pixel counting algorithm at 2 hr post treatment. No statistically significant difference in the expression of BAD between the tumor cores derived from vehicle- or QLT0267-treated animals was observed through visual inspection of the TMA or through performing digital quantification (Fig. 5D). This was consistent with results from 4, 6 and 24 hr (data not shown). Upon closer examination of the tumor tissue, however, a change in the sub-cellular localization of BAD was noted in tumor cores obtained from animals treated with QLT0267 for 2 hr. Figure 5E shows representative images of tumor cores from the 2-hr time-point for vehicle-treated and QLT0267-treated animals; in the tumor tissue from mice treated with the vehicle control, BAD is diffuse within the cytoplasm, whereas in tumor tissue from animals treated with QLT0267, BAD appears within distinct puncta. It is known that mitochondrial localization of BAD occurs when AKT is inactive (Le Bras et al. 2006; Thompson and Thompson 2004). Thus, we speculate that inhibition of AKT 2 hr after treatment with QLT0267 may lead to an activation of BAD, its localization to the mitochondrial membrane, and the beginning of the intrinsic apoptotic pathway. Scoring of tumor cores from the 2-hr time-point for the presence of puncta was done on a scale of 0 to 2. These data are summarized in Fig. 5F. Staining for p(136)BAD was attempted in order to determine whether treatment with QLT0267 was able to activate BAD; however, three commercially available antibodies for p(136)BAD could not be validated using the IHC methods described.
In vitro, cells were treated with QLT0267 for 12, 24, 46 and 72 hr and subjected to a luminescence-based Caspase-3/7 activity assay in vitro to confirm that treatment of cells using QLT0267 is capable of initiating apoptosis. The results summarized in Fig. 6A demonstrate that treated cells have higher Caspase-3 activity than untreated controls. However, the activity of Caspase-3 post-treatment is not sustained; rather, the highest level of Caspase-3 activity is observed at 12 hr after treatment. To determine whether apoptosis was actively occurring in treated tumors, TMA cores were examined for Caspase-3 expression. Quantification of Caspase-3 expression in vehicle- and QLT0267-treated tumors was accomplished across the entire time-course (2, 4, 6, 24, 72, 126, and 168 hr; Fig. 1) using the constructed TMA. A preliminary analysis revealed a significant (p=0.045) peak in Caspase-3 expression at 6 hr in tumors from QLT0267-treated mice when compared to tumors from mice treated with the vehicle control (Fig. 6B). Because tumor cores were collected from regions of the whole tissue that appear seemingly healthy, the analysis of Caspase-3 activity at the 6-hr time-point was also subjected to evaluation in whole tumor tissue sections from vehicle- and QLT0267-treated animals. Figure 6C is a representative image of a whole tumor section, where the analyzed ROI included the viable tumor tissue outlined in red but excluded necrotic tissue (and a tumor margin) outlined in green. The colorized image from Fig. 6C represents the mark-up of the pixels performed by Aperio ImageScope software, where blue represents negative pixels, and orange/red represents Caspase-3-positive pixels. Figure 6D shows the average pixel positivity in whole tumor sections from vehicle-treated and QLT0267-treated animals at the 6-hr time-point after the first dose, illustrating that higher Caspase-3 expression was observed in tumor samples obtained from QLT0267-treated animals compared with vehicle controls (p=0.055). Figure 6E shows a close-up of the representative images from the positive control tonsil tissue, tumors from vehicle-treated animals, and tumors from animals treated with QLT0267.
In Vivo Expression of Two ILK Effectors, TWIST and pGSK3, Is Suppressed following a Single Dose of QLT0267
In order to determine whether the inhibitory effects of QLT0267 are specific to ILK signaling, the expression of two additional downstream effectors of ILK—TWIST and GSK—was evaluated on the TMA. TWIST is a transcription factor that is up-regulated in breast cancers and has been shown to influence Y-box protein binding 1 (YB-1) expression (Shiota et al. 2008). Recently, it was demonstrated that ILK influences TWIST expression; more specifically, application of QLT0267 to breast cancer cells in culture led to suppression of TWIST, YB-1 and Her2/neu (Kalra et al. 2010). To determine whether a QLT0267-mediated suppression of TWIST also occurs in vivo, the TMA was evaluated for TWIST expression using digital quantification as well as visual scoring across all time-points. As seen in Fig. 7A, tonsil tissue was negative for TWIST expression. Representative images from vehicle- and QLT0267-treated tumors (Fig. 7A) demonstrated that TWIST expression is decreased in QLT0267-treated tumors at the 24-hr time-point. Figure 7B and 7C show the quantification of IHC data, illustrating that TWIST is significantly lower in QLT0267-treated tumors as compared to vehicle controls at 24 hr after the first treatment. As validation for digital quantification, visual inspection and scoring of the tumor cores was done on a scale of 0 to 3. These data are summarized in Fig. 7D.
GSK-3 is a serine/threonine kinase involved in glycogen metabolism, cell development, gene transcription, protein translation, cytoskeletal organization, cell cycle regulation, proliferation, and apoptosis (Phukan et al. 2010). GSK-3 is active in resting cells and its activity is inhibited when phosphorylated by ILK and AKT (McDonald et al. 2008; Yoganathan et al. 2000). TMA sections were assessed for p(ser9/21)GSK-3αβ expression by IHC at the 24-hr time-point following treatment initiation. As seen in Fig. 8A, tonsil tissue was mostly negative for p(ser9/21)GSK-3αβ. Figure 8B shows a colorized mark-up of the IHC image, differentiating negative (blue), moderately positive (orange), and strongly positive (red) pixels. Representative images from vehicle- (Fig. 8C and 8D) and QLT0267 (Fig. 8E and 8F)-treated tumors demonstrate that pGSK expression is decreased in QLT0267-treated tumors at the 24-hr time-point. Figure 8G and 8H represent the quantification of overall positivity and only strongly positive staining, respectively. The quantification results indicate that tumors from QLT0267-treated animals exhibit a small, albeit significant, decrease in overall positivity (Fig. 8G). On visual inspection, we noted that the staining of p(ser9/21)GSK-3αβ was generally very positive but showed some variability in intensity between treatment groups. This observation gave us an opportunity to use the intensity function of the digital quantification software. Using the Aperio ImageScope positive pixel algorithm, it is possible to stratify pixels according to the intensity of staining; therefore it was possible to separately quantify strongly positive p(ser9/21)GSK-3αβ staining from moderate or weak staining. A significant (p=0.0113) decrease in strongly positive staining (Fig. 8H) was seen in the QLT0267-treated tumors compared to vehicle-treated tumors.
Discussion
The primary objective for this study was to establish that several tools, such as non-invasive imaging, pharmacokinetics, TMA, IHC, and digital image analysis, can be used to rapidly evaluate multiple endpoints in a preclinical drug efficacy study. Potentially, these same tools could be used to assess the action of this drug when used to treat patients. It is recognized that analysis of protein expression by IHC methods (similar to western blot analysis) are qualitative in nature. In an attempt to address this, digital scanning and image analysis software were used to evaluate IHC staining. TMA cores were evaluated by visual scoring procedures using a microscope, and digitized images of the TMA were analyzed using pixel-counting algorithms from Aperio ImageScope software to quantify alterations in molecular expression as well as changes in staining intensity. Visual scoring proved time-consuming and was less effective at picking up the subtle changes in marker expression, whereas digital quantification enabled the use of high-resolution, high-magnification images to count positive pixels and to assess for changes in marker localization. Together, the use of TMA and digital quantification enabled a streamlined and more quantitative approach to the assessment of marker expression following the delivery of QLT0267 in animals with established tumors. There has been some discussion regarding the use of digital quantification in clinical pathology. Although the utility of pixel counting algorithms has proven to be similar to pathologist visual scoring of clinical samples, the use of automated technology may, at this time, still require the input of a pathologist for region selection and quality assurance (Cantaloni et al. 2011; Lloyd et al. 2010; Potts 2009; Prasad and Prabhu 2011; Rexhepaj et al. 2008; Rojo et al. 2009; Slodkowska et al. 2011). Newer, trainable systems are being evaluated, and the technology is certainly improving (Nassar et al. 2011). To date, this technology has not been employed in a meaningful way to support preclinical studies looking at drug efficacy and mechanism of action. We believe that this type of mechanistic study is an important step in the design of a robust, preclinical study of a targeted treatment. We also believe that this analytical strategy can be employed in Phase 0 clinical studies to establish that drug-mediated effects on targeted pathways are maintained in patients. In this study, it is demonstrated that the digital scanning and pixel counting of IHC is a useful tool for evaluating shifts in signal transduction. Furthermore, we were able to optimize our data collection by using a systemic approach that considered time as a key variable: (1) BLI was employed to determine the earliest time-point at which treatment is effective at suppressing tumor growth; (2) Pharmacokinetic data helped to define the time-point at which drug concentration is highest in the blood compartment; and (3) TMA/IHC and digital analysis enabled the examination of marker expression as a function of time. This study provides a framework on which future drug efficacy studies, both preclinical and clinical, can be designed.
The secondary objective of this study was to elucidate the mechanism through which ILK inhibition may lead to reduced tumor burden in vivo. Using IHC, the signaling consequences of ILK inhibition was evaluated in an animal model of breast cancer. Although it is recognized from in vitro studies of breast tumor cells that ILK inhibition or ILK silencing can reduce p(ser473)AKT levels (Kalra et al. 2009; McDonald et al. 2008), it is not clear whether these changes are transient or whether lasting changes occur in vivo. As a precursor to the in vivo studies described here, we had noted that, in three different breast cancer cell lines (MDA-MB-435/LCC6, JIMT-1, and SKBR-3), silencing of ILK using ILK siRNA was able to abrogate phosphorylation of AKT at least initially (between 12 and 24 hr). However, levels of p(ser473)AKT would rise back to baseline within 24–48 hr with sustained loss of ILK expression in vitro (unpublished data). It should be noted that a previous study has also demonstrated that ablation of ILK signaling caused only a short-lived suppression of p(ser473)AKT in vitro (Verreault and Bally 2009). Many studies to date indicate that the AKT pathway, being central to a number of homeostatic mechanisms within the cell, is likely controlled by multiple redundant pathways (Logue and Morrison 2012). Where one pathway leading to the phosphorylation of AKT is inhibited (for example, ILK), another may be activated to compensate (such as mTORC2/Rictor or IGF). In this study, we illustrate that QLT0267 was able to inhibit tumor growth after one week of daily dosing, as measured by bioluminescence (Fig. 2B, 2C). However, tumors harvested on the last day of the study exhibited no significant changes in p(ser473)AKT expression, as determined using immunohistochemistry (Fig. 2H). A pharmacokinetic profile of QLT067 showed that plasma drug levels rapidly decline within the first 6 hr after administration (Fig. 3). In light of these results, the remaining studies focused on early changes in signaling that occur when the tumors are first exposed to QLT0267. A TMA was constructed from treated and untreated tumors collected in the first 24 hr after a single treatment and from samples collected after subsequent dosing for 7 days (Fig. 1). The rationale for using a TMA in this study was based on the need to evaluate the expression of selected markers in a large number of samples simultaneously. Using the TMA, we were able to establish a time course for changes in each of the markers examined. Interestingly, we observe that, within 24 hr of treatment, p(ser473)AKT declines, however, these changes are seemingly transient. For example, a significant decrease in p(ser473)AKT was noted within 2 hr after QLT0267 was administered (Fig. 4) and the effect was maintained, albeit at a reduced level, for 24 hr. This result confirms our expectations, but it is not clear why the effect was not sustainable after 7 days of daily treatment (Fig. 2H). We hypothesize that signal ablation follows the same kinetic pattern of QLT0267 pharmacokinetics; thus, the changes in signal transduction are only transient, peaking when the drug concentration is at its peak. In Fig. 3 we show that QLT0267 is cleared from the plasma compartment within 6 hr and, by 24 hr, little to no drug remains. Our study of the TMA shows that the abrogation of ILK effectors is observed in the first few hours after treatment. Thereafter, the changes are no longer significant. Each of the subsequent tumor harvests are performed 6 hr after dosing (at 30 hr, 78 hr 126 hr) and the final harvest is done 30 hr after the last treatment. For each of the subsequent doses, we believe that any suppression of ILK signaling would have already occurred within the critical 6-hr time-point and the suppression is not maintained at the time of tumor harvest.
As noted, QLT0267 is able to cause growth inhibition in the LCC6WT-Luc breast cancer model, and it is possible that early suppression of p(ser473)AKT is contributing to this effect. For this reason, the survival pathway downstream of AKT was evaluated for changes in the first 24 hr after treatment. Following first exposure to QLT0267, p(ser473)AKT levels were reduced, and evidence for initiation of the intrinsic pathway to apoptosis was observed (Figs. 5, 6). A change in the subcellular localization of BAD (Fig. 5), and activation of Caspase-3 (Fig. 6) has been documented to occur at 6 hr. This novel in vivo evidence supports: (1) a link between inhibition of ILK, suppression of p(ser473)AKT, and activation of BAD, and (2) induction of apoptosis as the mechanism of action for QLT0267. We also provide evidence that QLT0267 treatment affected pathways involving EMT (TWIST) (Fig. 7) and cell metabolism (GSK-3) (Fig. 8) in vivo. Taken together, the data suggests that the inhibition of ILK signaling had early effects on an orthotopic model of breast cancer that could be important in the drugs’ action to inhibit tumor progression.
Acknowledgments
This work would not have been possible if not for the excellent animal care support that is provided through the BC Cancer Agency’s Animal Resource Center. Dr Robert Clarke (Georgetown University, Washington, DC, USA) generously donated MDA-MB-435 (LCC6) estrogen receptor-negative breast cancer cells. Center for Translation and Applied Genomics did TMA construction and IHC. Monika Fetijak (Faculty of Medicine, UBC, British Columbia, Canada) digitized slides using the Aperio ScanScope System. Jiang-Song Tao at QLT Inc completed the pharmacokinetic studies. Corinna Warburton contributed the LCC6WT-Luc cell line. Hong Yan provided cell culture support. Dita Strutt, Young-Joo Yang, Maryam Oosely, and Dana Masin provided animal study support. This work was supported by the Canadian Institute for Health Research [grant number MOP 106638].
Footnotes
Competing Interests: The authors declared no potential competing interests with respect to the research, authorship, and/or publication of this article.
Author’s Contributions: JK designed and executed all experiments and data analysis and wrote as well as revised the manuscript. WD helped revise the original manuscript. MB is Chief Investigator, and assisted in the study designs, and reviewed the original manuscript.
References
- Ahmed N, Oliva K, Rice GE, Quinn MA. (2004). Cell-free 59 kDa immunoreactive integrin-linked kinase: a novel marker for ovarian carcinoma. Clin Cancer Res 10:2415-2420. [DOI] [PubMed] [Google Scholar]
- Ahmed N, Riley C, Oliva K, Stutt E, Rice GE, Quinn MA. (2003). Integrin-linked kinase expression increases with ovarian tumour grade and is sustained by peritoneal tumour fluid. J Pathol 201:229-237. [DOI] [PubMed] [Google Scholar]
- Assi K, Mills J, Owen D, Ong C, Arnaud R, Dedhar S, Salh B. (2008). Integrin-linked kinase regulates cell proliferation and tumour growth in murine colitis-associated carcinogenesis. Gut 57:931-940. [DOI] [PubMed] [Google Scholar]
- Bachmeier BE, Nerlich AG, Mirisola V, Jochum M, Pfeffer U. (2008). Lineage infidelity and expression of melanocytic markers in human breast cancer. Int J Oncol 33:1011-1015. [PubMed] [Google Scholar]
- Becker-Santos DD, Guo Y, Ghaffari M, Vickers ED, Lehman M, Altamirano-Dimas M, Oloumi A, Furukawa J, Sharma M, Wang Y, Dedhar S, Cox ME. (2012). Integrin-linked kinase as a target for ERG-mediated invasive properties in prostate cancer models. Carcinogenesis 33:2558-2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravou V, Klironomos G, Papadaki E, Stefanou D, Varakis J. (2003). Integrin-linked kinase (ILK) expression in human colon cancer. Br J Cancer 89:2340-2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravou V, Klironomos G, Papadaki E, Taraviras S, Varakis J. (2006). ILK over-expression in human colon cancer progression correlates with activation of beta-catenin, down-regulation of E-cadherin and activation of the Akt-FKHR pathway. J Pathol 208:91-99. [DOI] [PubMed] [Google Scholar]
- Brazdziute E, Laurinavicius A. (2011). Digital pathology evaluation of complement C4d component deposition in the kidney allograft biopsies is a useful tool to improve reproducibility of the scoring. Diagn Pathol 6 Suppl 1:S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabodi S, del Pilar Camacho-Leal M, Di Stefano P, Defilippi P. (2010). Integrin signalling adaptors: not only figurants in the cancer story. Nat Rev Cancer 10:858-870. [DOI] [PubMed] [Google Scholar]
- Cantaloni C, Tonini RE, Eccher C, Morelli L, Leonardi E, Bragantini E, Aldovini D, Fasanella S, Ferro A, Cazzolli D, Berlanda G, Dalla Palma P, Barbareschi M. (2011). Diagnostic Value of Automated Her2 Evaluation in Breast Cancer: A Study on 272 Equivocal (score 2+) Her2 Immunoreactive Cases Using an FDA Approved System. Appl Immunohistochem Mol Morphol 19:306-312. [DOI] [PubMed] [Google Scholar]
- Chambers AF. (2009). MDA-MB-435 and M14 cell lines: identical but not M14 melanoma? Cancer Res 69:5292-5293. [DOI] [PubMed] [Google Scholar]
- Chen D, Zhang Y, Zhang X, Li J, Han B, Liu S, Wang L, Ling Y, Mao S, Wang X. (2012). Overexpression of integrin-linked kinase correlates with malignant phenotype in non-small cell lung cancer and promotes lung cancer cell invasion and migration via regulating epithelial-mesenchymal transition (EMT)-related genes. Acta Histochem [DOI] [PubMed] [Google Scholar]
- Chen Z, Yang A, Xu C, Xing Y, Gong W, Li J. (2011). c-Jun N-terminal kinase is involved in the regulation of proliferation and apoptosis by integrin-linked kinase in human retinoblastoma cells. Graefes Arch Clin Exp Ophthalmol 249:1399-1407. [DOI] [PubMed] [Google Scholar]
- Chung DH, Lee JI, Kook MC, Kim JR, Kim SH, Choi EY, Park SH, Song HG. (1998). ILK (beta1-integrin-linked protein kinase): a novel immunohistochemical marker for Ewing’s sarcoma and primitive neuroectodermal tumour. Virchows Arch 433:113-117. [DOI] [PubMed] [Google Scholar]
- Conley FK. (1979). Development of a metastatic brain tumor model in mice. Cancer Res 39:1001-1007. [PubMed] [Google Scholar]
- Dai DL, Makretsov N, Campos EI, Huang C, Zhou Y, Huntsman D, Martinka M, Li G. (2003). Increased expression of integrin-linked kinase is correlated with melanoma progression and poor patient survival. Clin Cancer Res 9:4409-4414. [PubMed] [Google Scholar]
- Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. (1998). Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci U S A 95:11211-11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng JT, Sutherland C, Brautigan DL, Eto M, Walsh MP. (2002). Phosphorylation of the myosin phosphatase inhibitors, CPI-17 and PHI-1, by integrin-linked kinase. Biochem J 367:517-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobreva I, Fielding A, Foster LJ, Dedhar S. (2008). Mapping the integrin-linked kinase interactome using SILAC. J Proteome Res 7:1740-1749. [DOI] [PubMed] [Google Scholar]
- Edwards LA, Thiessen B, Dragowska WH, Daynard T, Bally MB, Dedhar S. (2005). Inhibition of ILK in PTEN-mutant human glioblastomas inhibits PKB/Akt activation, induces apoptosis, and delays tumor growth. Oncogene 24:3596-3605. [DOI] [PubMed] [Google Scholar]
- Edwards LA, Woo J, Huxham LA, Verreault M, Dragowska WH, Chiu G, Rajput A, Kyle AH, Kalra J, Yapp D, Yan H, Minchinton AI, Huntsman D, Daynard T, Waterhouse DN, Thiessen B, Dedhar S, Bally MB. (2008). Suppression of VEGF secretion and changes in glioblastoma multiforme microenvironment by inhibition of integrin-linked kinase (ILK). Mol Cancer Ther 7:59-70. [DOI] [PubMed] [Google Scholar]
- Eke I, Leonhardt F, Storch K, Hehlgans S, Cordes N. (2009). The small molecule inhibitor QLT0267 Radiosensitizes squamous cell carcinoma cells of the head and neck. PLoS One 4:e6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faralli JA, Newman JR, Sheibani N, Dedhar S, Peters DM. (2011). Integrin-linked kinase regulates integrin signaling in human trabecular meshwork cells. Invest Ophthalmol Vis Sci 52:1684-1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielding AB, Lim S, Montgomery K, Dobreva I, Dedhar S. (2011). A critical role of integrin-linked kinase, ch-TOG and TACC3 in centrosome clustering in cancer cells. Oncogene 30:521-534. [DOI] [PubMed] [Google Scholar]
- Graff JR, Deddens JA, Konicek BW, Colligan BM, Hurst BM, Carter HW, Carter JH. (2001). Integrin-linked kinase expression increases with prostate tumor grade. Clin Cancer Res 7:1987-1991. [PubMed] [Google Scholar]
- Guo W, Jiang H, Gray V, Dedhar S, Rao Y. (2007). Role of the integrin-linked kinase (ILK) in determining neuronal polarity. Dev Biol 306:457-468. [DOI] [PubMed] [Google Scholar]
- Hannigan GE, McDonald PC, Walsh MP, Dedhar S. (2011). Integrin-linked kinase: not so ‘pseudo’ after all. Oncogene 30:4375-4385. [DOI] [PubMed] [Google Scholar]
- Imanishi Y, Hu B, Jarzynka MJ, Guo P, Elishaev E, Bar-Joseph I, Cheng SY. (2007). Angiopoietin-2 stimulates breast cancer metastasis through the alpha(5)beta(1) integrin-mediated pathway. Cancer Res 67:4254-4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito R, Oue N, Zhu X, Yoshida K, Nakayama H, Yokozaki H, Yasui W. (2003). Expression of integrin-linked kinase is closely correlated with invasion and metastasis of gastric carcinoma. Virchows Arch 442:118-123. [DOI] [PubMed] [Google Scholar]
- Jones CI, Tucker KL, Sasikumar P, Sage T, Kaiser WJ, Moore C, Emerson M, Gibbins JM. (2014). Integrin-linked kinase regulates the rate of platelet activation and is essential for the formation of stable thrombi. J Thromb Haemost 12:1342-1352. [DOI] [PubMed] [Google Scholar]
- Joshi MB, Ivanov D, Philippova M, Erne P, Resink TJ. (2007). Integrin-linked kinase is an essential mediator for T-cadherin-dependent signaling via Akt and GSK3beta in endothelial cells. FASEB J 21:3083-3095. [DOI] [PubMed] [Google Scholar]
- Kalra J, Anantha M, Warburton C, Waterhouse D, Yan H, Yang YJ, Strut D, Osooly M, Masin D, Bally MB. (2011). Validating the use of a luciferase labeled breast cancer cell line, MDA435LCC6, as a means to monitor tumor progression and to assess the therapeutic activity of an established anticancer drug, docetaxel (Dt) alone or in combination with the ILK inhibitor, QLT0267. Cancer Biol Ther 11:826-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra J, Sutherland BW, Stratford AL, Dragowska W, Gelmon KA, Dedhar S, Dunn SE, Bally MB. (2010). Suppression of Her2/neu expression through ILK inhibition is regulated by a pathway involving TWIST and YB-1. Oncogene 29:6343-6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra J, Warburton C, Fang K, Edwards L, Daynard T, Waterhouse D, Dragowska W, Sutherland BW, Dedhar S, Gelmon K, Bally M. (2009). QLT0267, a small molecule inhibitor targeting integrin-linked kinase (ILK), and docetaxel can combine to produce synergistic interactions linked to enhanced cytotoxicity, reductions in P-AKT levels, altered F-actin architecture and improved treatment outcomes in an orthotopic breast cancer model. Breast Cancer Res 11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koul D, Shen R, Bergh S, Lu Y, de Groot JF, Liu TJ, Mills GB, Yung WK. (2005). Targeting integrin-linked kinase inhibits Akt signaling pathways and decreases tumor progression of human glioblastoma. Mol Cancer Ther 4:1681-1688. [DOI] [PubMed] [Google Scholar]
- Le Bras M, Rouy I, Brenner C. (2006). The modulation of inter-organelle cross-talk to control apoptosis. Med Chem 2:1-12. [DOI] [PubMed] [Google Scholar]
- Lee SL, Hsu EC, Chou CC, Chuang HC, Bai LY, Kulp SK, Chen CS. (2011). Identification and characterization of a novel integrin-linked kinase inhibitor. J Med Chem 54:6364-6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonessa F, Green D, Licht T, Wright A, Wingate-Legette K, Lippman J, Gottesman MM, Clarke R. (1996). MDA435/LCC6 and MDA435/LCC6MDR1: ascites models of human breast cancer. Br J Cancer 73:154-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tan X, Dai C, Stolz DB, Wang D, Liu Y. (2009). Inhibition of integrin-linked kinase attenuates renal interstitial fibrosis. J Am Soc Nephrol 20:1907-1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yang J, Dai C, Wu C, Liu Y. (2003). Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J Clin Invest 112:503-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang J, Yan H. (2013). Integrin-linked kinase inhibition attenuates permeability of the streptozotocin-induced diabetic rat retina. Cell Biochem Biophys 67:1467-1472. [DOI] [PubMed] [Google Scholar]
- Lim S, Kawamura E, Fielding AB, Maydan M, Dedhar S. (2013). Integrin-linked kinase regulates interphase and mitotic microtubule dynamics. PLoS One 8:e53702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Costello PC, Pham NA, Pintillie M, Jabali M, Sanghera J, Tsao MS, Johnston MR. (2006). Integrin-linked kinase inhibitor KP-392 demonstrates clinical benefits in an orthotopic human non-small cell lung cancer model. J Thorac Oncol 1:771-779. [PubMed] [Google Scholar]
- Liu Q, Xiao L, Yuan D, Shi X, Li P. (2012). Silencing of the integrin-linked kinase gene induces the apoptosis in ovarian carcinoma. J Recept Signal Transduct Res 32:120-127. [DOI] [PubMed] [Google Scholar]
- Lloyd MC, Allam-Nandyala P, Purohit CN, Burke N, Coppola D, Bui MM. (2010). Using image analysis as a tool for assessment of prognostic and predictive biomarkers for breast cancer: How reliable is it? J Pathol Inform 1:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue JS, Morrison DK. (2012). Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev 26:641-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maydan M, McDonald PC, Sanghera J, Yan J, Rallis C, Pinchin S, Hannigan GE, Foster LJ, Ish-Horowicz D, Walsh MP, Dedhar S. (2010). Integrin-linked kinase is a functional Mn2+-dependent protein kinase that regulates glycogen synthase kinase-3beta (GSK-3beta) phosphorylation. PLoS One 5:e12356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald PC, Fielding AB, Dedhar S. (2008). Integrin-linked kinase–essential roles in physiology and cancer biology. J Cell Sci 121:3121-3132. [DOI] [PubMed] [Google Scholar]
- Mills J, Digicaylioglu M, Legg AT, Young CE, Young SS, Barr AM, Fletcher L, O’Connor TP, Dedhar S. (2003). Role of integrin-linked kinase in nerve growth factor-stimulated neurite outgrowth. J Neurosci 23:1638-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montel V, Suzuki M, Galloy C, Mose ES, Tarin D. (2009). Expression of melanocyte-related genes in human breast cancer and its implications. Differentiation 78:283-291. [DOI] [PubMed] [Google Scholar]
- Muranyi AL, Dedhar S, Hogge DE. (2010). Targeting integrin linked kinase and FMS-like tyrosine kinase-3 is cytotoxic to acute myeloid leukemia stem cells but spares normal progenitors. Leuk Res 34:1358-1365. [DOI] [PubMed] [Google Scholar]
- Muranyi AL, Dedhar S, Hogge DE. (2009). Combined inhibition of integrin linked kinase and FMS-like tyrosine kinase 3 is cytotoxic to acute myeloid leukemia progenitor cells. Exp Hematol 37:450-460. [DOI] [PubMed] [Google Scholar]
- Naska S, Park KJ, Hannigan GE, Dedhar S, Miller FD, Kaplan DR. (2006). An essential role for the integrin-linked kinase-glycogen synthase kinase-3 beta pathway during dendrite initiation and growth. J Neurosci 26:13344-13356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar A, Cohen C, Agersborg SS, Zhou W, Lynch KA, Albitar M, Barker EA, Vanderbilt BL, Thompson J, Heyman ER, Lange H, Olson A, Siddiqui MT. (2011). Trainable Immunohistochemical HER2/ neu Image Analysis: A Multisite Performance Study Using 260 Breast Tissue Specimens. Arch Pathol Lab Med 135:896-902. [DOI] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW. (2006). A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10:515-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara S, Nakata M, Takeshima H, Katagiri H, Asano T, Oka Y, Maruyama I, Kuratsu J. (2004). Integrin-linked kinase (ILK) regulation of the cell viability in PTEN mutant glioblastoma and in vitro inhibition by the specific COX-2 inhibitor NS-398. Cancer Lett 208:115-122. [DOI] [PubMed] [Google Scholar]
- Oloumi A, Syam S, Dedhar S. (2006). Modulation of Wnt3a-mediated nuclear beta-catenin accumulation and activation by integrin-linked kinase in mammalian cells. Oncogene 25:7747-7757. [DOI] [PubMed] [Google Scholar]
- Persad S, Attwell S, Gray V, Mawji N, Deng JT, Leung D, Yan J, Sanghera J, Walsh MP, Dedhar S. (2001). Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase: critical roles for kinase activity and amino acids arginine 211 and serine 343. J Biol Chem 276:27462-27469. [DOI] [PubMed] [Google Scholar]
- Phukan S, Babu VS, Kannoji A, Hariharan R, Balaji VN. (2010). GSK3beta: role in therapeutic landscape and development of modulators. Br J Pharmacol 160:1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts SJ. (2009). Digital pathology in drug discovery and development: multisite integration. Drug Discov Today 14:935-941. [DOI] [PubMed] [Google Scholar]
- Prasad K, Prabhu GK. (2011). Image Analysis Tools for Evaluation of Microscopic Views of Immunohistochemically Stained Specimen in Medical Research-a Review. J Med Syst [DOI] [PubMed] [Google Scholar]
- Rae JM, Creighton CJ, Meck JM, Haddad BR, Johnson MD. (2007). MDA-MB-435 cells are derived from M14 melanoma cells–a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res Treat 104:13-19. [DOI] [PubMed] [Google Scholar]
- Rexhepaj E, Brennan DJ, Holloway P, Kay EW, McCann AH, Landberg G, Duffy MJ, Jirstrom K, Gallagher WM. (2008). Novel image analysis approach for quantifying expression of nuclear proteins assessed by immunohistochemistry: application to measurement of oestrogen and progesterone receptor levels in breast cancer. Breast Cancer Res 10:R89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo MG, Bueno G, Slodkowska J. (2009). Review of imaging solutions for integrated quantitative immunohistochemistry in the Pathology daily practice. Folia Histochem Cytobiol 47:349-354. [DOI] [PubMed] [Google Scholar]
- Rosano L, Spinella F, Di Castro V, Dedhar S, Nicotra MR, Natali PG, Bagnato A. (2006). Integrin-linked kinase functions as a downstream mediator of endothelin-1 to promote invasive behavior in ovarian carcinoma. Mol Cancer Ther 5:833-842. [DOI] [PubMed] [Google Scholar]
- Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, Van de, Rijn M, Waltham M, Pergamenschikov A, Lee JC, Lashkari D, Shalon D, Myers TG, Weinstein JN, Botstein D, Brown PO. (2000). Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet 24:227-235. [DOI] [PubMed] [Google Scholar]
- Santos ND, Habibi G, Wang M, Law JH, Andrews HN, Wei D, Triche T, Dedhar S, Dunn SE. (2007). Urokinase-type plasminogen activator (uPA) is inhibited with QLT0267 a small molecule targeting integrin-linked kinase (ILK). Transl Oncogenomics 2:85-97. [PMC free article] [PubMed] [Google Scholar]
- Sawai H, Okada Y, Funahashi H, Matsuo Y, Takahashi H, Takeyama H, Manabe T. (2006). Integrin-linked kinase activity is associated with interleukin-1 alpha-induced progressive behavior of pancreatic cancer and poor patient survival. Oncogene 25:3237-3246. [DOI] [PubMed] [Google Scholar]
- Sellappan S, Grijalva R, Zhou X, Yang W, Eli MB, Mills GB, Yu D. (2004). Lineage infidelity of MDA-MB-435 cells: expression of melanocyte proteins in a breast cancer cell line. Cancer Res 64:3479-3485. [DOI] [PubMed] [Google Scholar]
- Serrano I, McDonald PC, Lock F, Muller WJ, Dedhar S. (2013). Inactivation of the Hippo tumour suppressor pathway by integrin-linked kinase. Nat Commun 4:2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota M, Izumi H, Onitsuka T, Miyamoto N, Kashiwagi E, Kidani A, Hirano G, Takahashi M, Naito S, Kohno K. (2008). Twist and p53 reciprocally regulate target genes via direct interaction. Oncogene 27:5543-5553. [DOI] [PubMed] [Google Scholar]
- Sikkema WK, Strikwerda A, Sharma M, Assi K, Salh B, Cox ME, Mills J. (2014). Regulation of mitotic cytoskeleton dynamics and cytokinesis by integrin-linked kinase in retinoblastoma cells. PLoS One 9:e98838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slodkowska J, Markiewicz T, Grala B, Kozlowski W, Papierz W, Pleskacz K, Murawski P. (2011). Accuracy of a remote quantitative image analysis in the whole slide images. Diagn Pathol 6 Suppl 1:S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbrunn T, Siegmund D, Andrulis M, Grella E, Kortum M, Einsele H, Wajant H, Bargou RC, Stuhmer T. (2012). Integrin-linked kinase is dispensable for multiple myeloma cell survival. Leuk Res 36:1165-1171. [DOI] [PubMed] [Google Scholar]
- Tabe Y, Jin L, Tsutsumi-Ishii Y, Xu Y, McQueen T, Priebe W, Mills GB, Ohsaka A, Nagaoka I, Andreeff M, Konopleva M. (2007). Activation of integrin-linked kinase is a critical prosurvival pathway induced in leukemic cells by bone marrow-derived stromal cells. Cancer Res 67:684-694. [DOI] [PubMed] [Google Scholar]
- Takanami I. (2005). Increased expression of integrin-linked kinase is associated with shorter survival in non-small cell lung cancer. BMC Cancer 5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C, Cruet-Hennequart S, Troussard A, Fazli L, Costello P, Sutton K, Wheeler J, Gleave M, Sanghera J, Dedhar S. (2004). Regulation of tumor angiogenesis by integrin-linked kinase (ILK). Cancer Cell 5:79-90. [DOI] [PubMed] [Google Scholar]
- Thompson JE, Thompson CB. (2004). Putting the rap on Akt. J Clin Oncol 22:4217-4226. [DOI] [PubMed] [Google Scholar]
- Troussard AA, McDonald PC, Wederell ED, Mawji NM, Filipenko NR, Gelmon KA, Kucab JE, Dunn SE, Emerman JT, Bally MB, Dedhar S. (2006). Preferential dependence of breast cancer cells versus normal cells on integrin-linked kinase for protein kinase B/Akt activation and cell survival. Cancer Res 66:393-403. [DOI] [PubMed] [Google Scholar]
- Verreault M, Bally MB. (2009). siRNA-mediated integrin-linked kinase suppression: nonspecific effects of siRNA/cationic liposome complexes trigger changes in the expression of phosphorylated-AKT and mTOR independently of ILK silencing. Oligonucleotides 19:129-140. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhang Z, Yao C. (2011). Targeting integrin-linked kinase increases apoptosis and decreases invasion of myeloma cell lines and inhibits IL-6 and VEGF secretion from BMSCs. Med Oncol 28:1596-1600. [DOI] [PubMed] [Google Scholar]
- Watzka SB, Rauscher-Potsch I, Stubenberger E, Getman V, Setinek U, Totsch M, Potschger U, Muller MR. (2010). Immunoreactivity of integrin-linked kinase in primary non-small-cell lung cancer and survival after curative resection. Eur J Cardiothorac Surg 38:254-259. [DOI] [PubMed] [Google Scholar]
- Watzka SB, Setinek U, Huber M, Cantonati H, Lax F, Watson S, Weigel G, Muller MR. (2008). Reactivity of integrin-linked kinase in human mesothelial cell proliferation. Interact Cardiovasc Thorac Surg 7:107-110. [DOI] [PubMed] [Google Scholar]
- White DE, Cardiff RD, Dedhar S, Muller WJ. (2001). Mammary epithelial-specific expression of the integrin-linked kinase (ILK) results in the induction of mammary gland hyperplasias and tumors in transgenic mice. Oncogene 20:7064-7072. [DOI] [PubMed] [Google Scholar]
- White DE, Coutu P, Shi YF, Tardif JC, Nattel S, Arnaud R, Dedhar S, Muller WJ. (2006). Targeted ablation of ILK from the murine heart results in dilated cardiomyopathy and spontaneous heart failure. Genes Dev 20:2355-2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RP, Ng P, Dedhar S, Li G. (2007). The role of integrin-linked kinase in melanoma cell migration, invasion, and tumor growth. Mol Cancer Ther 6:1692-1700. [DOI] [PubMed] [Google Scholar]
- Xie D, Yin D, Tong X, O’Kelly J, Mori A, Miller C, Black K, Gui D, Said JW, Koeffler HP. (2004). Cyr61 is overexpressed in gliomas and involved in integrin-linked kinase-mediated Akt and beta-catenin-TCF/Lef signaling pathways. Cancer Res 64:1987-1996. [DOI] [PubMed] [Google Scholar]
- Yamaji S, Suzuki A, Sugiyama Y, Koide Y, Yoshida M, Kanamori H, Mohri H, Ohno S, Ishigatsubo Y. (2001). A novel integrin-linked kinase-binding protein, affixin, is involved in the early stage of cell-substrate interaction. J Cell Biol 153:1251-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau CY, Wheeler JJ, Sutton KL, Hedley DW. (2005). Inhibition of integrin-linked kinase by a selective small molecule inhibitor, QLT0254, inhibits the PI3K/PKB/mTOR, Stat3, and FKHR pathways and tumor growth, and enhances gemcitabine-induced apoptosis in human orthotopic primary pancreatic cancer xenografts. Cancer Res 65:1497-1504. [DOI] [PubMed] [Google Scholar]
- Yoganathan TN, Costello P, Chen X, Jabali M, Yan J, Leung D, Zhang Z, Yee A, Dedhar S, Sanghera J. (2000). Integrin-linked kinase (ILK): a “hot” therapeutic target. Biochem Pharmacol 60:1115-1119. [DOI] [PubMed] [Google Scholar]
- Younes MN, Kim S, Yigitbasi OG, Mandal M, Jasser SA, Dakak Yazici Y, Schiff BA, El-Naggar A, Bekele BN, Mills GB, Myers JN. (2005). Integrin-linked kinase is a potential therapeutic target for anaplastic thyroid cancer. Mol Cancer Ther 4:1146-1156. [DOI] [PubMed] [Google Scholar]
- Younes MN, Yigitbasi OG, Yazici YD, Jasser SA, Bucana CD, El-Naggar AK, Mills GB, Myers JN. (2007). Effects of the integrin-linked kinase inhibitor QLT0267 on squamous cell carcinoma of the head and neck. Arch Otolaryngol Head Neck Surg 133:15-23. [DOI] [PubMed] [Google Scholar]
- Yu J, Shi R, Zhang D, Wang E, Qiu X. (2011). Expression of integrin-linked kinase in lung squamous cell carcinoma and adenocarcinoma: correlation with E-cadherin expression, tumor microvessel density and clinical outcome. Virchows Arch 458:99-107. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Xiao Y, Li Q, Liu Z, Zhang X, Qin C, Xie J, Wang X, Xu T. (2013). In vitro and in vivo effects of short hairpin RNA targeting integrin-linked kinase in prostate cancer cells. Mol Med Rep 8:419-424. [DOI] [PubMed] [Google Scholar]
- Zhao G, Guo LL, Xu JY, Yang H, Huang MX, Xiao G. (2011). Integrin-linked kinase in gastric cancer cell attachment, invasion and tumor growth. World J Gastroenterol 17:3487-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]