

Supplemental Digital Content is Available in the Text.
Key Words: atazanavir, efficacy, HIV-1, raltegravir, switch study, tolerability, resistance
Abstract:
This open-label, multinational, pilot study randomized (1:2 ratio) adults with HIV-1 RNA <40 copies per milliliter and nucleos(t)ide-related safety/tolerability issues to switch to ritonavir-boosted atazanavir (ATV/r) plus tenofovir disoproxil fumarate/emtricitabine (n = 37) or the nucleos(t)ide reverse transcriptase inhibitor-sparing regimen of ATV/r plus raltegravir (RAL) (n = 72). At 24 weeks, 35/37 (94.6%) and 58/72 (80.6%) of patients, respectively, maintained virological suppression, the primary endpoint, and 1 (2.7%) and 7 (9.7%), respectively, experienced virological rebound. Corresponding 48-week proportions were 86.5%, 69.4%, 2.7%, and 12.5%, respectively. Adherence was lower and treatment discontinuation was higher with ATV/r+RAL. In conclusion, switching to ATV/r+RAL resulted in a higher virological rebound rate than switching to ATV/r plus tenofovir disoproxil fumarate/emtricitabine.
INTRODUCTION
Highly active antiretroviral therapy has transformed HIV-1 infection into a long-term manageable disease.1,2 However, treatment-related toxicities and safety issues can limit the use of some drug components of antiretroviral regimens.3–5 In particular, safety concerns may result in treatment discontinuation of nucleos(t)ide reverse transcriptase inhibitors (NRTIs),3,6–11 which has led to the development of NRTI-sparing regimens.
NRTI-sparing regimens have been evaluated in treatment-naive,12–15 virologically suppressed,16–18 and treatment-experienced patients with previous virological failure.19–21 However, controversy remains as to whether NRTI-sparing regimens are beneficial.22
Atazanavir (ATV) combined with the integrase strand transfer inhibitor (INSTI) raltegravir (RAL) was effective in treatment-naive patients in the SPARTAN trial,13 although RAL resistance emerged in 4 patients and adverse events (AEs) of grade 4 hyperbilirubinemia occurred in 20.6% of patients.
Here, we report the findings of the HARNESS pilot study, that evaluated the efficacy and safety of switching from a triple-drug antiretroviral regimen that included 2 NRTIs to ritonavir-boosted ATV (ATV/r) plus tenofovir disoproxil fumarate/emtricitabine (TDF/FTC) or to the NRTI-sparing regimen of ATV/r plus RAL in treatment-experienced HIV-1–infected adults who required a treatment change due to safety/tolerability issues.
METHODS
HARNESS was a prospective, randomized, open-label, parallel-group, multinational, 48-week pilot study conducted from October, 2011 to February, 2014 at 30 sites in 7 countries (ClinicalTrials.gov NCT01332227). The study was conducted in accordance with Good Clinical Practice, and with ethical principles issued by the European Union and contained in the United States Code of Federal Regulations. The protocol was approved by the institutional review board for each site. Study participants provided written informed consent before entering the study.
Eligible patients were medically stable HIV-1–infected adults aged ≥18 years with an HIV-1 RNA level <50 copies per milliliter for ≥3 months (and a single measurement of <40 copies per milliliter during the preceding 30 days) during which time they were on a stable regimen that comprised of 2 NRTIs with a third antiretroviral agent (excluding ATV). In addition, patients were required to be experiencing treatment-related safety/tolerability issues.
Patients with a history of switching highly active antiretroviral therapy regimens for virological failure, resistance to any component of the study regimen, previous or current exposure to ATV or RAL, safety/tolerability issues with TDF/FTC or ritonavir, and those using proton pump inhibitors were excluded.
Patients were randomized (1:2 ratio) by an interactive voice response system to receive oral ATV/r 300/100 mg plus TDF/FTC 300/200 mg once daily (ATV/r+TDF/FTC) or ATV/r 300/100 mg once daily plus RAL 400 mg twice daily (ATV/r+RAL).
An independent Data Monitoring Committee (DMC) was responsible for stopping the study if ≥15% of patients in the ATV/r+RAL group had 2 consecutive on-treatment HIV-1 RNA levels ≥400 copies per milliliter by week 12 (Stopping Rule 1); or if ≥5 patients in the ATV/r+RAL group developed genotypic substitutions and phenotypic resistance to RAL or ATV by week 24 (Stopping Rule 2).
The primary endpoint was the proportion of patients maintaining virological suppression (HIV-1 RNA <40 copies per milliliter, Abbott m2000rt PCR assay; Abbot Laboritories, Des Plaines, IL) at week 24, assessed using an intention-to-treat algorithm. Patients who discontinued therapy before week 24, or experienced virological rebound (2 consecutive on-treatment HIV-1 RNA levels of ≥40 copies per milliliter or the last on-treatment HIV-1 RNA level of ≥40 copies per milliliter followed by discontinuation), or those with missing week 24 measurements were regarded as nonresponders. Treatment response was analyzed cumulatively at weeks 4, 8, 12, 16, 24, 36, and 48 by intention-to-treat algorithm and observed values approaches (number of responders divided by the number of patients randomized or number of patients with on-treatment HIV-1 RNA values, respectively).
Secondary endpoints were the proportion of patients maintaining virological suppression at week 48, and the number of patients with virological rebound and associated genotypic or phenotypic resistance. Genotypic resistance profiles were assessed in patients with suitable isolates (HIV-1 RNA ≥500 copies per milliliter) using recognized databases.23,24 Adherence was assessed using the modified antiviral medication adherence questionnaire at baseline and at weeks 4, 12, 24, and 48. Safety/tolerability reasons for switching from prestudy regimens were assessed at baseline using a questionnaire designed for this study, and were reassessed at weeks 12, 24, and 48. Safety and tolerability of study treatment was assessed by AE reports and laboratory tests.
No formal sample size calculation or statistical hypothesis testing was performed in this pilot study. Exact binomial 95% confidence intervals were calculated for the proportions of patients maintaining virological suppression.
RESULTS
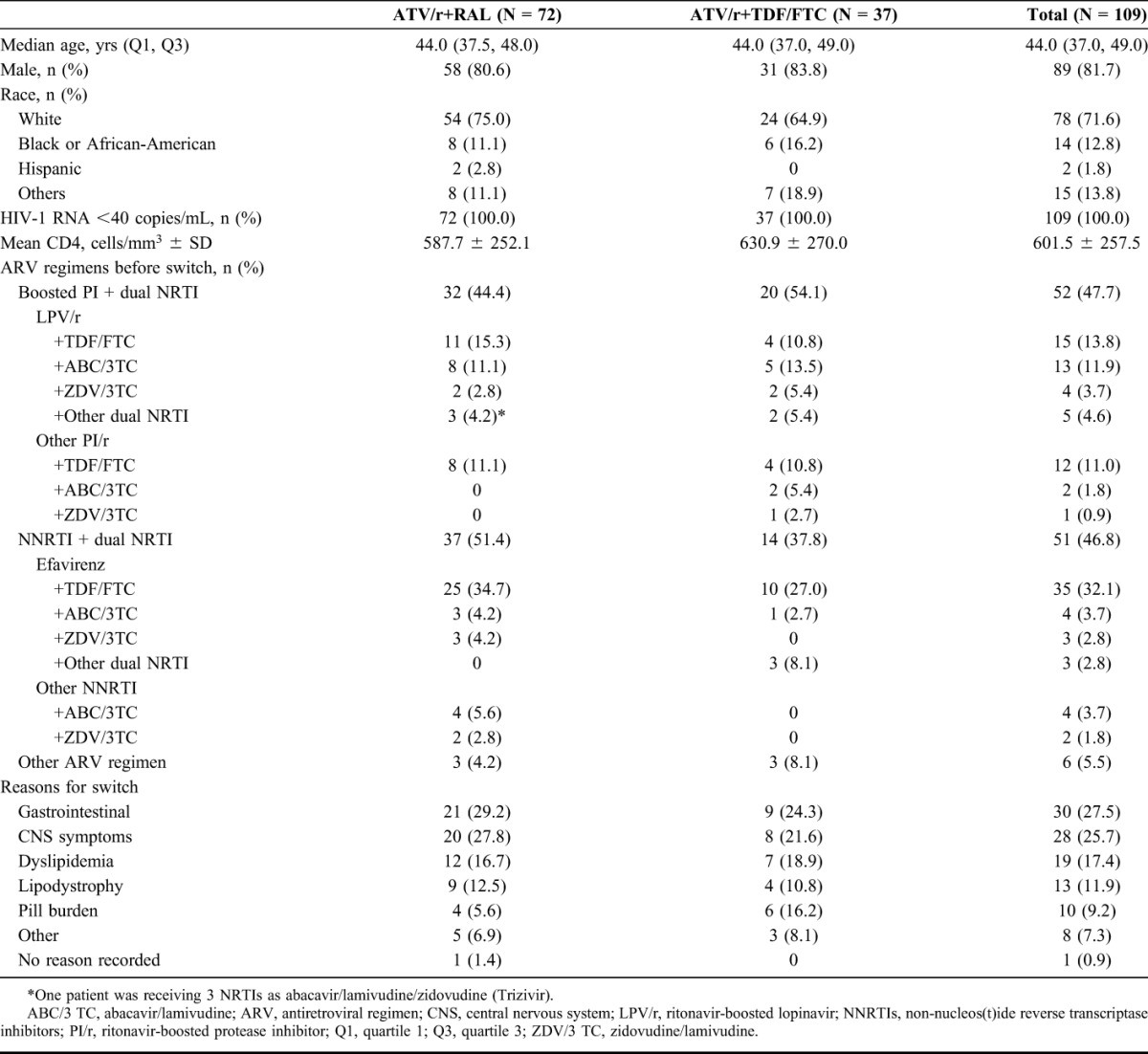
Of 132 patients screened, 109 were randomized and treated with ATV/r+RAL (n = 72) or ATV/r+TDF/FTC (n = 37). Reasons for switching, in descending order of frequency, were gastrointestinal symptoms, central nervous system symptoms, dyslipidemia, lipdystrophy and pill burden (Table 1).
TABLE 1.
Patient Demographics and Baseline Characteristics
A lower proportion of ATV/r+RAL recipients (56/72; 77.8%) than ATV/r+TDF/FTC recipients (32/37; 86.5%) completed therapy due to discontinuation for AEs (n = 4 vs 1, respectively), lack of efficacy (n = 3 vs 1), withdrawal of consent (n = 4 vs 1), noncompliance (n = 1 vs 1), and other reasons (n = 4 vs 1) (for CONSORT flow diagram, see Supplemental Digital Content 1, http://links.lww.com/QAI/A771).
A lower proportion of ATV/+RAL recipients than ATV/r+TDF/FTC recipients maintained virological suppression at week 24 (primary endpoint) and at week 48 (Table 2).
TABLE 2.
Results at Weeks 24 and 48
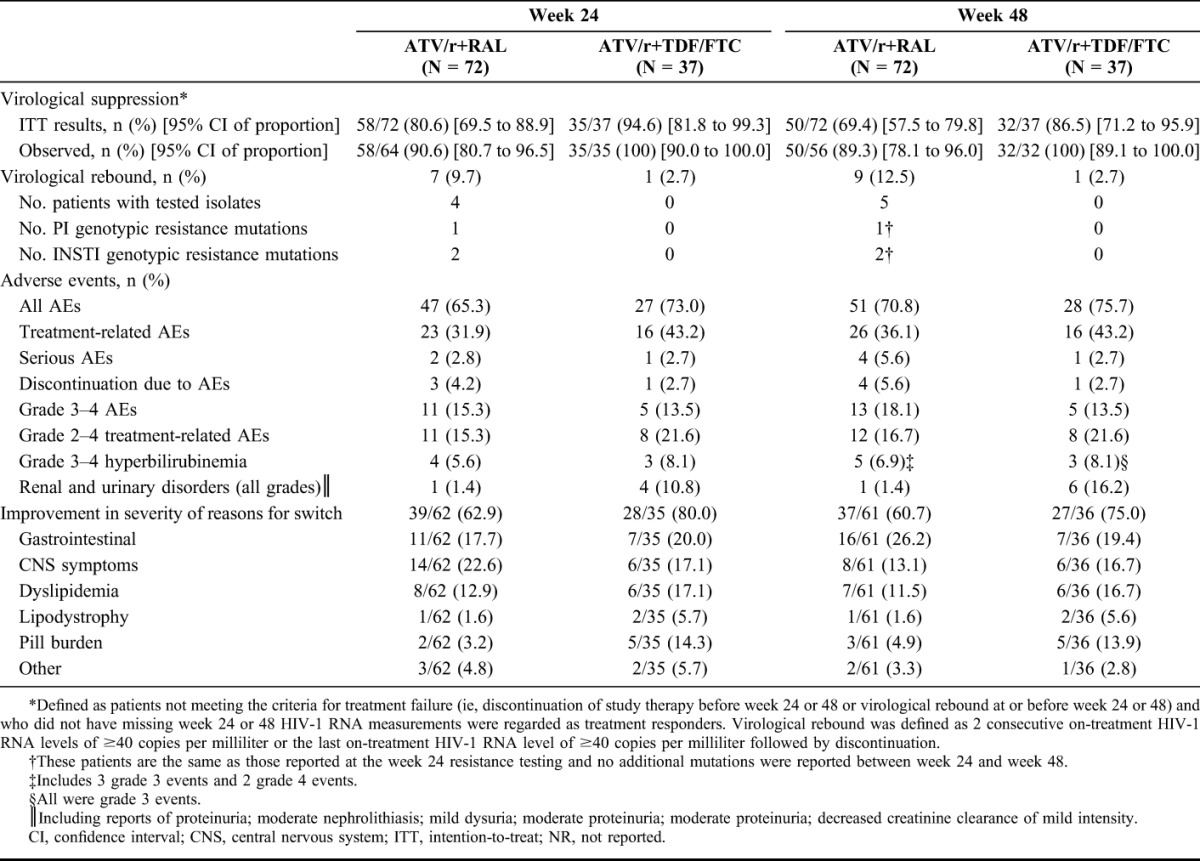
Virological rebound was documented in 10 patients. By week 24, virological rebound had occurred in 7/72 (9.7%) patients on ATV/r+RAL and 1/37 (2.7%) on ATV/r+TDF/FTC. Because of delays in obtaining resistance data, Stopping Rule 2 could not be assessed at the 24-week DMC meeting. Despite not meeting either of the predefined Stopping Rules, the DMC recommended stopping the trial, given the divergent rebound rate and absence of resistance data. However, by that time all patients had reached week 48 of treatment.
Two further patients had rebound between weeks 24 and 48, one of whom had HIV-1 RNA levels of 129 and 85 copies per milliliter at weeks 36 and 48, respectively, and the other who had an HIV-1 RNA level of 797 copies per milliliter at week 36 and <40 copies per milliliter by week 48. (For time-to-event analysis of treatment response, see Supplemental Digital Content 2, http://links.lww.com/QAI/A771).
CD4+ counts increased from baseline at week 24 by 31 and 24 cells per cubic millimeter with ATV/r+RAL and ATV/r+TDF/FTC, respectively, and at week 48, by 54 and 10 cells per cubic millimeter, respectively.
One of 4 ATV/r+RAL-treated patients with rebound at week 24 who underwent resistance testing had HIV-1 isolates with INSTI mutations Y143C and N155H and phenotypic resistance to RAL. Another patient had isolates with INSTI mutation F121Y without phenotypic resistance, but had numerous mutations that confer resistance to protease inhibitors (PIs), including ATV. No evidence of genotypic resistance was detected in the remaining 2 patients. No evidence of INSTI or PI resistance was observed in the 1 ATV/r+TDF/FTC-treated patient who experienced rebound at week 24.
The proportion of patients who never missed medication in the ATV/r+RAL and ATV/r+TDF/FTC groups was comparable at week 24 [77.0% (47/61) and 75.8% (25/33), respectively], but was lower in recipients of ATV/r+RAL [75.9% (41/54)] than ATV/r+TDF/FTC [81.3% (26/32)] at week 48. One ATV/r+RAL-treated patient interrupted treatment for 49 days because of Legionella pneumonia, which resulted in rebound. No other patients had interruptions for >3 days.
Tolerability issues identified at baseline improved in the majority of subjects in both groups. However, the proportion of ATV/r+TDF/FTC-treated patients reporting improvement was higher than that in the ATV/r+RAL group, largely driven by improvements in dyslipidemia and pill burden in the ATV/r+TDF/FTC group (Table 2; for the range of responses and their categorization, see Supplemental Digital Content 2, http://links.lww.com/QAI/A771).
More ATV/r+RAL than ATV/r+TDF/FTC recipients discontinued treatment due to AEs (n = 4 vs 1). In contrast, more patients on ATV/r+TDF/FTC had renal and urinary disorders (Table 2). One ATV/r+RAL-treated patient developed proteinuria on day 56, which resolved without treatment interruption and was considered unrelated to study treatment.
Similar proportions of patients had AEs of grade 3–4 hyperbilirubinemia. Overall rates of grade 3–4 changes in total bilirubin (not necessarily reported as an AE) in the ATV/r+RAL and ATV/r+TDF/FTC groups were 49.3% and 40.5%, respectively. Two patients discontinued ATV/r+RAL and 1 discontinued ATV/r+TDF/FTC for hyperbilirubinemia-related events.
Lipid profiles showed opposite trends during treatment, with all parameters increasing in the ATV/r+RAL group and all parameters decreasing in the ATV/r+TDF/FTC group.
DISCUSSION
Among patients switched to ATV/r+RAL, maintenance of virological suppression was lower and virological rebound was higher than those switched to ATV/r+TDF/FTC. Although safety/tolerability issues identified at baseline improved with ATV/r+RAL, these improved to a lesser extent than with ATV/r+TDF/FTC. In addition, treatment discontinuation occurred more frequently and adherence was lower with ATV/r+RAL. These findings, therefore, do not support switching for safety/tolerability reasons to ATV/r+RAL in virologically-suppressed patients.
Other randomized controlled trials have evaluated RAL-containing NRTI-sparing regimens in treatment-naive patients13,15,25 and treatment-experienced patients with virological suppression switched either because of tolerability concerns17,18,26 or because of virological failure.19–21 In patients switched because of virological failure, 3 substantive noninferiority trials have established similar virological efficacy in patients randomized to receive RAL plus a boosted PI with or without a third agent compared with NRTI-containing regimens.19–21 However, evidence for efficacy of NRTI-sparing regimens in other contexts is less certain. In treatment-naive patients, INSTI resistance mutations occurred more frequently with ritonavir-boosted darunavir (DRV/r)+RAL than with DRV/r+TDF/FTC, and virological outcomes were worse in DRV/r+RAL recipients with CD4+ counts <200 cells per microliter in the NEAT001/ANRS143 study15; and in the SPARTAN study, unboosted ATV+RAL provided better virological suppression than ATV/r+TDF/FTC, although RAL resistance occurred.13 In treatment-experienced patients switched for tolerability concerns, only limited evidence is available from small pilot studies.17,18,26 The results of the small BATAR study suggested that switching to ATV/r+RAL was equivalent to remaining on ATV/r+TDF/FTC with respect to virological suppression17; however, these preliminary findings have not been supported by the larger HARNESS pilot study.
Potential reasons for the lower maintenance of virological suppression with ATV/r+RAL compared with ATV/r+TDF/FTC may include differences in regimens at baseline, treatment-emergent resistance mutations, reduced drug exposures, adherence issues, and treatment discontinuations.
More patients in the ATV/r+RAL group (51.4%) than in the ATV/r+TDF/FTC group (37.8%) switched from NNRTI-based regimens, which are associated with a lower genetic barrier to resistance than PI-based regimens,27 and this could have potentially biased the results.
Of the 9 patients in the ATV/r+RAL group with virological rebound up to week 48 (2 of whom resuppressed), only 2 had isolates with clinically relevant resistance mutations. One patient had 2 major INSTI resistance mutations and the second had multiple mutations associated with resistance to PIs, including ATV. This second patient was virologically suppressed with DRV/r+TDF/FTC at baseline, developed virological rebound after switching to ATV/r+RAL and then resuppressed after resuming DRV/r+TDF/FTC. The pattern in this patient might be explained by archived resistance to ATV and the lower genetic barrier to resistance of RAL. However, the development of resistance mutations in just 2 patients receiving ATV/r+RAL is insufficient to explain the between-group difference in virological suppression rates.
Intensive pharmacokinetic and C-trough analyses did not identify reduced ATV exposure in the ATVr+RAL group; however C-trough values were not necessarily obtained at the time of virologic failure (see Supplemental Digital Content 4, http://links.lww.com/QAI/A771).
Self-reported adherence was poorer with ATV/r+RAL than ATV/r+TDF/FTC after week 24 and may have contributed to virological rebound occurring during weeks 24–48. Twice-daily RAL dosing and lesser improvements in pill burden in the ATV/r+RAL than in ATV/r+TDF/FTC recipients may have also adversely affected adherence.
More recipients of ATV/r+RAL than ATV/r+TDF/FTC discontinued treatment, thereby contributing to the difference in virological suppression rates. However, there were no unexpected safety signals and no differences in bilirubin-related AEs or bilirubin-related treatment discontinuations. Interestingly, grade 4 hyperbilirubinemia was less frequent in the current study (2.8% at week 48) than in SPARTAN (20.6% at week 24), possibly because of the higher ATV exposures with unboosted twice-daily ATV+RAL in SPARTAN.13 Renal AEs were more common and lipid abnormalities were less common in ATV/r+TDF/FTC recipients, consistent with the safety profile of TDF.
Study limitations include small sample size, lack of formal statistical comparisons between the 2 groups, potential bias associated with observed value analysis, absence of pretreatment genotype data for patients who failed previous treatment regimens, self-reporting of treatment adherence, and lack of a continuation of baseline treatment arm.
In conclusion, this pilot study did not support switching to ATV/r+RAL for safety/tolerability reasons in treatment-experienced patients with virological suppression. The lower virological suppression rate with ATV/r+RAL than ATV/r+TDF/FTC was likely due to lower adherence with twice-daily RAL, tolerability issues, and increased treatment discontinuation. It remains to be established whether ATV/r plus RAL, in a fully-powered study, or ATV/r plus once-daily dolutegravir may improve outcomes for patients needing an NRTI-sparing regimen.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the study participants, study investigators, and study staff. In addition, the authors thank Miao Yu for the provision of statistical support for this study while she was an employee of Bristol-Myers Squibb, and thank Luna Zaru, an employee of Bristol-Myers Squibb, for subsequent statistical support. The present study was supported by Bristol-Myers Squibb. The authors also thank Simone Boniface, Caroline Perry, and Julian Martins of inScience Communications, Springer Healthcare, who provided medical writing assistance, which was funded by Bristol-Myers Squibb.
Footnotes
Presented at the 20th International AIDS Conference, July 20–25, 2014, Melbourne, Australia (abstract and poster).
J.v.L. was a study investigator and his institution received a research grant from Bristol-Myers Squibb for the conduct of this study; he has received personal fees from Bristol-Myers Squibb for consultancy during the planning of this study, and for attendance at investigator meetings, advisory boards, and conferences to present study data; his institution has received or is currently receiving research grants from Abbvie, Bionor AS, Boehringer, Bristol-Myers Squibb, Genetic Immunity, Gilead Sciences, GlaxoSmithKline, Janssen-Cilag, Merck Sharp & Dohme, Roche, Vision7, and ViiV Healthcare; he has received personal fees for speakers' bureaus from Abbvie, Bionor AS, Boehringer, Bristol-Myers Squibb, GlaxoSmithKline, Janssen-Cilag, Merck Sharp & Dohme, and ViiV Healthcare; he has received personal fees for the development of educational materials from Abbvie, Bristol-Myers Squibb, and ViiV Healthcare; he has also received personal fees for conference attendance from Gilead Sciences (at manuscript submission); he is now an employee of ViiV Healthcare (at manuscript acceptance). A.P. was a study investigator and his institution received a research grant from Bristol-Myers Squibb for the conduct of this study and for attendance at investigator meetings; he has received personal fees for attendance at advisory boards from Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, Merck, and ViiV Healthcare; his institution is receiving ongoing research grants from Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, Merck, and ViiV Healthcare; and he has received personal fees for speakers' bureaus from Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, Merck, and ViiV Healthcare. J.M.G. was a study investigator and his institution received a research grant from Bristol-Myers Squibb for the conduct of this study and for attendance at investigator meetings; he has received personal fees for attendance at advisory boards from Abbvie, Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, Merck Sharpe & Dohme, and ViiV Healthcare; and his institution has received research grants from Abbvie, Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, Merck Sharpe & Dohme, and ViiV Healthcare. A.A. was a study investigator and his institution received a research grant from Bristol-Myers Squibb for the conduct of this study and for attendance at investigator meetings; he has received personal consultancy fees from Abbvie, Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, Meck, and ViiV Healthcare; his institution is currently receiving research grants from Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, and ViiV Healthcare; and he has received personal fees from Abbvie and ViiV Healthcare for conference attendance. O.O. was a study investigator and his institution received a research grant from Bristol-Myers Squibb for the conduct of this study and for attendance at investigator meetings; he has received personal fees for attendance at advisory/review boards from Abbvie, Bristol-Myers Squibb, Durata, Gilead Sciences, and Janssen-Cilag; his institution currently receives research grants from Forrest and Gilead Sciences; and he has received personal fees for speakers' bureaus from Gilead Sciences and Janssen-Cilag. I.K., O.S., A.B., and H.S. are employees of and own stock in Bristol-Myers Squibb, the manufacturers of atazanavir. P.-M.G. was a study investigator and his institution received a research grant from Bristol-Myers Squibb for the conduct of this study and for attendance at investigator meetings; he has received personal fees for attendance at advisory boards from Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, and Merck Sharp & Dohme; his institution is receiving ongoing research grants from Gilead Sciences and Roche; he has received personal fees for speakers' bureaus from Bristol-Myers Squibb, Janssen-Cilag, and ViiV Healthcare; he has received personal fees for the development of educational materials from ViiV Healthcare; and his institution has received payment for conference attendance from Gilead Sciences.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.jaids.com).
REFERENCES
- 1.Mocroft A, Ledergerber B, Katlama C, et al. Decline in the AIDS and death rates in the EuroSIDA study: an observational study. Lancet. 2003;362:22–29. [DOI] [PubMed] [Google Scholar]
- 2.Le Douce V, Janossy A, Hallay H, et al. Achieving a cure for HIV infection: do we have reasons to be optimistic? J Antimicrob Chemother. 2012;67:1063–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monteiro P, Perez I, Laguno M, et al. Dual therapy with etravirine plus raltegravir for virologically suppressed HIV-infected patients: a pilot study. J Antimicrob Chemother. 2014;69:742–748. [DOI] [PubMed] [Google Scholar]
- 4.Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. Department of Health and Human Services; Available at: http://aidsinfo.nih.gov/contentfiles/AdultandAdolescentGL.pdf. Accessed June 5, 2015. [Google Scholar]
- 5.European AIDS Clinical Society. Guidelines: Version 7.1, November 2014. Available at: http://www.eacsociety.org/guidelines/eacs-guidelines/eacs-guidelines.html. Accessed June 5, 2015. [Google Scholar]
- 6.Walker UA, Setzer B, Venhoff N. Increased long-term mitochondrial toxicity in combinations of nucleoside analogue reverse-transcriptase inhibitors. AIDS. 2002;16:2165–2173. [DOI] [PubMed] [Google Scholar]
- 7.Lewis W, Dalakas MC. Mitochondrial toxicity of antiviral drugs. Nat Med. 1995;1:417–422. [DOI] [PubMed] [Google Scholar]
- 8.Goicoechea M, Liu S, Best B, et al. Greater tenofovir-associated renal function decline with protease inhibitor-based versus nonnucleoside reverse-transcriptase inhibitor-based therapy. J Infect Dis. 2008;197:102–108. [DOI] [PubMed] [Google Scholar]
- 9.Kiser JJ, Carten ML, Aquilante CL, et al. The effect of lopinavir/ritonavir on the renal clearance of tenofovir in HIV-infected patients. Clin Pharmacol Ther. 2008;83:265–272. [DOI] [PubMed] [Google Scholar]
- 10.Zimmermann AE, Pizzoferrato T, Bedford J, et al. Tenofovir-associated acute and chronic kidney disease: a case of multiple drug interactions. Clin Infect Dis. 2006;42:283–290. [DOI] [PubMed] [Google Scholar]
- 11.Bedimo R, Drechsler H, Turner D, et al. RADAR study: raltegravir combined with boosted darunavir has similar safety and antiviral efficacy as tenofovir/emtricitabine combined with boosted darunavir in antiretroviral-naïve patients [Abstract MOPE214]. Paper presented at: 6th International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention; 17–20 July 2011; Rome, Italy. Available at: http://pag.ias2011.org/abstracts.aspx?aid=370. Accessed June 5, 2015.
- 12.Reynes J, Lawal A, Pulido F, et al. Examination of noninferiority, safety, and tolerability of lopinavir/ritonavir and raltegravir compared with lopinavir/ritonavir and tenofovir/emtricitabine in antiretroviral-naive subjects: the progress study, 48-week results. HIV Clin Trials. 2011;12:255–267. [DOI] [PubMed] [Google Scholar]
- 13.Kozal MJ, Lupo S, DeJesus E, et al. A nucleoside- and ritonavir-sparing regimen containing atazanavir plus raltegravir in antiretroviral treatment-naive HIV-infected patients: SPARTAN study results. HIV Clin Trials. 2012;13:119–130. [DOI] [PubMed] [Google Scholar]
- 14.Taiwo B, Zheng L, Gallien S, et al. Efficacy of a nucleoside-sparing regimen of darunavir/ritonavir plus raltegravir in treatment-naive HIV-1-infected patients (ACTG A5262). AIDS. 2011;25:2113–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raffi F, Babiker AG, Richert L, et al. Ritonavir-boosted darunavir combined with raltegravir or tenofovir-emtricitabine in antiretroviral-naive adults infected with HIV-1: 96 week results from the NEAT001/ANRS143 randomised non-inferiority trial. Lancet. 2014;384:1942–1951. [DOI] [PubMed] [Google Scholar]
- 16.Ghosn J, Slama L, Chermak A, et al. Switching to darunavir/ritonavir 800/100 mg once-daily containing regimen maintains virological control in fully suppressed pre-treated patients infected with HIV-1. J Med Virol. 2013;85:8–15. [DOI] [PubMed] [Google Scholar]
- 17.Cohen C, Green J, Olivet H, et al. A randomized pilot study of tenofovir/emtricitabine (TDF/FTC) + boosted atazanavir (ATV/r) vs. raltegravir (RAL BID) + ATV/r vs. RAL BID + ATV BID [Poster Abstract—P286]. J Int AIDS Soc. 2012;15(suppl 4):18279. [Google Scholar]
- 18.Ofotokun I, Sheth AN, Sanford SE, et al. A switch in therapy to a reverse transcriptase inhibitor sparing combination of lopinavir/ritonavir and raltegravir in virologically suppressed HIV-infected patients: a pilot randomized trial to assess efficacy and safety profile: the KITE study. AIDS Res Hum Retroviruses. 2012;28:1196–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.SECOND-LINE Study Group, Boyd MA, Kumarasamy N, et al. Ritonavir-boosted lopinavir plus nucleoside or nucleotide reverse transcriptase inhibitors versus ritonavir-boosted lopinavir plus raltegravir for treatment of HIV-1 infection in adults with virological failure of a standard first-line ART regimen (SECOND-LINE): a randomised, open-label, non-inferiority study. Lancet. 2013;381:2091–2099. [DOI] [PubMed] [Google Scholar]
- 20.Paton N, Kityo C, Hoppe A, et al. A pragmatic randomised controlled strategy trial of three second-line treatment options for use in public health rollout programme settings: the Europe-Africa Research Network for Evaluation of Second-line Therapy (EARNEST) Trial. Paper presented at: 7th IAS Conference on HIV Pathogenesis Treatment and Prevention; June 30, 2013–July 3, 2013; Kuala Lumpur, Malaysia. Available at: http://pag.ias2013.org/abstracts.aspx?aid=3024. Accessed June 5, 2015.
- 21.Tashima K, Smeaton L, Andrade A, et al. Omitting NRTI from ARV regimens is not inferior to adding NRTI in treatment-experienced HIV+ subjects failing a protease inhibitor regimen: the ACTG OPTIONS study [CROI abstract 153LB]. Paper presented at: 20th Conference on Retroviruses and Opportunistic Infections; March 3–6, 2013; Atlanta, GA.
- 22.Di Biagio A, Ricci E, Viscoli C, et al. The use of nucleoside reverse transcriptase inhibitors sparing regimens in treatment-experienced HIV-1 infected patients. Curr HIV Res. 2013;11:179–186. [DOI] [PubMed] [Google Scholar]
- 23.Johnson VA, Calvez V, Gunthard HF, et al. Update of the drug resistance mutations in HIV-1. Top Antivir Med. 2013;21:6–14. [PMC free article] [PubMed] [Google Scholar]
- 24.Stanford University. HIV Drug Resistance Database. Available at: http://sierra2.stanford.edu/sierra/servlet/JSierra?action=mutationsInput. Accessed June 5, 2015. [Google Scholar]
- 25.Reynes J, Trinh R, Pulido F, et al. Lopinavir/ritonavir combined with raltegravir or tenofovir/emtricitabine in antiretroviral-naive subjects: 96-week results of the progress study. AIDS Res Hum Retroviruses. 2013;29:256–265. [DOI] [PubMed] [Google Scholar]
- 26.Carey D, Pett SL, Bloch M, et al. A randomized study of pharmacokinetics, efficacy, and safety of 2 raltegravir plus atazanavir strategies in ART-treated adults. J Acquir Immune Defic Syndr. 2012;60:143–149. [DOI] [PubMed] [Google Scholar]
- 27.Tang MW, Shafer RW. HIV-1 antiretroviral resistance: scientific principles and clinical applications. Drugs. 2012;72:e1–e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.