

Abstract
Sequence control in polymers, well‐known in nature, encodes structure and functionality. Here we introduce a new architecture, based on the nucleophilic aromatic substitution chemistry of cyanuric chloride, that creates a new class of sequence‐defined polymers dubbed TZPs. Proof of concept is demonstrated with two synthesized hexamers, having neutral and ionizable side chains. Molecular dynamics simulations show backbone–backbone interactions, including H‐bonding motifs and pi–pi interactions. This architecture is arguably biomimetic while differing from sequence‐defined polymers having peptide bonds. The synthetic methodology supports the structural diversity of side chains known in peptides, as well as backbone–backbone hydrogen‐bonding motifs, and will thus enable new macromolecules and materials with useful functions.
Keywords: biomimetics, macromolecules, sequence-defined polymers, simulations, solid-phase synthesis
Sequence control in synthetic polymers has gained renewed interest,1 because sequence leads to structure and function. Sequence‐controlled polymers have repeated sequence motifs (e.g., (ABC)n) whereas sequence‐defined polymers have monomers in any predetermined order (e.g. ABCADC). The latter polymers are epitomized by natural biopolymers such as polypeptides and poly(nucleic acids), where pendant side chains distinguish one monomer from another. In polypeptides, the sequencing leads to diverse structures and functions that are vital to life, including material architectures, biocatalysis, molecular recognition, and transport across membranes. Sequence in polymers was discussed in a critical review by Lutz et al. in 2013, including discussions of relevance to materials science.1b Some sequence‐defined oligomers and polymers fold into conformational structures such as helices, and hence are called “foldamers”, which have potential applications in materials science, catalysis, and molecular recognition.2
Construction of synthetic poly(peptides) using biomolecular machinery, solution synthesis, or solid‐phase synthesis, is well‐established.1b Synthetic sequence‐defined polymers typically have monomers selected from natural and unnatural alpha‐amino acids, non‐alpha‐amino acids, and pseudo‐amino acids. Peptoids have amide bonds in the backbone where the side chains are attached to a nitrogen atom instead of a carbon atom, prepared by the solid‐phase submonomer synthesis approach without using amino acid monomers or protecting groups.3 Until recently, most synthetic sequence‐defined polymers have a similarity to polypeptides by virtue of having amide bonds.
With resurgent interest in sequence‐controlled and sequence‐defined polymers, additional bond‐forming reactions are being implemented to create new polymer architectures1g–1r Lutz et al. described alternating cycloaddition and amidation reactions in 2009 to create (AB)n sequence‐controlled polymer segments.1h This group employed the same or similar strategies 2014 and 2105 to design sequence‐defined oligomers that encode digital information.1i–1l The sequencing could be decoded with tandem mass spectrometry.1k, 1l In 2013, Madder et al. described sequence‐defined trimers and tetramers where the polymer is extended by a method that attaches a cyclic thiolactone, and a side chain is added by nucleophilic aminolysis of the thiolactone.1n Han et al. described sequence‐controlled polymers prepared by a radical initiated step‐growth thiol‐yne approach with functional groups alternately arranged along the chain, which could be further functionalized after polymerization.1o Han asserted in this 2014 article that “all of the artificial SCPs [sequence‐controlled polymers] have no independent functional groups suspended on backbone”, citing twenty five references. In 2014, Solleder and Meier created a sequence‐defined tetramer using Passerini 3‐component reactions and thiol–ene chemistry, without protecting groups, where each monomer unit contains a different amide‐containing side chain.1p Also in 2014, Porel and Alabi described two sequence‐defined 10‐mers using allyl acrylamide building blocks and dithiols in a fluorous‐assisted synthesis approach.1q In this case, amide bond formation is not used for chain extension reactions, but is present in the backbone. A number of these recent approaches for sequence‐defined polymers were described by Lutz in 2015 in a review on iterative approaches without protecting groups.1m Recently, a unique photochemical strategy for defining sequence in polymers, with a thermal deprotection step, was described and demonstrated with symmetrical sequencing n(BA)C(AB)n.1r Gutekunst and Hawker described a relay metathesis method to polymerize sequence‐defined macromonomers.1s
Here we introduce a new macromolecular architecture, based on the nucleophilic aromatic substitution chemistry of cyanuric chloride, that we call TZPs, for triazine‐based polymers. Selected examples shown in Scheme 1 illustrate a general architecture, 1, a homopolymer of defined length, 2, and two sequence‐defined heteropolymers, 3 and 4. There have been a few reports describing triazine chemistry to make pseudo‐amino acids for incorporation into oligomeric peptides.4 In one case, the peptides were constructed entirely of triazine‐based pseudo‐amino acids with different side chains into small cyclic peptides containing up to four monomers.4b Our architecture is distinct from peptides based on triazine‐containing pseudo‐amino acids and amide bond chemistry.
Scheme 1.
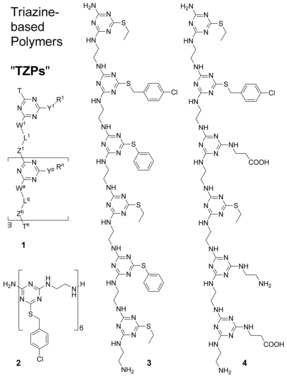
Triazine‐based polymers.
We show that these new polymers meet three requirements for a new biomimetic sequence‐defined architecture: 1) diverse side‐chain‐functionalized monomer units can be prepared, 2) the units can be sequenced into chains, and 3) potential backbone–backbone interactions exist that could influence conformational folding or intermolecular assembly. The pendant Y‐R groups in the general structure 1 are side chains, while W‐L‐Z sections connecting triazine rings in the backbone will be called linkers. The generalized atoms Y, W, and Z are typically heteroatoms derived from reactive amines or thiols.
A submonomer synthesis method, shown in Scheme 2, provides a solid‐phase iterative method to define sequence. Linkers in Scheme 2 and the examples 2–4 are all ethylene diamine, but need not be. The submonomer method for TZPs supports sequence definition in both the Y‐R side chains and the W‐L‐Z backbone linker sections. Groups labeled T and Te are terminal groups, where T can be an amine arising from cleavage from the Rink amide solid phase, and Te is typically a hydrogen atom on an unreacted end of a linker. In another variation, not shown, the terminal end may be another amine group arising from final displacement of the last ring chloride with ammonium hydroxide rather than adding a final linker.
Scheme 2.
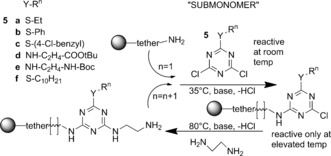
Submonomer solid‐phase synthesis of a triazine‐based polymer.
Diverse side‐chain functionalized molecular precursors and monomers can be prepared taking advantage of the stepwise reactivity in the nucleophilic aromatic substitution of the three chlorine atoms of cyanuric chloride, a cheap industrial chemical. Each substitution deactivates remaining sites such that higher temperatures are required for each substitution around the ring,5 from −20 to 5 °C for first reaction, rising from 18 °C to 35 °C for the next reaction, and temperatures above 60 °C for the third. Degrees of substitution, and substitution with two or three different substituents, are both readily controlled. Accordingly, we have synthesized a variety of molecular precursors, including those indicated as 5 a–f in Scheme 2, as well as additional precursors, monomers, and protected monomers described in the Supporting Information.
While a variety of polymer syntheses can follow from these types of molecules, we were inspired by the submonomer method for peptoids3 to develop a submonomer approach for sequence‐defined TZPs, shown in Scheme 2. This approach does not require any protecting groups in the main chain assembly, and it takes advantage of the sequential reactivity of triazine chlorine substituents to provide control. The solid‐phase method has the potential for automation. The monosubstituted triazine ring submonomer 5 reacts at room temperature or slightly elevated temperatures (35 °C), while the linker section, using ethylene diamine in 20‐fold excess, reacts with the remaining chlorine‐substituted site at elevated temperatures, typically 80 °C. Use of the 35 °C temperature for the addition of 5 enables shorter reaction times than at room temperature. In this reaction, the submonomer begins with two identical reaction sites, but the second of these becomes different after the first is substituted. Rink amide resin was selected for our synthetic scheme because the tether chemistry is stable to the synthesis conditions. The release of final product from the resin can be achieved using 50 % trifluoroacetic acid (TFA) in dichloromethane. (For detailed methods and characterization of products, see the Supporting Information.) For TZP 2, using monomer 5 c, the crude product was purified by reversed‐phase HPLC on an analytical C‐18 column to give, after evaporation of solvents under reduced pressure, an amorphous white solid (35 mg, 43 %, based on initial loading of the resin). The product was characterized by HRMS, found m/z: 1776.3343, calcd for [M+H]+/C72H75Cl6N31S6, 1776.3350.
TZP 3 was prepared using triazine compounds 5 a, 5 b and 5 c in a defined sequence. The crude product was purified by reversed‐phase HPLC (Figure 1) on a semipreparative C‐18 column to give, after evaporation of solvents under reduced pressure, 3 (48 mg, 75 %, based on initial loading of the resin) as an amorphous white solid. The high‐resolution mass spectrum of compound 3 (Figure 2) clearly shows the multiply charged adducts and we found that the observed m/z of the parent [M+H]+ adduct (molecular formula C55H70ClN31S6) is 1392.4537 which agrees well with the calculated value. TZP 4 was prepared to demonstrate the ability to append side chains with ionizable groups such as amines or carboxylic acids, as would be desired for a truly biomimetic polymer architecture. These were achieved using protected R groups, such as a tBu ester‐protected carboxylic acid in 5 d, and the Boc‐protected (Boc=tert‐butoxycarbonyl) amino group in 5 e. Both of these protecting groups remain on the functional group under the conditions of polymer synthesis, while cleaving in one step when the polymers are released from the Rink amide resin with TFA. We obtained 4 (19 mg, 31 %, based on initial loading of the resin) as an amorphous white solid. After purification, characterization by HRMS found m/z: 1348.5527, calcd [M+H]+ C49H74ClN35O4S3: 1348.5586.
Figure 1.
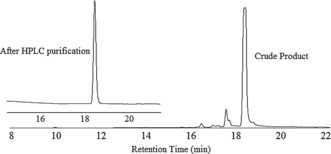
HPLC chromatogram of crude and pure TZP 3.
Figure 2.
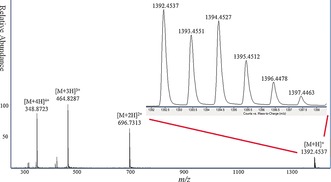
High‐resolution mass spectrum and isotopic pattern of TZP 3. The expanded spectrum shows the adduct [M+H]+.
The presence of various side chains in the sequence‐defined TZPs 3 and 4 can be verified by NMR spectroscopy as shown in Figure 3. The triazine groups have no protons and the linkers have only methylene groups. Therefore, the NMR spectra of TZPs with diverse side chains show peaks with chemical shifts and integrated areas indicative of the side‐chain content of the synthesized structures. The relative peak areas and assignments in Figure 3 show that the prepared TZPs have the intended content. Tandem mass spectrometry6 of TZPs 3 and 4, shown in Figure 4, confirm that the monomers are in the intended order. Bond breaking of the TZPs occurs between the aliphatic C−N bonds in ethylene diamine linkers, and by degradation of the triazine rings to give predictable fragments, the vast majority of which are found. The mass spectra are shown in Figure 4, while the expected and observed masses for the M and N fragments (indicated by the arrows in Figure 4 ) are in Table 1. The complementary fragments to the indicated M and N fragments are typically much less intense but are found (see the Supporting Information). Fragments from degradation and cleavage at triazine rings, dubbed T, are observed from both sides of the ring (indicate by L or R); more on these T fragments is given in the Supporting Information. In addition, some peaks are seen corresponding to the loss of a side chain from the parent ion. The NMR and tandem MS data for each sequence‐defined TZP confirm that the intended structures and sequences were actually synthesized.
Figure 3.
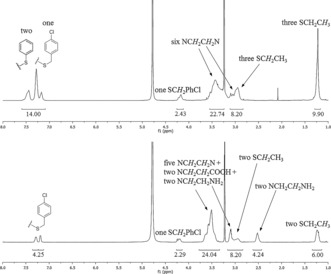
1H NMR spectra (500 MHz) of TZPs 3 (upper) and 4 (bottom) in deuterated methanol.
Figure 4.
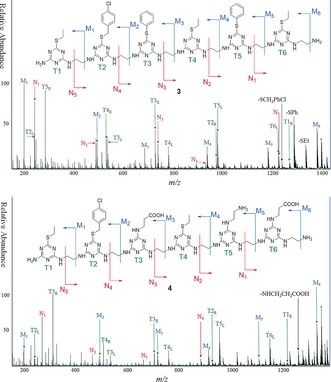
Tandem MS spectra of the [M+H]+ adduct of TZPs 3 (upper) and 4 (bottom). Fragments from breaking triazine rings(T) are indicated by ring number and left(L) or right(R) fragments. See the Supporting Information for details.
Table 1.
Calculated and observed masses of M and N fragment ions from sequence‐defined TZPs 3 and 4 obtained by tandem mass spectrometry.
| TZP 3 | TZP 4 | |||
|---|---|---|---|---|
| calculated | observed | calculated | observed | |
| M1 | 198.0808 | 198.0797 | 198.0808 | 198.0796 |
| N1 | 241.1230 | 241.1222 | 268.1516 | 268.1515 |
| M2 | 491.1310 | 491.1325 | 491.1310 | 491.1323 |
| N2 | 486.1965 | 486.1974 | 463.2749 | 463.2753 |
| M3 | 683.2700 | 683.2698 | 715.2332 | 715.2329 |
| N3 | 736.2045 | 736.2055 | 660.3480 | 660.3484 |
| M4 | 933.2780 | 933.2786 | 912.3067 | 912.3060 |
| N4 | 928.3435 | 928.3423 | 884.4506 | 884.4386 |
| M5 | 1178.3515 | 1178.3551 | 1107.4294 | 1107.4299 |
| N5 | 1221.3837 | 1221.3851 | 1177.5008 | not found |
| M6 | 1375.4251 | 1375.4252 | 1331.5315 | 1331.5349 |
The Supporting Information presents additional synthesis examples and approaches, as well as detailed experimental methods and characterization. This content includes a 12‐mer synthesis by the submomomer method, an alternative synthesis approach using protected monomers as proof of principle, and various additional conceptual synthetic schemes for sequenced polymers from triazine‐based molecular precursors.
Given that the new TZP polymers are synthetically accessible, we have performed molecular dynamics (MD) simulations, revealing a variety of possible hydrogen‐bonding motifs as well as pi–pi interactions. Detailed methods and additional examples are given in the Supporting Information.
Figure 5a shows the simulation of two interacting all‐trans tetramers, after two tetramers with S‐ethyl side chains were started in extended conformations in explicit solvent (see the Supporting Information). Parameters for the triazine residues and linkers were generated using the generalized amber force field (GAFF) and the programs ACPYPE and Ante‐chamber,7 and the simulation was carried out for 500 ns using GROMACS 4.6.4.8 The found conformer, which represents 28.2 % of population, shows 1) single hydrogen bonds between ring nitrogen atoms and linker amino hydrogen atoms that occur between the two chains (e.g. 2.3 Å), 2) single hydrogen bonds connecting a ring nitrogen to the distal amino group of an immediate linker section, stabilizing a turn (e.g. 2.2 Å), 3) pairs of hydrogen bonds between chains involving ring nitrogen atoms and the adjacent amino group on one chain interacting with the corresponding atoms of the other chain in an antiparallel fashion (e.g. 2.0 and 2.2 Å, as well as 2.1 and 2.0 Å), and 4) interchain pi–pi interactions with ring–ring distances of less than 4 Å. Turn and paired hydrogen bonds are shown in Figure 6. The paired hydrogen bonds indicate an intrinsic self‐complementarity in this polymer architecture, when the linker sections are derived from amino‐based nucleophiles that leave an alpha‐amino group attached to the triazine ring.
Figure 5.
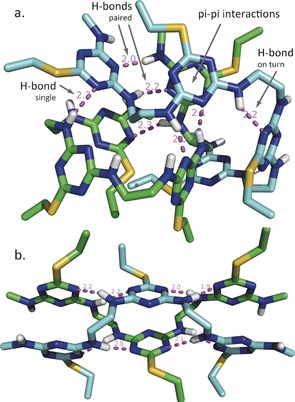
MD simulations of a) two tetramers and b) two trimers interacting by hydrogen bonding and pi–pi interactions. Side chains are S‐ethyl, and nonpolar hydrogen atoms are not shown.
Figure 6.
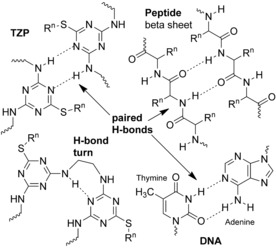
Backbone–backbone hydrogen‐bonding motifs found in molecular dynamics simulations of triazine‐based polymers. Paired hydrogen bonds in DNA and peptide beta sheets are shown for comparison.
Simulation of two interacting all‐cis trimers, shown in Figure 5b, revealed a more extended pattern of cross‐strand paired hydrogen bonds (1.9–2.2 Å) and pi–pi interactions (<4 Å). The found conformer, which represents 100 % of population, is a nanorod with a repeating pattern of side chains with a periodicity of 11–12 Å, where central rings on each strand are stabilized by two pairs of hydrogen bonds and a pi–pi interaction. In addition, as described in more detail in the Supporting Information, this type of structure is independently observed in folding of a related hexamer. When the nanorod structure is extended to a dimer of pentamers, it remains stable thereafter for 500 ns of explicit solvent simulations.
The simulations show the possibilities for multiple backbone–backbone interactions, and hence potential conformational behavior of single macromolecules or self‐assembly between macromolecules. The paired hydrogen bonds are reminiscent base‐pair interactions in DNA, and also the pairs of hydrogen bonds along interacting peptide strands in beta sheets (Figure 6). The regularity and three‐dimensional ordering of side chains along the nanorod are reminiscent of the side chains projecting from the outer surfaces of peptide alpha‐helix samples found in the literature on molecular recognition and foldamers indicating that structures with pyridine, pyrimidine or triazine architectures may be expected to participate in hydrogen‐bonding interactions, create helical foldamers, or assemble into ribbons.9 Paired hydrogen bonds, consisting of complementary ring nitrogen atoms and the protons on alpha‐amino groups, as seen in our simulations and shown in Figure 6, are present in examples of triazine‐containing self‐complementary molecules and triazine‐containing oligomers that self‐assemble into ribbons or tapes.9d,9e (Opportunities afforded by arrangements of multiple hydrogen bonds in assembly processes and polymers have been reviewed.10) Therefore, our simulations, in combination with past experimental work on related structures, support the idea that these new triazine‐based sequence‐defined polymers have the potential for conformational structure or self‐assembly behavior because of backbone–backbone interactions. We may anticipate that side‐chain structures and sequences may modulate the assembly processes.
This report describes the first general approach for converting cyanuric chloride to side‐chain functionalized sequence‐defined polymers, creating an architecture that is distinctive from other sequence‐defined polymer architectures in nature or synthesis. The submonomer approach offers tremendous opportunity for structural diversity, synthetically varying and sequencing both the side chains and the linkers. Heteroatoms can be selected that participate in hydrogen bonding (NH), or those that do not (S), thus enabling or limiting backbone/backbone interactions. As succinctly put by Archer and Krische, “The capability of defining structure carries with it the potential to engineer functionality”.9d The simulations show that noncovalent interactions are available that are similar to those in biopolymers, foldamers, and self‐assembled ribbons. The backbone has no readily hydrolyzed structures such as esters or peptide bonds. This new sequence‐defined architecture and synthetic methodology will thus be enabling for new macromolecules and materials with useful functions.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported as part of the Materials Synthesis and Simulation across Scales (MS3) initiative by the Laboratory Directed Research and Development program at Pacific Northwest National Laboratory (PNNL), a multi‐program national laboratory operated by Battelle for the U.S. Department of Energy(DOE).
J. W. Grate, K.-F. Mo, M. D. Daily, Angew. Chem. Int. Ed. 2016, 55, 3925.
References
- 1.
- 1a. Sequence-Controlled Polymers: Synthesis Self-Assembly, and Properties, Vol. 1170, American Chemical Society, Washington, D.C., 2014; [Google Scholar]
- 1b. Lutz J.-F., Ouchi M., Liu D. R., Sawamoto M., Science 2013, 341, 1238149; [DOI] [PubMed] [Google Scholar]
- 1c. Stayshich R. M., Weiss R. M., Li J., Meyer T. Y., Macromol. Rapid Commun. 2011, 32, 220–225; [DOI] [PubMed] [Google Scholar]
- 1d. Norris B. N., Zhang S., Campbell C. M., Auletta J. T., Calvo-Marzal P., Hutchison G. R., Meyer T. Y., Macromolecules 2013, 46, 1384–1392; [Google Scholar]
- 1e. Hartmann L., Krause E., Antonietti M., Börner H. G., Biomacromolecules 2006, 7, 1239–1244; [DOI] [PubMed] [Google Scholar]
- 1f. Sun J., Zuckermann R. N., ACS Nano 2013, 7, 4715–4732; [DOI] [PubMed] [Google Scholar]
- 1g. Yan J.-J., Wang D., Wu D.-C., You Y.-Z., Chem. Commun. 2013, 49, 6057–6059; [DOI] [PubMed] [Google Scholar]
- 1h. Pfeifer S., Zarafshani Z., Badi N., Lutz J.-F., J. Am. Chem. Soc. 2009, 131, 9195–9197; [DOI] [PubMed] [Google Scholar]
- 1i. Thanh Tam T., Oswald L., Chan-Seng D., Lutz J.-F., Macromol. Rapid Commun. 2014, 35, 141–145; [DOI] [PubMed] [Google Scholar]
- 1j. Thanh Tam T., Oswald L., Chan-Seng D., Charles L., Lutz J.-F., Chem. Eur. J. 2015, 21, 11961–11965; [DOI] [PubMed] [Google Scholar]
- 1k. Roy R. K., Meszynska A., Laure C., Charles L., Verchin C., Lutz J.-F., Nat. Commun. 2015, 6, 7237; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1l. Roy R. K., Laure C., Fischer-Krauser D., Charles L., Lutz J.-F., Chem. Commun. 2015, 51, 15677–15680; [DOI] [PubMed] [Google Scholar]
- 1m. Trinh T. T., Laure C., Lutz J.-F., Macromol. Chem. Phys. 2015, 216, 1498–1506; [Google Scholar]
- 1n. Espeel P., Carrette L. L. G., Bury K., Capenberghs S., Martins J. C., Du Prez F. E., Madder A., Angew. Chem. Int. Ed. 2013, 52, 13261–13264; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 13503–13506; [Google Scholar]
- 1o. Han J., Zheng Y. C., Zhao B., Li S. P., Zhang Y. C., Gao C., Sci. Rep. 2014, 4, 4387; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1p. Solleder S. C., Meier M. A. R., Angew. Chem. Int. Ed. 2014, 53, 711–714; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 729–732; [Google Scholar]
- 1q. Porel M., Alabi C. A., J. Am. Chem. Soc. 2014, 136, 13162–13165; [DOI] [PubMed] [Google Scholar]
- 1r. Zydziak N., Feist F., Huber B., Mueller J. O., Barner-Kowollik C., Chem. Commun. 2015, 51, 1799–1802; [DOI] [PubMed] [Google Scholar]
- 1s. Gutekunst W. R., Hawker C. J., J. Am. Chem. Soc. 2015, 137, 8038–8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Gellman S. H., Acc. Chem. Res. 1998, 31, 173–180; [Google Scholar]
- 2b. Guichard G., Huc I., Chem. Commun. 2011, 47, 5933–5941; [DOI] [PubMed] [Google Scholar]
- 2c. Hecht S., Huc I., Foldamers: Structure, Properties, and Applications, Wiley-VCH, Weinheim, 2007; [Google Scholar]
- 2d. Hill D. J., Mio M. J., Prince R. B., Hughes T. S., Moore J. S., Chem. Rev. 2001, 101, 3893–4012; [DOI] [PubMed] [Google Scholar]
- 2e. Cheng R. P., Curr. Opin. Struct. Biol. 2004, 14, 512–520. [DOI] [PubMed] [Google Scholar]
- 3. Zuckermann R. N., Kerr J. M., Kent S. B. H., Moos W. H., J. Am. Chem. Soc. 1992, 114, 10646–10647. [Google Scholar]
- 4.
- 4a. Bourguet E., Correia I., Dorgeret B., Chassaing G., Sicsic S., Ongeri S., J. Pept. Sci. 2008, 14, 596–609; [DOI] [PubMed] [Google Scholar]
- 4b. Zerkowski J. A., Hensley L. M., Abramowitz D., Synlett 2002, 0557–0560; [Google Scholar]
- 4c. Chaleix V., Sol V., Krausz P., Tetrahedron Lett. 2011, 52, 2977–2979. [Google Scholar]
- 5. Blotny G., Tetrahedron 2006, 62, 9507–9522. [Google Scholar]
- 6. Mutlu H., Lutz J.-F., Angew. Chem. Int. Ed. 2014, 53, 13010–13019; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13224–13233. [Google Scholar]
- 7.
- 7a. Sousa da Silva A., Vranken W., BMC Res. Notes 2012, 5, 367; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Wang J. M., Wolf R. M., Caldwell J. W., Kollman P. A., Case D. A., J. Comput. Chem. 2004, 25, 1157–1174; [DOI] [PubMed] [Google Scholar]
- 7c. Wang J., Wang W., Kollman P. A., Case D. A., J. Mol. Graphics Modell. 2006, 25, 247–260. [DOI] [PubMed] [Google Scholar]
- 8. Pronk S., Pall S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., Lindahl E., Bioinformatics 2013, 29, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Schmitt J.-L., Stadler A.-M., Kyritsakas N., Lehn J.-M., Helv. Chim. Acta 2003, 86, 1598–1624; [Google Scholar]
- 9b. Zhao H., Ong W. Q., Fang X., Zhou F., Hii M. N., Li S. F. Y., Su H., Zeng H., Org. Biomol. Chem. 2012, 10, 1172–1180; [DOI] [PubMed] [Google Scholar]
- 9c. Liu S., Zavalij P. Y., Lam Y.-F., Isaacs L., J. Am. Chem. Soc. 2007, 129, 11232–11241; [DOI] [PubMed] [Google Scholar]
- 9d. Archer E. A., Krische M. J., J. Am. Chem. Soc. 2002, 124, 5074–5083; [DOI] [PubMed] [Google Scholar]
- 9e. Beijer F. H., Kooijman H., Spek A. L., Sijbesma R. P., Meijer E. W., Angew. Chem. Int. Ed. 1998, 37, 75–78; [Google Scholar]; Angew. Chem. 1998, 110, 79–82. [Google Scholar]
- 10. Wilson A. J., Soft Matter 2007, 3, 409–425. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary