

Abstract
The base‐catalyzed allylic borylation of tertiary allylic alcohols allows the synthesis of 1,1‐disubstituted allyl boronates, in moderate to high yield. The unexpected tandem performance of the Lewis acid–base adduct, [Hbase]+[MeO‐B2pin2]− favored the formation of 1,2,3‐triborylated species from the tertiary allylic alcohols and 1‐propargylic cyclohexanol at 90 °C.
Keywords: allyl boronates, allylic alcohols, allylic borylation, metal-free, polyboronates
Allyl boron compounds have become one of the most important reagents for the regio‐ and stereoselective allylation of carbonyl compounds.1 Recently, many excellent applications have been reported, including examples for asymmetric synthesis.2 Of course, there is a considerable demand for developing efficient selective synthetic procedures that can access a wide variety of functionalized allyl boron compounds.
Despite the fact that the hydroxy group is one of the most reluctant leaving groups in substitution reactions, the borylation of allylic alcohols has become one of the most suitable methods for preparing the corresponding allyl boronates.3 From an economic perspective, allylic alcohols are stable and readily available substrates. However, the main allylic borylation reactions have been performed using activated allylic substrates, such as allylic carboxylates, carbonates, and halides, which are themselves usually prepared from the corresponding allylic alcohols.4 In this context, some of us were able to develop a palladium‐catalyzed allylic displacement reactions from allylic alcohols, opening a new perspective towards the synthesis and one‐pot functionalization of the allyl boronates.3a,3b, 5 In addition, the mechanism of the palladium‐catalyzed synthesis of allylic boronates from allylic alcohols has also been investigated in detail.6 In the last decade, the discovery of the alkoxide‐catalyzed β‐boration,7 diboration8, 9, 10 of unsaturated substrates from diboron reagents, as well as borylation of diaryliodonium salts,11 led to the development of conceptually new metal‐free borylation reactions with total control of the regio‐, diastereo‐, and enantioselectivity through quaternized diboron reagents.12
Herein, we aim to converge the two challenges into one, by carrying out the catalytic borylation of allylic alcohols only in the presence of the diboron reagents, base, and MeOH. This idea is based on the finding that alkoxides (formed from MeOH and base) might activate the diboron reagent through direct quaternization of one boryl unit to generate the corresponding Lewis acid–base adduct [Hbase]+[MeO‐B2pin2]−, which is characterized by an enhanced nucleophilic character of the B(sp2) unit.12a–12d
To experimentally verify the above hypothesis, we selected the commercially available tertiary allylic alcohol 1‐vinyl‐1‐cyclohexanol (1) as a model substrate to be borylated in the presence of a series of diboron reagents, such as bis(pinacolato)diboron (B2pin2), bis(neopentylglycolato) diboron (B2nep2), and bis(hexyleneglycolato)diboron (B2hex2), by the sole addition of a catalytic amount of base and MeOH (Scheme 1). We initially conducted several reactions with B2pin2 to identify the optimal base source, as well as the temperature required for high yields. The best yields and selectivities for borylation of 1 were achieved with Cs2CO3 at 70 °C, leading to 2 a in an isolated yield of 85 %, which represents a significant improvement in comparison with the alternative copper‐catalyzed hydroboration of the corresponding allene substrate (66 %).13
Scheme 1.

Optimized reaction conditions for the metal‐free allylic borylation of 1, with B2pin2, B2nep2, and B2hex2. % NMR yields [% Isolated yields].
Owing to the low efficiency of B2nep2 and B2hex2, we maintained B2pin2 as the reagent of choice and Cs2CO3 as the optimal base in our subsequent investigation into the generality of the reaction. Under the optimal reaction conditions for 1 (Table 1, entry 1) we examined the cyclic tertiary allylic alcohol 1‐vinyl‐1‐cycloheptanol (3), 1‐vinyl‐1‐cyclooctanol (5), and 1‐vinyl‐1‐cyclododecanol (7), which were prepared following a general procedure for the synthesis of tertiary allylic alcohols.14 The metal‐free borylation produced the corresponding cyclic allyl boronates 4, 6, and 8 in quantitative NMR yield and high isolated yields (Table 1, entries 2–4). These allyl boronates were synthesized for the first time in this work.
Table 1.
Substrate scope of the transition‐metal‐free allylic borylation of tertiary allylic alcohols.[a]
| Entry | Substrate | Product | NMR yield [%][b] | Isolated yield [%] |
|---|---|---|---|---|
| 1 |
 1
n=1
1
n=1 |
 2 a
n=1
2 a
n=1 |
99 | 85 |
| 2 | 3 n=2 | 4 n=2 | 92 | 84 |
| 3 | 5 n=3 | 6 n=3 | 97 | 79 |
| 4 | 7 n=7 | 8 n=7 | 99 | 79 |
| 5 |
 9
9
|
 10
10
|
97 | 55 |
| 6 |
 11
11
|
 12
12
|
95 | 60 |
| 7 |
 13
13
|
 14
14
|
71 | 42 |
[a] Reaction conditions: tertiary allylic alcohols (0.3 mmol), B2pin2 (2 equiv), Cs2CO3 (15 mol %), THF (0.5 mL), MeOH (10 equiv), at 70 °C for 16 h. [b] Yields were determined by 1H NMR analysis of the crude reaction mixture with naphthalene as an internal standard, which was added after the reaction. [c] E/Z ratio for 14 is 1:1.
Considering the potentially useful reactivity observed with the cyclic allylic alcohols, we next conducted a series of allylic borylation with acyclic allylic alcohols (Table 1, entries 5–7). Interestingly, under the same reaction conditions, we were able to transform substrates 9, 11, and 13 into the corresponding allyl boronates 10, 12 (as a single diastereoisomer), and 14 (E/Z=1:1). The last reaction was particularly interesting as the allylic borylation was selectively performed over the diboration of the concurrent double bond in substrate 13 (Table 1, entry 7). The synthetic efficiency of the allylboration of tertiary allylic alcohols under the transition‐metal‐free conditions is comparable to the corresponding methods with palladium catalysis.3a,3b
Next, we turned our attention to the substituted cyclohexyl allylic alcohols14 to study the influence in the reaction outcome, of the substituents on the cyclic ring (Scheme 2). For these 4‐substituted substrates, the two diastereoisomeric alcohols were isolated in pure form,15 and in the case of 19 b we obtained the crystal diffraction data to conclude its relative configuration (Supporting Information). Both diastereoisomers (15 a and b) were independently subjected to allylic borylations under the same reaction conditions. Scheme 2 shows an identical product 16 formed in both reactions with similar yields, independently of the relative configuration of 15 a or 15 b. Similar observations were made for the epimeric compounds 17 a and 17 b, as well as 19 a and 19 b. When the cyclohexyl allylic alcohol was 4‐substituted with iPr, the mixture of diastereomers could not be separated, so the borylation was carried out directly from substrate 21, to form the corresponding allyl boronate 22 in moderate yield (Scheme 2). These allyl boronates (16, 18, 20, 22) have been reported for the first time in this work.
Scheme 2.

a) Scope of transition‐metal‐free allylic borylation of substituted cyclohexyl allylic alcohols (0.3 mmol), B2pin2 (2 equiv), Cs2CO3 (15 mol %), THF (2.0 mL), MeOH (10 equiv), at 70 °C for 16 h. IY: [% Isolated yields].
Moving from 4‐substituted to 3‐ or 2‐substituted cyclohexyl allylic alcohols, the transition‐metal‐free allylic boryl‐ation provided mixtures of cis/trans allylic boronates. Scheme 2 shows the transformation of the two diastereoisomers of 3‐methyl‐1‐vinyl‐1‐cyclohexanol (23 a and 23 b), into the allylic boronate 24 as a 50:50 mixture of isomers. A similar trend was observed for 2‐methyl‐1‐vinyl‐1‐cyclohexanol (25) and 2‐phenyl‐1‐vinyl‐1‐cyclohexanol (27; Scheme 2). The bicyclic substrate 29, synthesized from decalone in its chiral form, was subjected to the metal‐free allylic borylation, and the allylic boronate 30 was isolated in a yield of 65 %. The metal‐free allylic borylation was also performed for the chiral substrate 31, prepared from the 5‐α‐cholestan‐3‐one, and the corresponding allylic boronate 32 was provided in a 1:1 mixture of cis/trans isomers (Scheme 2) with 80 % NMR yield, despite the fact it was isolated only in a yield of 33 %. All of these allyl boronates (24, 26, 28, 30, 32) were synthesized for the first time in this work.
The tolerance of other functional groups was also studied. We were delighted to see that the allylic borylation was selectively carried out in substrate 33 a, versus the potential metal‐free diboration8a of the concurrent double bond (Scheme 3). The reaction was performed under the same optimized reaction conditions, and in this case the cis/trans ratio of the product 34 a was 4:1. We also explored the transition‐metal‐free allylic borylation of N‐ and O‐containing tertiary alcohols. The reactions indeed performed well and showed a high level of functional group compatibility, providing 34 b and 34 c in moderate yield as single isomers (Scheme 3).
Scheme 3.
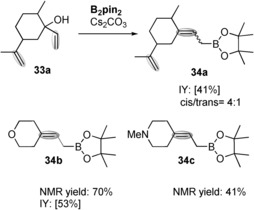
Transition‐metal‐free allylic borylation of polyfunctional substituted cyclohexyl allylic alcohols (0.3 mmol), B2pin2 (2 equiv), Cs2CO3 (15 mol %), THF ((0.5 mL), MeOH (10 equiv), at 70 °C for 16 h. [% Isolated yields].
The mechanism of this reaction can be best described as an SN2′‐type process (Scheme 4). Accordingly, the initial step is a base‐mediated nucleophilic attack by the methoxy group of MeOH on one of the Bpin moieties of B2pin2 to give adduct (a). As a consequence, the other Bpin unit becomes nucleo‐philic,12a–12d and attacks the terminal position of alcohol 1, which leads to C−O bond cleavage in intermediate (b). The OH group reacts with the conjugated acid of the base (BaseH+) to form water and regenerate the base catalyst. Simultaneous formation of the new carbon−boron bond and cleavage of the carbon−oxygen bond of 1 leads to the formation of allyl boronate 2 a. The addition of MeOH and base is a prerequisite for the formation of adduct a, and thus for formation of the allyl‐Bpin product 2 a. In the absence of MeOH and base, we did not observe any formation of 2 a from 1. The presence of the unprotected hydroxy group in substrate 1 is also essential. We could not observe any formation of 2 a using benzyl‐protected substrate 1(OBn) (Scheme 4). A possible explanation for this is that the C−O bond cleavage and/or protonation of the oxygen by BaseH+ is more difficult in benzyl derivative 1(OBn) than in 1 with a free hydroxy group. When we carried out the borylation reaction with the secondary alcohol 1‐octen‐3‐ol, the isolated yield was moderate (17 %), while the primary alcohol (E)‐2‐octen‐1‐ol did not react at all. This could be the consequence of the better leaving group ability of a protonated tertiary hydroxy group (such as in 1) compared to primary or secondary ones. Accordingly, the metal‐free borylation method is the most efficient for tertiary allyl alcohols and is limited in scope with primary and secondary allyl alcohol substrates.
Scheme 4.

Suggested mechanism for the metal‐free allylic borylation.
To complement the study, we carried out the metal‐free allylic borylation of 1‐propargylic cyclohexanol (35) and, to our delight, we observed that the main product was the corresponding hydroborated triple bond (36), working at 50 °C, with an isolated yield of 91 % (Scheme 5). Interestingly, by increasing the temperature to 70 or 90 °C, the alkenyl borane 37 was selectively formed, possibly as a consequence of the diboration of the allene intermediate.16 Working at 90 °C, it was also possible to observe the allyl boronate 2 a up to 15 % of NMR yield.
Scheme 5.

Transition‐metal‐free allylic borylation of 1‐propargyl cyclohexanol (35, 0.3 mmol), B2pin2, Cs2CO3 (15 mol %), THF (0.5 mL), MeOH (10 equiv), % NMR yields, [% Isolated yields].
Remarkably, the double bond, formed in the allyl boronates during the transition‐metal‐free allylic borylation, might be susceptible to diboration in the same reaction conditions. Experimentally, when the allylic borylation of the model substrate 1‐vinyl‐1‐cyclohexanol (1), is carried out within 90 h at 70 °C, a new product can be identified as the triborated 38 (Scheme 6). Our explanation to understand the in situ formation of 38 is based on the plausible metal‐free diboration of product 2 a. This fact was confirmed by a separate study of the metal‐free diboration of isolated compound 2 a with 2 equiv of B2 pin2 and 15 mol % of Cs2CO3 at 70 °C for 90 h, leading to 38 in high yields (Supporting Information). This unexpected tandem performance of the Lewis acid–base adduct, [Hbase]+[MeO‐B2pin2]−, has not been observed before and it serves as the basis of a new metal‐free tandem borylation reaction towards polyborated compounds. Interestingly, when we explored the metal‐free tandem allylic borylation/diboration of 1‐vinyl‐cyclopentanol (39) and 1‐vinyl‐1‐cyclobutanol (42), the triborylated products were probably mainly formed as a consequence of the more congested cyclobutane and cyclopentane systems (Scheme 6). The X‐ray diffraction structure of 1,2,3‐poly‐borated product 44 is shown in Scheme 6.17
Scheme 6.

Transition‐metal‐free tandem allylic borylation/diboration (0.3 mmol), B2pin2, Cs2CO3 (15 mol %), THF (0.5 mL), MeOH (10 equiv), % NMR yields, [% Isolated yields]. X‐Ray diffraction structure of 44.
To conclude, we have developed an unprecedented metal‐free allylic borylation of tertiary allylic alcohols that provides access to new allylic boronates. The reactions are carried out in the presence of 15 mol % of Cs2CO3 and MeOH to promote the formation of the Lewis acid–base adduct, [Hbase]+[MeO‐B2pin2]−, which may also be responsible for the in situ diboration of the allylic double bond that generates 1,2,3‐polyborated products.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The present research was supported by the Spanish Ministerio de Economia y Competitividad (MINECO) through project CTQ2013‐43395P. KJS thanks the Swedish Reserach Council and the KAW foundation for support. We thank AllyChem for the gift of diboranes.
N. Miralles, R. Alam, K. J. Szabó, E. Fernández, Angew. Chem. Int. Ed. 2016, 55, 4303.
References
- 1.
- 1a. Hall D. G., Boronic Acids, Wiley-VCH, Weinheim, 2011; [Google Scholar]
- 1b. Hall D. G., Lachance H., Allylboration of Carbonyl Compounds, Wiley, Hoboken, New Jersey, 2012; [Google Scholar]
- 1c. Yus M., González-Gómez J. C., Foubelo F., Chem. Rev. 2013, 113, 5595. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Ding J. Y., Hall D. G., Angew. Chem. Int. Ed. 2013, 52, 8069; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8227; [Google Scholar]
- 2b. Ito H., Okura T., Matsuura K., Sawamura M., Angew. Chem. Int. Ed. 2010, 49, 560; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 570; [Google Scholar]
- 2c. Hesse M., Essafi S., Watson C., Harvey J., Hirst D., Willis C., Aggarwal V. K., Angew. Chem. Int. Ed. 2014, 53, 6145; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6259; [Google Scholar]
- 2d. Ferris G. E., Hong K., Roundtree I. A., Morken J. P., J. Am. Chem. Soc. 2013, 135, 2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Olsson V. J., Sebelius S., Selander N., Szabó K. J., J. Am. Chem. Soc. 2006, 128, 4588; [DOI] [PubMed] [Google Scholar]
- 3b. Selander N., Kipke A., Sebelius S., Szabó K. J., J. Am. Chem. Soc. 2007, 129, 13723; [DOI] [PubMed] [Google Scholar]
- 3c. Dutheuil G., Selander N., Szabó K. J., Aggarwal V. K., Synthesis 2008, 2293; [Google Scholar]
- 3d. Selander N., Szabó K. J., J. Org. Chem. 2009, 74, 5695; [DOI] [PubMed] [Google Scholar]
- 3e. Selander N., Szabó J., Dalton Trans. 2009, 6267; [DOI] [PubMed] [Google Scholar]
- 3f. Selander N., Paasch J. R., Szabó K. J., J. Am. Chem. Soc. 2011, 133, 409. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Horino Y., Aimono A., Abe H., Org. Lett. 2015, 17, 2824; [DOI] [PubMed] [Google Scholar]
- 4b. Atack T. C., Lecker R. M., Cook S. P., J. Am. Chem. Soc. 2014, 136, 9521; [DOI] [PubMed] [Google Scholar]
- 4c. Zhang P., Roundtree I. A., Morken J. P., Org. Lett. 2012, 14, 1363; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Ito H., Kawakami C., Sawamura M., J. Am. Chem. Soc. 2005, 127, 16034; [DOI] [PubMed] [Google Scholar]
- 4e. Ito H., Ito S., Sasaki Y., Matsuura K., Sawamura M., J. Am. Chem. Soc. 2007, 129, 14856; [DOI] [PubMed] [Google Scholar]
- 4f. Sebelius S., Wallner O. A., Szabó K. J., J. Org. Chem. 2003, 5, 3065; [DOI] [PubMed] [Google Scholar]
- 4g. Sebelius S., Szabó K. J., Eur. J. Org. Chem. 2005, 5, 2539; [Google Scholar]
- 4h. Yamamoto E., Takenouchi Y., Ozaki T., Miya T., Ito H., J. Am. Chem. Soc. 2014, 136, 16515. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Sebelius S., Olsson V. J., Wallner O. A., Szabó K. J., J. Am. Chem. Soc. 2006, 128, 8150; [DOI] [PubMed] [Google Scholar]
- 5b. Selander N., Sebelius S., Estay C., Szabó K. J., Eur. J. Org. Chem. 2006, 4085; [Google Scholar]
- 5c. Aydin J., Kumar K. S., Sayah M. J., Wallner O. A., Szabó K. J., J. Org. Chem. 2007, 72, 4689. [DOI] [PubMed] [Google Scholar]
- 6. Larsson J. M., Szabó K. J., J. Am. Chem. Soc. 2013, 135, 443. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Bonet A., Gulyás H., Fernández E., Angew. Chem. Int. Ed. 2010, 49, 5130; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5256; [Google Scholar]
- 7b. Pubill-Ulldemolins C., Bonet A., Bo C., Gulyás H., Fernández E., Chem. Eur. J. 2012, 18, 1121; [DOI] [PubMed] [Google Scholar]
- 7c. Cid J., Carbó J. J., Fernández E., Chem. Eur. J. 2014, 20, 3616; [DOI] [PubMed] [Google Scholar]
- 7d. Cascia E. L., Sanz X., Bo C., Whiting A., Fernández E., Org. Biomol. Chem. 2015, 13, 1328. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Bonet A., Pubill-Ulldemolins C., Bo C., Gulyás H., Fernández E., Angew. Chem. Int. Ed. 2011, 50, 7158; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7296; [Google Scholar]
- 8b. Bonet A., Solé C., Gulyás H., Fernández E., Org. Biomol. Chem. 2012, 10, 6621; [DOI] [PubMed] [Google Scholar]
- 8c.diboration of C=N bonds: Solé C., Gulyás H., Fernández E., Chem. Commun. 2012, 48, 3769; [DOI] [PubMed] [Google Scholar]
- 8d. Miralles N., Cid J., Cuenca A. B., Carbó J. J., ernández E., Chem. Commun. 2015, 51, 1693. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Nagashima Y., Hirano K., Takita R., Uchiyama M., J. Am. Chem. Soc. 2014, 136, 8532; [DOI] [PubMed] [Google Scholar]
- 9b. Blaisdell T. P., Caya T. C., Zhang L., Sanz-Marco A., Morken J. P., J. Am. Chem. Soc. 2014, 136, 9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.1,1-diboration:
- 10a. Li H., Shangguan X., Zhang Z., Huang S., Zhang Y., Wang J., Org. Lett. 2014, 16, 448; [DOI] [PubMed] [Google Scholar]
- 10b. Cuenca A. B., Cid J., García-López D., Carbó J. J., Fernández E., Org. Biomol. Chem. 2015, 13, 9659. [DOI] [PubMed] [Google Scholar]
- 11. Miralles N., Romero M., Fernández E., Muñiz K., Chem. Commun. 2015, 51, 14068. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Cid J., Carbó J. J., Fernández E., Chem. Eur. J. 2012, 18, 12794; [DOI] [PubMed] [Google Scholar]
- 12b. Cid J., Gulyás H., Carbó J. J., Fernández E., Chem. Soc. Rev. 2012, 41, 3558; [DOI] [PubMed] [Google Scholar]
- 12c. Dewhurst R. D., Neeve E. C., Braunschweig H., Marder T. B., Chem. Commun. 2015, 51, 9594; [DOI] [PubMed] [Google Scholar]
- 12d. Pietsch S., Neeve E. C., Apperley D. C., Bertermann R., Mo F., Qiu D., Cheung M. S., Dang L., Wang J., Radius U., Lin Z., Kleeberg C., Marder T. B., Chem. Eur. J. 2015, 21, 7082; [DOI] [PubMed] [Google Scholar]
- 12e. Yamamoto E., Ukigai S., Ito H., Chem. Sci. 2015, 6, 2943; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12f. Lee K.-S., Zhugralin A. R., Hoveyda A. H., J. Am. Chem. Soc. 2009, 131, 7253; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12g.correction: Lee K., Zhugralin A. R., Hoveyda A. H., J. Am. Chem. Soc. 2010, 132, 12766; [Google Scholar]
- 12h. Wu H., Garcia J. M., Haeffner F., Radomkit S., Zhugralin A. R., Hoveyda A. H., J. Am. Chem. Soc. 2015, 137, 10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Semba K., Shinomiya M., Fujihara T., Terao J., Tsuji Y., Chem. Eur. J. 2013, 19, 7125. [DOI] [PubMed] [Google Scholar]
- 14. Albrecht U., Langer P., Tetrahedron 2007, 63, 4648. [Google Scholar]
- 15. Ouellette R. J., Liptak K., Booth G. E., J. Org. Chem. 1966, 31, 546. [Google Scholar]
- 16.
- 16a. Ishiyama T., Kitano T., Miyaura N., Tetrahedron Lett. 1998, 39, 2357; [Google Scholar]
- 16b. Ito H., Sasaki Y., Sawamura M., J. Am. Chem. Soc. 2008, 130, 15774. [DOI] [PubMed] [Google Scholar]
- 17.CCDC 1455256 and 1455255 contain the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary