

Abstract
A study of P4 transformations at low‐valent iron is presented using β‐diketiminato (L) FeI complexes [LFe(tol)] (tol=toluene; L=L1 (1 a), L2 (1 b), L3 (1 c)) with different combinations of aromatic and backbone substituents at the ligand. The products [(LFe)4(μ4‐η2:η2:η2:η2‐P8)] (L=L1 (2 a), L2 (2 b)) containing a P8 core were obtained by the reaction of 1 a,b with P4 in toluene at room temperature. Using a slightly more sterically encumbered ligand in 1 c results in the formation of [(L3Fe)2(μ‐η4:η4‐P4)] (2 c), possessing a cyclo‐P4 moiety. Compounds 2 a–c were comprehensively characterized and their electronic structures investigated by SQUID magnetization and 57Fe Mössbauer spectroscopy as well as by DFT methods.
Keywords: Mössbauer spectroscopy, paramagnetic compounds, small-molecule activation, substituent effects, white phosphorus
The activation of white phosphorus (P4) with main‐group1 and transition‐metal2 compounds is an ongoing area of research. The latter topic is dominated by CpR containing transition‐metal complexes.2 More recently, complexes of the β‐diketiminato (nacnac=L) ligand have been employed for P4 activation as well. For early transition‐metal compounds, exclusively Group 5 complexes were used,3 whereas for electron‐rich metals Group 8–10 complexes have been applied so far.4 Selected examples of Pn complexes A–D with β‐diketiminato ligands of late transition metals are shown in Figure 1. Recently, we reported on the CuI compounds [(LCu)2(μ‐η2:η2‐E4)] (E=P (D), As) and [LCu(η2‐P4)], respectively, containing intact E4 moieties,5 while all other examples (A–C) contain transformed P4 units. Also, we investigated the reaction of FeI complexes [LFe(tol)] with P4. When the Driess group recently reported on the formation of the FeIII complex [(L0Fe)2(μ‐η2:η2‐P2)2] (A), containing two dianionic P2 ligands,4a we were surprised as our investigations showed quite different results. Since the reaction conditions were identical, we supposed that the reason for the different P4 activation pathways (and products) was due to the slightly different aromatic flanking groups and α‐backbone substituents of our [LFe(tol)] precursors. Therefore, we systematically studied the driving forces for the different outcome of P4 activation by FeI centers.
Figure 1.
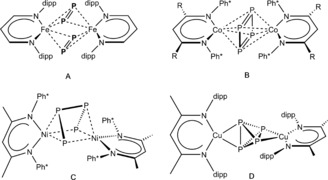
Selected examples of Pn complexes with late transition metals Fe, Co, Ni, and Cu supported by the β‐diketiminato ligand.4, 5
Herein, we present a comparative study of P4 activation by FeI β‐diketiminato (L) complexes [LFe(tol)] (L=L1 (1 a), L2 (1 b), L3 (1 c)) with toluene (tol) as a labile leaving group. The starting materials [LFe(tol)] (L=L1 (1 a), L2 (1 b), L3 (1 c)) were synthesized in a one‐pot synthesis (see the Supporting Information) and characterized by single‐crystal X‐ray crystallography (1 b and 1 c, see the Supporting Information).
The reaction of [L1Fe(tol)] (1 a) with 0.5 equivalent of P4 in toluene at room temperature leads to the formation of a tetranuclear complex, namely [(L1Fe)4(μ4‐η2:η2:η2:η2‐P8)] (2 a), which displays a realgar‐type6 P8 moiety. Changing the stoichiometry of the reaction does not affect the product formation (ratio [L1Fe(tol)]/P4=2:1 and 1:2). The formation of a P8 moiety in 2 a is in contrast to the recently reported product, [(L0Fe)2(μ‐η2:η2‐P2)2] (A), published by the Driess group,4a which contains two [P2]2− ligands (Scheme 1). A comparison of ligand L0 with L1, however, displays only small differences in the aromatic (Ph*=dipp (=2,6‐diisopropylphenyl) or dmp (=2,6‐dimethylphenyl)) and in the backbone (R) substituents. In both cases the reaction conditions were identical. Therefore, we were interested to understand whether the steric demand or the electronic properties of the aromatic flanking groups Ph* and backbone α‐substituents R cause the different reactivity of the FeI precursors towards P4. According to DFT calculations at the BP86//def2‐SVP/def2‐TZVP (N, Fe, P) level, the dimerization of the hypothetical complex [(L1Fe)2(μ‐η4:η4‐P4)] (quintet spin state) to 2 a (nonet spin state) is endothermic (91.5 kJ mol−1). This seems to be in contrast with the experimental results. However, considering that the unrestricted singlet spin state of 2 a is more stable than the nonet spin state (102.1 kJ mol−1), the reaction becomes exothermic. Furthermore, the natural population analyses (NPA) clearly indicates the presence of FeII centers and [P8]4− ligand in 2 a.
Scheme 1.
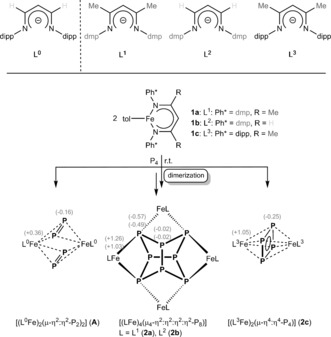
Top: Comparison of L0 with ligands L1, L2, and L3, containing a variety of different substituents. Bottom: Coordinated Pn moieties obtained by P4 transformation with different FeI precursors. The gray numbers in brackets represent the NPA charges at the corresponding atoms.7 For 2 a,b the upper value corresponds to 2 a.
Accordingly, we decided to additionally synthesize ligand L2 (see Scheme 1, top), representing the missing combination between ligands L0 and L1, to investigate the steric and electronic effects induced by the different substitution of the chelating N atoms and the ligand backbone. Conducting the reaction of [L2Fe(tol)] (1 b) and P4 under identical conditions (RT, toluene) and same stoichiometries (2:1 and 1:2) facilitates the clean and selective formation of the P8 moiety containing complex [(L2Fe)4(μ4‐η2:η2:η2:η2‐P8)] (2 b) (Figure 2). Even if a higher local concentration of P4 was used by the dropwise addition of 1 equivalent of 1 b to a solution of 2 equiv of P4 in toluene, 2 b is the only product of the reaction. Comparing 2 a and 2 b, we assume that the methyl flanking groups in dmp are not able to prevent the dimerization reaction to the P8 moiety, as the dipp substituents did in [(L0Fe)2(μ‐η2:η2‐P2)2] (A). Along with A,4a possessing two separate P2 units, compounds 2 a,b are different activation steps of P4 (Scheme 1).
Figure 2.
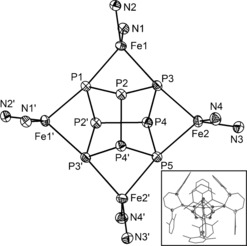
Core structure of 2 b in crystals of 2 b⋅toluene (hydrogen and carbon atoms are omitted for clarity; ellipsoids are set at 50 % probability).15 A representation of 2 b with its complete ligands is shown in the inset.
A single‐crystal X‐ray structural analysis reveals that compounds 2 a⋅2 toluene and 2 b⋅toluene are isostructural (Figure 2 for 2 b). Both compounds contain a realgar‐type P8 ligand coordinating to four [LFe] (L=L1 (2 a), L=L2 (2 b)) fragments. All P−P distances are in the range of 2.1991(8) to 2.2813(7) Å in 2 a and 2.2111(6) to 2.2792(6) Å in 2 b; and therefore, are in line with P−P single bonds (for comparison: P−P single bond in white phosphorus determined by electron diffraction: 2.1994(3) Å,8 Raman spectroscopy: 2.2228(5) Å,9 and DFT calculations: 2.1994(3) Å8). The coordination geometry of the Fe metal centers in 2 a and 2 b, respectively, is best described as distorted tetrahedral. The torsion angles between the Fe‐P‐P and Fe‐N‐N planes are between 74.66(6)° and 84.74(5)° in 2 a and 83.45(4)° and 84.91(6)° in 2 b. There are no significant differences in the P−P bond distances in 2 a,b and those of previously reported related P8 ligands in [(NNfcSc)4P8], [(Cp*Sm)4P8] (Cp*=C5Me5), [CpMe 4Fe4(CO)6P8] (CpMe=C5H4Me), [(CpMe 4Fe6(CO)13P8], and [Cp*2Ir2Cr3(CO)17P8].10
The Fe−N distances lie between 1.983(2) and 2.006(2) Å in 2 a and between 1.982(2) and 1.990(2) Å in 2 b. The distances of Fe and the coordinating phosphorus atoms are in the range of 2.4559(6) and 2.5006(6) Å in 2 a and 2.4583(3) and 2.4807(5) Å in 2 b, respectively.
No signals were detected in the 31P{1H} NMR spectra of 2 a,b. These solutions (2 a in C6D6 and 2 b in [D8]toluene) are also EPR‐silent at RT as well as at 10 K, suggesting a higher spin multiplicity or antiferromagnetically coupled iron centers that result in a non‐magnetic (EPR‐silent) ground state at low temperature. However, the 1H NMR spectra of 2 a and 2 b reveal signals in the range from 273 ppm to −29 ppm; thus indicating a paramagnetic spin state for 2 a,b. The careful analysis of the spectra enabled us to assign all resonances (see the Supporting Information). The effective magnetic moment (μ eff) at room temperature was determined to be 6.79 μB for 2 a in C6D6 and 6.71 μB for 2 b in [D8]THF solution (Evans method). These values are well‐confirmed by temperature‐dependent SQUID measurements in the solid state. Both complexes exhibit a similar magnetic behavior with a strong temperature dependency of their effective magnetic moments over a temperature range between 2 and 300 K. At 2 K, the effective magnetic moments amount to 1.14 μB (2 a) and 0.54 μB (2 b). With increasing temperature, the magnetic moments gradually increase until effective magnetic moments of 7.04 μB (2 a) and 6.92 μB (2 b) are reached at 300 K (see the Supporting Information). This magnetic behavior is likely caused by an antiferromagnetic coupling. The zero‐field 57Fe Mössbauer spectrum of 2 b at 77 K shows a doublet with an isomer shift δ of 0.73(1) mm s−1 and a quadrupole splitting ΔE Q of 1.93(1) mm s−1, which is in agreement with a high‐spin iron(II) complex. Similar Mössbauer parameters have been observed in the four‐coordinate iron(II) complex [PhB(MesIm)3Fe(N=PPh3)].11 The presence of iron(II) centers in 2 b is also indicated by NPA analysis.
So far, we assume that the aromatic dmp substituents at the coordinating N atoms of the ligand play a crucial role for the formation of the P8 ligand moieties in 2 a and 2 b, and the α‐substituent of the ligand backbone does not have much influence on the outcome of P4 activation. Regardless, to conclusively address this point, the ligand L3H was synthesized (Scheme 1). While L3 features aromatic dipp groups at the coordinating N atoms (like L0), its ligand backbone is substituted with two Me α‐substituents (like L1); and hence, represents the missing hybrid ligand between L0 and L1. Owing to steric reasons, the Me substituents at the ligand backbone are restricting the rotational flexibility of the iPr groups in dipp, thus increasing their steric pressure.12
The reaction of 1 c with 0.5 equivalent of P4 in toluene at RT leads to the formation of [(L3Fe)2(μ‐η4:η4‐P4)] (2 c), containing a cyclo‐P4 moiety. Again, changing the stoichiometry of the reaction does not have an effect on the product formation ([L3Fe(tol)]/P4=2:1 and 1:2). Different from our experience with the complexes of the dmp containing ligands L1 and L2, we now obtain a cyclo‐P4 unit in the product 2 c, which is also in contrast to Driess’ product A, featuring two separated P2 units (Scheme 1).
Single crystals of 2 c suitable for X‐ray diffraction were grown from a saturated toluene solution (Figure 3). Compound 2 c is a centrosymmetric dinuclear iron complex that consists of two [L3Fe] fragments bridged by a planar cyclo‐P4 ligand. The middle deck displays weak disorder (occupancy 97:3; see the Supporting Information). In the following, only the major component of the middle deck is discussed. The P−P distances within the central P4 moiety (P1−P2 and P1−P2′) in 2 c amount to 2.178(1) and 2.207(1) Å, respectively. These distances are longer than those reported for cyclo‐[P4]2− ligands (2.146(1)–2.1484(9) Å)13 and shorter than those reported for cyclo‐[P4]4− moieties (2.230(2)–2.259(2) Å).14 The angles of P2′‐P1‐P2 and P1‐P2‐P1′ are 91.73(3)° and 88.27(3)°, respectively, indicating a slightly distorted ring conformation. The Fe−P distances are between 2.4376(6) and 2.5163(6) Å, comparable to those observed in 2 a and 2 b. Similarly, the Fe−N distances in 2 c (2.018(2) and 2.025(2) Å) are comparable to A (2.023(3) and 2.025(3) Å),4a but slightly elongated compared to 2 a (1.983(2) and 2.006(2) Å) and 2 b (1.982(2) and 1.990(2) Å). The Fe1−Fe1′ distance in 2 c is 3.902 Å, being significantly elongated compared to compound A (2.777 Å). One of the most remarkable differences between 2 c and A is the torsion angle θ between the Fe−Fe axis and the plane formed by the nitrogen atoms and the methine carbon atom in the ligand backbone, which is considerably smaller in 2 c (15°) compared to A (33°; Figure 4).
Figure 3.
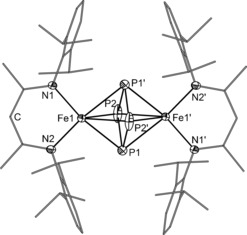
Molecular structure of 2 c (hydrogen atoms are omitted for clarity; ellipsoids are set at 50 % probability).15 Selected bond lengths [Å] and angles [°]: P1–P2 2.178(1), P1–P2′ 2.207(1), Fe1–P1 2.4376(6), Fe1–P2 2.5064(6), Fe1–P1′ 2.5163(6), Fe1–P2′ 2.5064(6), Fe1–N1 2.018(2), Fe1–N2 2.025(2), Fe1–Fe1′ 3.902; P2′‐P1‐P2 91.73(3), P1‐P2‐P1′ 88.27(3).
Figure 4.
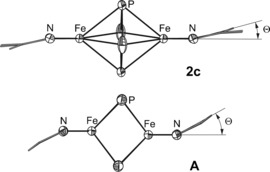
Comparison of the coordination geometry in 2 c and A.4a
Like in the tetranuclear complexes 2 a,b, no resonances were detected in the 31P{1H} NMR spectra of 2 c and solutions of 2 c are EPR‐silent at room temperature and at 10 K. However, the 1H NMR spectra of 2 c in [D8]THF reveals signals in the range from 7 ppm to −2 ppm. The magnetic moment of 2 c in [D8]THF at RT was determined to be 3.09 μB (Evans method). Temperature‐dependent SQUID measurements in the solid state are in agreement with this result with an effective magnetic moment of 3.46 μB at 300 K. The magnetism of complex 2 c is strongly temperature‐dependent. At 2 K, the effective magnetic moment was determined to be 0.54 μB, and is rising to 1.00 μB at 20 K. Between 20 and 80 K, it remains roughly constant. Increasing the temperature to 300 K leads to a gradual increase of the effective magnetic moment up to a value of 3.46 μB at 300 K (see the Supporting Information). This magnetic behavior is explained by a S tot=0 ground state between 0 and 80 K and antiferromagnetic coupling of the two iron nuclei at higher temperatures. The zero‐field 57Fe Mössbauer spectrum of 2 c at 77 K features a doublet with an isomer shift δ of 0.74(1) mm s−1 and a quadrupole splitting ΔE Q of 1.74(1) mm s−1, which is very similar to the Mössbauer parameters of 2 b and is in accordance with a high‐spin iron(II) complex.
The optimized geometry of 2 c in the quintet spin state obtained from DFT calculations (BPW91/def2‐SVP) is in good agreement with the experimentally found geometric parameters, with a slightly shorter Fe−Fe distance (3.827 Å) and slightly longer P−P distances (2.203–2.250 Å).15 Notably, the geometry optimization in the unrestricted singlet spin state instead leads to further shortening of the Fe−Fe distance (3.712 Å) and to a planar P4 ring with two shorter and two longer P−P distances (2.181 Å and 2.325 Å, respectively). Since the Fe−Fe distance in A (2.777 Å) is significantly shorter than in 2 c, the geometry of 2 c (quintet spin state) was optimized with a fixed Fe−Fe distance of 2.777 Å. In the optimized geometry, the cyclo‐P4 unit is cleaved into two P2 units and the nacnac ligand shows the same type of folding like the one reported for A. The energy difference between both isomers is 29.19 kJ mol−1, favoring the relaxed geometry of 2 c. This points towards a flat energy surface and suggests that the outcome of the P4 transformation is mostly determined by the Fe−Fe distance. Broken symmetry calculations (BPW91//def2‐SVP/aug‐cc‐pVTZ (Fe, P)) indicate an antiferromagnetic coupling between the two Fe centers, which increases with the decrease of the Fe−Fe distance.15 The Mulliken population analysis for the quintet spin state of 2 c shows that the spin density is localized on iron atoms, but no considerable spin density was found on the P4 or nacnac ligands. The Mayer bond order for the P−P bonds vary from 0.81 to 0.87; thus, indicating P−P single bonds.
In conclusion, we have shown that the different reactivity of β‐diketiminato FeI complexes [LFe(tol)] (L=L1 (1 a), L2 (1 b), L3 (1 c)) towards P4 is sensitive to minimal changes in the ligand: its flanking groups (Ph*) and its backbone α‐substituents (R). By conducting the reactions under similar conditions (RT) in the same solvent (toluene), and using exact stoichiometric amounts of P4 ([LFe(tol)]/P4=2:1) or even larger amounts of P4 ([LFe(tol)]/P4=1:2), a different outcome of P4 activation is realized. By employing the aromatic dmp flanking groups as substituents of the coordinating N atoms, the formation of a [P8]4− structural motif in the iron(II) compounds [(LFe)4(μ4‐η2:η2:η2:η2‐P8)] (L=L1 (2 a), L=L2 (2 b)) is observed.7b Employing the sterically more demanding dipp substituents leads to the formation of an iron(II) compound [(L3Fe)2(μ‐η4:η4‐P4)] (2 c), containing a cyclo‐[P4]2− moiety. This finding is in contrast to the formation of two separate [P2]2− units observed in the iron(III) complex A, with two H α‐substituents being located in the ligand backbone instead of Me atoms in 2 c. This demonstrates the additional steric influence of the Me groups as α‐substituents to push the dipp substituents closer together, thereby preventing the opening of the cyclo‐P4 ring by relaxing the Fe⋅⋅⋅Fe distance in 2 c in comparison with the rather short distance in A. The discussed ligand dependencies in the β‐diketiminato ligand complexes may foster the systematic study of such dependencies in other metal systems for the activation of small molecules in general and in particular for the controlled Pn ligand formation from white phosphorus.
Dedicated to Professor Hangeorg Schnöckel on the occasion of his 75th birthday
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft. The European Research Council (ERC) is acknowledged for the support in the SELFPHOS AdG‐339072 project.
F. Spitzer, C. Graßl, G. Balázs, E. M. Zolnhofer, K. Meyer, M. Scheer, Angew. Chem. Int. Ed. 2016, 55, 4340.
References
- 1.
- 1a. Scheer M., Balázs G., Seitz A., Chem. Rev. 2010, 110, 4236–4256; [DOI] [PubMed] [Google Scholar]
- 1b. Khan S., Sen S. S., Roesky H. W., Chem. Commun. 2012, 48, 2169–2179; [DOI] [PubMed] [Google Scholar]
- 1c. Giffin N. A., Masuda J. D., Coord. Chem. Rev. 2011, 255, 1342–1359. [Google Scholar]
- 2.
- 2a. Cossairt B. M., Piro N. A., Cummins C. C., Chem. Rev. 2010, 110, 4164–4177; [DOI] [PubMed] [Google Scholar]
- 2b. Caporali M., Gonsalvi L., Rossin A., Peruzzini M., Chem. Rev. 2010, 110, 4178–4235. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Tran B. L., Singhal M., Park H., Lam O. P., Pink M., Krzystek J., Ozarowski A., Telser J., Meyer K., Mindiola D. J., Angew. Chem. Int. Ed. 2010, 49, 9871–9875; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 10067–10071; [Google Scholar]
- 3b. Camp C., Maron L., Bergman R. G., Arnold J., J. Am. Chem. Soc. 2014, 136, 17652–17661; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Pintér B., Smith K. T., Kamitani M., Zolnhofer E. M., Tran B. L., Fortier S., Pink M., Wu G., Manor B. C., Meyer K., Baik M.-H., Mindiola D. J., J. Am. Chem. Soc. 2015, 137, 15247–15261. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Yao S., Szilvasi T., Lindenmaier N., Xiong Y., Inoue S., Adelhardt M., Sutter J., Meyer K., Driess M., Chem. Commun. 2015, 51, 6153–6156; [DOI] [PubMed] [Google Scholar]
- 4b. Yao S., Lindenmaier N., Xiong Y., Inoue S., Szilvási T., Adelhardt M., Sutter J., Meyer K., Driess M., Angew. Chem. 2015, 127, 1266–1270; [DOI] [PubMed] [Google Scholar]
- 4c. Yao S., Xiong Y., Milsmann C., Bill E., Pfirrmann S., Limberg C., Driess M., Chem. Eur. J. 2010, 16, 436–439. [DOI] [PubMed] [Google Scholar]
- 5. Spitzer F., Sierka M., Latronico M., Mastrorilli P., Virovets A. V., Scheer M., Angew. Chem. Int. Ed. 2015, 54, 4392–4396; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4467–4472. [Google Scholar]
- 6.The term “realgar-type” P8 moiety for the tricyclo[3.3.0.03,7]octaphosphane ligand comes from its analogy to the isostructural realgar (As4S4) molecule.
- 7.
- 7a.For 2 a,b,c calculated at the BP86/def2-SVP level. The NPA charges for A were taken from Ref. [4a]. It should be noted that different basis sets were used;
- 7b.For the relationship of the formal oxidation states and the spectroscopic and structural parameters (see the Supporting Information, Table S3).
- 8. Cossairt B. M., Cummins C. C., Head A. R., Lichtenberger D. L., Berger R. J. F., Hayes S. A., Mitzel N. W., Wu G., J. Am. Chem. Soc. 2010, 132, 8459–8465. [DOI] [PubMed] [Google Scholar]
- 9. Brassington N. J., Edwards H. G. M., Long D. A., J. Raman Spectrosc. 1981, 11, 346–348. [Google Scholar]
- 10.
- 10a. Konchenko S. N., Pushkarevsky N. A., Gamer M. T., Köppe R., Schnöckel H., Roesky P. W., J. Am. Chem. Soc. 2009, 131, 5740–5741; [DOI] [PubMed] [Google Scholar]
- 10b. Huang W., Diaconescu P. L., Chem. Commun. 2012, 48, 2216–2218; [DOI] [PubMed] [Google Scholar]
- 10c. Barr M. E., Adams B. R., Weller R. R., Dahl L. F., J. Am. Chem. Soc. 1991, 113, 3052–3060; [Google Scholar]
- 10d. Scheer M., Becker U., Matern E., Chem. Ber. 1996, 129, 721–724; for structurally similar As8 complexes, see: [Google Scholar]; Schwarzmaier C., Timoshkin A. Y., Balázs G., Scheer M., Angew. Chem. Int. Ed. 2014, 53, 9077–9081; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9223–9227. [Google Scholar]
- 11. Scepaniak J. J., Harris T. D., Vogel C. S., Sutter J., Meyer K., Smith J. M., J. Am. Chem. Soc. 2011, 133, 3824–3827. [DOI] [PubMed] [Google Scholar]
- 12. Spencer D. J. E., Reynolds A. M., Holland P. L., Jazdzewski B. A., Duboc-Toia C., Le Pape L., Yokota S., Tachi Y., Itoh S., Tolman W. B., Inorg. Chem. 2002, 41, 6307–6321. [DOI] [PubMed] [Google Scholar]
- 13. Kraus F., Aschenbrenner J. C., Korber N., Angew. Chem. Int. Ed. 2003, 42, 4030–4033; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 4162–4165. [Google Scholar]
- 14. Velian A., Cummins C. C., Chem. Sci. 2012, 3, 1003–1006. [Google Scholar]
- 15.See the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary