

Abstract
The ability of the cellular prion protein (PrPC) to bind copper in vivo points to a physiological role for PrPC in copper transport. Six copper binding sites have been identified in the nonstructured N-terminal region of human PrPC. Among these sites, the His111 site is unique in that it contains a MKHM motif that would confer interesting CuI and CuII binding properties. We have evaluated CuI coordination to the PrP(106–115) fragment of the human PrP protein, using NMR and X-ray absorption spectroscopies and electronic structure calculations. We find that Met109 and Met112 play an important role in anchoring this metal ion. CuI coordination to His111 is pH-dependent: at pH >8, 2N1O1S species are formed with one Met ligand; in the range of pH 5–8, both methionine (Met) residues bind to CuI, forming a 1N1O2S species, where N is from His111 and O is from a backbone carbonyl or a water molecule; at pH <5, only the two Met residues remain coordinated. Thus, even upon drastic changes in the chemical environment, such as those occurring during endocytosis of PrPC (decreased pH and a reducing potential), the two Met residues in the MKHM motif enable PrPC to maintain the bound CuI ions, consistent with a copper transport function for this protein. We also find that the physiologically relevant CuI-1N1O2S species activates dioxygen via an inner-sphere mechanism, likely involving the formation of a copper(II) superoxide complex. In this process, the Met residues are partially oxidized to sulfoxide; this ability to scavenge superoxide may play a role in the proposed antioxidant properties of PrPC. This study provides further insight into the CuI coordination properties of His111 in human PrPC and the molecular mechanism of oxygen activation by this site.
Short abstract
CuI coordination to the His111 site in the HuPrP protein is highly dependent on the pH: at pH <5, two methionine (Met) residues bind CuI; in the range of pH 5−8, both Met residues remain coordinated, forming a 1N1O2S species, with N from His111 and O from a backbone carbonyl or a water molecule; at pH >8, 2N1O1S species are formed with only a Met ligand. The CuI-1N1O2S species activates dioxygen, and in this process, the Met residues are partially oxidized to sulfoxide.
1. Introduction
Transmissible spongiform encephalopathies are a group of neurodegenerative disorders that include scrapie in sheep, bovine spongiform encephalopathy, chronic wasting disease in elk and deer, and Creutzfeldt–Jakob disease in humans.1 The infectious agent is an abnormally folded isoform of cellular prion protein (PrPC), designated as PrPSc. This misfolded protein is rich in β-sheet structure, insoluble, and protease-resistant and possesses a tendency to polymerize into amyloid aggregates.2,3 These fibrils accumulate as plaques in the nervous system and have been associated with the induction of neuronal death.4,5 PrPSc propagates itself by autocatalytic conversion of the normal cellular form.6 PrPC is a cell surface glycoprotein expressed throughout the body but mainly found in the central nervous system of all mammals and avian species.1,7
After posttranslational modification, human PrP comprises 209 amino acids PrP(23–231).8,9 The N-terminal region of PrPC is largely unstructured, while the C-terminal region has a globular structure with three α-helices and two short β-sheets.10,11 The physiological role of PrPC has not been determined; however, it has been proposed to participate in the allosteric function, signal transduction, cell–cell adhesion, and suppression of apoptosis.12−17 The ability of PrPC to bind CuII in vivo has led to the proposals that it may play a role in copper sensing, buffering, and/or transport.16−20 Furthermore, cellular studies show that copper or zinc binding to PrPC induces its endocytosis.21−23 It has been established that PrPC can bind up to six CuII ions in its flexible N-terminal region.24,25 CuII coordination to the N-terminal region of PrPC as well as related synthetic peptides have been extensively studied.26 Because these copper binding sites are unstructured, the study of peptide fragments has been a successful approach to studying copper binding to PrP. In the octarepeat region (OR), a domain comprised of four tandem repeats of eight amino acids with the sequence PHGGGWGQ, CuII sites are populated in response to the pH and CuII concentration.27
Outside the OR region, two sites have been identified; these are associated with His96 and His111. The His96 CuII site can adopt a 3N1O or 4N equatorial coordination mode, with a pKa value of 7.8 for conversion between these species.28,29 Regarding CuII binding to His111, several proposals of coordination modes have been suggested.30−32 In our previous study, CuII-PrP(106–115) complexes were characterized using different spectroscopic techniques [absorption, circular dichroism (CD), and electron paramagnetic resonance (EPR)] in combination with electronic structure calculations.33 This study shows a pH-dependent CuII-PrP(106–115) coordination mode with a pKa value of 7.5 (structures given in Scheme 1).
Scheme 1. Physiologically Relevant CuII-PrP(106–115) Species at pH 7.433.

Several reports have suggested that copper bound to PrP is redox-active. Electrochemical measurements of copper bound to the OR region have concluded that the low-occupancy copper binding mode [E1/2(CuII-OR/CuI-OR) = 0.323 V vs NHE] cannot reduce dioxygen to hydrogen peroxide [E1/2(O2/H2O2) = 0.296 V vs NHE], while the high-occupancy copper binding mode [E1/2(CuII4-OR/CuI4-OR) = 0.172 V vs NHE] can.34 With respect to the N-terminal His111 site, spin-trapping and NBT/Formazan experiments with the PrP(106–126) and PrP(106–114) fragments suggested that these peptides are also capable of hydrogen peroxide or superoxide production in the presence of CuII, ascorbic acid, and dioxygen.35,36 While the nature of the CuII coordination properties of the N-terminal region of the PrP protein has been widely explored, studies of its CuI binding properties have been limited.35,37 Unlike the other sites, the His111 binding site contains two adjacent methionine (Met) residues, which could provide good ligands for CuI and promote interesting redox behavior for this site. In fact, extended X-ray absorption fine structure (EXAFS) studies have demonstrated CuI–S interactions in the CuI complexes of PrP(106–114) and PrP(91–126) fragments at pH 7.4.35 However, at this pH, the EXAFS spectra would have contributions from different protonation states of the complex (vide infra), obscuring the coordination mode assignments.
In this study, we used different spectroscopic techniques (X-ray absorption spectroscopy and NMR) in combination with electronic structure calculations to elucidate the coordination modes involved in CuI binding to His111 in the PrP(106–115) fragment. A detailed pH study of the CuI binding properties of this fragment has allowed us to identify species that would be physiologically relevant. We have also evaluated the oxygen reactivity of these CuI species, identifying specific sites for copper-catalyzed oxidation. The roles of Met109 and Met112 in CuII and CuI coordination, as well as oxygen activation have been evaluated.
2. Experimental Section
2.1. Peptide Synthesis
Peptides KTNMKHMAGA [PrP(106–115)], KTNAKHMAGA [PrP(106–115)M109A], KTNMKHAAGA [PrP(106–115)M112A], and KTNAKHAAGA [PrP(106–115)M109&M112A] were prepared by solid-phase fluorenylmethoxycarbonyl (Fmoc) methods, using a Fmoc-Rink amide MBHA resin, as previously described.33,38,39 All peptides were acetylated at the amino terminus and amidated in the carboxyl terminal. Peptides were purified by semipreparative reversed-phase high-performance liquid chromatography (HPLC). The final purity was determined by analytical HPLC and was found to be >95%. The molecular mass of each peptide was confirmed by electrospray ionization mass spectroscopy.
2.2. K-Edge X-ray Absorption Spectroscopy (XAS)
Sample Preparation
Copper(II) peptide complexes were prepared either at a 2 mM peptide concentration with 0.5 equiv of CuII (at pH 6.5) or 1 mM peptide with 0.8 equiv of CuII at pH 8.5, in a mixture of 20 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer with 20 mM N-ethylmorpholine (NEM) and 50% glycerol; the pH was adjusted by adding small volumes of NaOH or HCl. The nature of the copper(II) peptide complexes was probed by electron paramagnetic resonance (EPR) spectroscopy, yielding g∥ and A∥ values that are consistent with those previously reported.33 The copper(I) peptide complexes were obtained upon reduction of the copper(II) peptide complexes with 100 equiv of ascorbic acid (adjusted to the corresponding pH), under anaerobic conditions. The reduced complexes were characterized by EPR, and in all cases, they were found to contain less than 3% of the residual CuII species (Figure S1 in the Supporting Information, SI).
XAS Data Collection
The Cu K-edge XAS data were collected at the SSRL on the unfocused 20-pole, 2.0-T wiggler beamline 7-3 under storage ring parameters of 3 GeV and 300–350 or 500 mA. A rhodium-coated premonochromator flat bent mirror was used for harmonic rejection and vertical collimation. A Si(220) double-crystal monochromator was used for energy selection. The samples were loaded into 2 mm Lucite XAS cells with 38 μm Kapton windows and maintained at a constant temperature of ∼10 K during data collection using an Oxford Instruments CF 1208 continuous-flow liquid-helium cryostat. A Canberra Ge 30-element solid-state array detector was used to collect Cu Kα fluorescence signals, using a Soller slit and a zinc filter inserted between the sample and detector. The sample was positioned at 45° to the incident beam. Internal energy calibration was accomplished by simultaneous measurement of the absorption of a copper foil placed between two ionization chambers located after the sample. The first inflection point of the foil spectrum was assigned to 8980.3 eV. Extended X-ray absorption fine structure (EXAFS) data are reported to k = 12.8 Å–1 in order to avoid interference from the Zn K-edge. No photodamage was observed to CuI samples, and thus all of the scans were used in the final average. Each data set includes an average of 7–27 scans. Data from CuII samples were collected on four physically separate spots on the sample cells with 2–4 scans/spot to minimize the effect of photoreduction.
XAS Data Analysis
The energy-calibrated and averaged data were processed by fitting a second-order polynomial to the preedge region and subtracting this from the entire spectrum as a background. A three-region polynomial spline of orders 2, 3, and 3 was used to model the smoothly decaying postedge region. The data were normalized by scaling the spline function to an edge jump of 1.0 at 9000 eV. This background subtraction and normalization was done using PySpline.40 The least-squares fitting program OPT in EXAFSPAK(41) was used to fit the data. Initial ab initio theoretical phase and amplitude functions were generated in FEFF 7.0(42) using calculated structures of CuI models as the starting structures. Atomic coordinates were adjusted as necessary as fits were improved. During the fitting process, the bond distance (R) and the mean-square deviation or bond variance in R arising from thermal and static disorder (σ2) were varied for all components. The threshold energy (E0) was also allowed to vary for each fit but was constrained to the same value for all components in a given fit. Coordination numbers (N) were systematically varied to provide the best chemically viable agreement to the EXAFS data and their Fourier transform (FT) but were fixed within a given fit. The fits were evaluated based on a comparison of the normalized error (Fn) of each fit along with inspection of individual fits to the data and the agreement of the FTs and of individual wave components.
2.3. NMR Spectroscopy
CuI-PrP(106–115) complexes were prepared as described above but at a lower peptide concentration (0.3 mM) and without glycerol, and the pH was varied from 3.4 to 9.2. Homonuclear assignment of PrP peptides was achieved by 1H–1H total correlated spectroscopy (TOCSY), and 1H–13C heteronuclear single quantum coherence (HSQC) experiments were performed at pH 6.5 and 8.5. 1H–1H TOCSY and 1H–13C HSQC cross peaks affected by CuI (0.8 equiv) were identified by comparing their chemical shift values in the absence and presence of the metal ion. All spectra were acquired at 288 K in NMR tubes sealed under a N2 atmosphere, using a Bruker Avance II 600 MHz spectrometer with a triple-resonance probe equipped with z-axis self-shielded gradient coils. Acquisition, processing, and visualization of the NMR spectra were performed using TOPSPIN 2.1 (Bruker) and Sparky.
2.4. Electronic Structure Calculations
In all calculations, the structure of the PrP(106–113) peptide with sequence KTNMKHMA was used, with an acetylated N-terminus and amidated C-terminus. The structures had a total of 141–161 atoms, depending on the protonation state of the backbone amides and the number of explicit water molecules included in each model. Each initial copper(I) peptide complex model was built in GaussView 4.1.2, starting from the previously reported CuII models.33 Using a restricted Kohn–Sham (RKS) approach, all copper(I) peptide structures with a spin multiplicity of 1 (singlet) were fully optimized without geometry constraints, and the stationary points of selected CuI structures were characterized by a harmonic analysis (Table S1 in the SI). Open-shell calculations were done with the unrestricted Kohn–Sham (UKS) approach. All geometry optimizations were performed using the deMon2k code43 with the functional OPBE (which combines Handy and Cohen’s OPTX exchange functional44 with the PBE45 correlation functional). It should be noted that calculations at the LDA level and with the nonempirical PBE functional were also performed for all models; however, only the results obtained with OPBE are presented here because this functional provides the best description and distances for CuI–S bonds. The orbital and auxiliary basis sets used were TZVP46,47 and GEN-A2,48,49 respectively. Solvent effects on selected optimized structures were evaluated in the deMon2k code, including six explicit water molecules in the gas phase and reoptimizing with OPBE and TZVP; in these cases, after reoptimization, the water molecules remained outside the coordination sphere (Figure S2 in the SI). Implicit solvation of selected models was also calculated using the model COSMO,50,51 as implemented in the ORCA(52) program. Because explicit solvation with six water molecules provides a geometric description very similar to that of implicit solvation (Table S2 in the SI), herewith we report the results corresponding to explicit solvation with six water molecules for all of the structures.
The inner-sphere reorganization energy for the electron-transfer step was computed using the following expression, which depends on the energies of the oxidized (ox) and reduced (red) geometries at the selected structures that best reproduce the copper complex at pH 6.5 and 8.5:53
where Eox(redgeom) is the energy of the oxidized state in the reduced structure and Ered(oxgeom) is the energy of the reduced state in the oxidized structure.
2.5. Stopped-Flow Kinetic Measurements
Kinetic studies of the reduction of the copper(II) peptide complexes by ascorbate were carried out on a SX20 stopped-flow system operated by the Pro-data software (Applied Photophysics) with a 150 W xenon light source and equipped with a photodiode array for multiwavelength analysis. All experiments were performed in a single-mixing mode of the instrument, with a 1:1 (v/v) mixing ratio in a 1 cm optical path. The temperature was maintained at 25.0 ± 0.1 °C using a water bath and monitored via the internal sensor of the mixing unit. Data analysis was performed with Pro-data viewer software. CuII-PrP(106–115) complexes were degassed in a vacuum line and loaded into the driving syringes anaerobically. All flow lines of the instrument were extensively washed with degassed sodium dithionite and 20 mM NEM/MES buffer, before charging the driving syringes with reactant solutions. The final concentration of the copper(II) peptide complexes in the cell was 0.15 mM. The reduction was followed under pseudo-first-order conditions with a 20-fold excess of reductant, and the loss of absorbance was monitored at 570 nm (pH 8.5) or 600 nm (pH 6.5) as a function of time. The dead time of the measurements was 1 ms. The reported rate constants represent the average values and standard deviations of three independent runs.
2.6. Reoxidation Kinetic Measurements
Kinetic studies of the reoxidation of the copper(I) peptide complexes by oxygen were carried out on an Agilent 8453 diode-array spectrometer. All experiments were performed in a screw-cap quartz cuvette with a 1 cm optical path. Data analysis was performed with Origin 6.1 software. The CuI-PrP(106–115) complexes at pH 6.5 were prepared by adding 1 equiv of ascorbate to a degassed solution of the copper(II) peptide complex (0.8 mM peptide with 0.5 equiv of copper) in 20 mM NEM/MES; full reduction under these conditions was confirmed by UV–vis absorption. Reoxidation of the CuI complexes was followed by the appearance of a characteristic d–d band at 600 nm after the addition of an air-saturated buffer (∼0.5 equiv of dioxygen). The reported rate constants represent average values and standard deviations of three independent runs.
3. Results and Analysis
3.1. Structure of Oxidized and Reduced Sites
3.1.1. XAS of CuII-PrP(106–115) Complexes
The CuII-PrP(106–115) complexes have been spectroscopically characterized previously.33 The coordination mode of the CuII-PrP(106–115) complex is pH-dependent with a pKa of 7.5. At low pH, the equatorial coordination is the 3N1O mode, while at high pH, a 4N coordination mode is stabilized with a third deprotonated amide of the backbone (Scheme 1). Here, we use XAS to evaluate these models and the role of Met109 and Met112 in copper coordination. The Cu K-edge spectra of the CuII-PrP(106–115) complexes at pH 6.5 and 8.5 are presented in Figure 1. The extremely weak signal around 8979 eV is the 1s → 3d transition, associated with the CuII d9 ion. Figure 1 shows that the spectra are not affected by the replacement of Met109 and Met112 by Ala. Thus, at both pH values, the thioether groups of the Met residues are not involved in the coordination of CuII. EXAFS fits for the data shown in Figure 1 are consistent with 4N/O tetracoordinated complexes with Cu–ligand distances between 1.96 and 1.98 Å (Table 1 and Figure S3 in the SI). These results are in a good agreement with the previously proposed coordination models (Scheme 1).33 The effect of the pH on the copper coordination is not reflected in XAS because of the similar light-atom (N/O) ligands at both pH values. However, the difference between the two coordination modes (3N1O and 4N) is clearly observed by CD and EPR.33 The reoptimized structures [from density functional theory (DFT) calculations] of the 3N1O and 4N models reported in ref (33) were used to simulate the EXAFS spectra at pH 6.5 and 8.5, respectively, without any fits against the experimental data. The simulated EXAFS spectra show reasonable frequency agreement with the experimental data (Figure S4 in the SI), thus validating the proposed models for the CuII-PrP(106–115) complexes in Scheme 1.
Figure 1.

XANES (A and D), EXAFS (B and E), and FT (C and F) spectra of CuII-PrP(106–115) (black line), CuII-PrP(106–115)M09A (green line), CuII-PrP(106–115)M112A (red line), and CuII-PrP(106–115)M109&M112A (blue line, at pH 6.5 (A–C) and 8.5 (D–F). The parameters obtained for the best fits for these data (Figure S3 in the SI) are listed in Table 1.
Table 1. EXAFS Fits of the Spectra of CuII-PrP(106-115) and Its Met-to-Ala Variants at pH 6.5 and 8.5a.
| pH 6.5 |
pH 8.5 |
|||||
|---|---|---|---|---|---|---|
| CuII complex | coord no. | path | R (Å) | σ2(Å2) | R (Å) | σ2(Å2) |
| CuII-PrP(106–115) | 4 | Cu–O/N | 1.96 | 499 | 1.97 | 544 |
| CuII-PrP(106–115)M109A | 4 | Cu–O/N | 1.96 | 516 | 1.96 | 486 |
| CuII-PrP(106–115)M112A | 4 | Cu–O/N | 1.97 | 504 | 1.98 | 355 |
| CuII-PrP(106–115)M109&M112A | 4 | Cu–O/N | 1.97 | 505 | 1.96 | 549 |
The goodness-of-fit Fn ranges between 0.19 and 0.31 depending on the signal-to-noise ratio of the data set.
3.1.2. XAS of CuI-PrP(106–115) Complexes
The Cu K-edge X-ray absorption near-edge structure (XANES) spectra of the CuI-PrP(106–115) complexes show an intense signal assigned to the electric dipole-allowed Cu 1s → 4p transition at 8984 eV, characteristic of CuI complexes (Figures 2A,D).54 Both XANES and EXAFS spectra (Figure 2) of the CuI-PrP(106–115) complexes change with the pH (Figure S5 in the SI), while the most drastic changes are observed upon substitution of the Met residues by Ala. Regardless of the pH, the XANES spectra of the CuI-PrP(106–115) complex (black line) and the M109A and M112A complexes (red and green lines) show a characteristic signal of four-coordinate complexes (Figures 2A,D), while the CuII complex without Met residues (CuII-PrP(106–115)M109&M112A, blue line) displays a spectrum indicative of a two-coordinate complex.
Figure 2.

XANES (A and D), EXAFS (B and E), and FT (C and F) spectra of CuI-PrP(106–115) (black line), CuI-PrP(106–115)M09A (green line), CuI-PrP(106–115)M112A (red line), and CuI-PrP(106–115)M109&M112A (blue line) at pH 6.5 (A–C) and 8.5 (D–F).
At pH 6.5, the EXAFS spectra (Figure 2B) of the CuI-PrP(106–115) complex change significantly upon each Met-to-Ala substitution, suggesting that both Met109 and Met112 participate in CuI coordination. Consistently, the FTs of the EXAFS spectra (Figure 2C) show a drastic loss in intensity in the Met-to-Ala variants compared to those of the CuI-PrP(106–115) complex; the low intensity of these signals is best modeled by a lower number of sulfur atoms in the coordination sphere (see below). These changes are most dramatic for the CuI-PrP(106–115)M109&M112A complex, consistent with the XANES results. In contrast, at pH 8.5, the EXAFS spectra of the CuI-PrP(106–115) complex (black line) and the single Met-to-Ala variants (red and green lines) are practically identical (Figure 2E), while only the spectrum for the CuI-PrP(106–115)M109&M112A (blue line), where there are no sulfur atoms available for coordination, differs from the original complex. Consistently, the FTs of the EXAFS spectra of CuI-PrP(106–115) (black line) and the M109A (green line) and M112A (red line) variants (Figure 2F) are also very similar in intensity, indicating that they have the same sulfur content in the first coordination sphere, while the spectrum of the CuI-PrP(106–115)M109&M112A complex (blue line) shows essentially no intensity at the R value of a Cu–S contribution.
Overall, the EXAFS results indicate that at pH 8.5 only one Met residue participates in CuI coordination, while at pH 6.5, both Met residues play an important role. Consistently, a comparison of the spectra of the CuI-PrP(106–115) complexes at pH 6.5 and 8.5 in Figure S5 in the SI shows differences that can be attributed to a larger contribution of sulfur atoms in the first coordination sphere at low pH. The best fits of the CuI-PrP(106–115) complexes at pH 6.5 and 8.5 are shown in Figure 3, and the corresponding parameters are listed in Table 2. At pH 6.5, the best fit is achieved with two sulfur atoms at 2.37 Å and two nitrogen or oxygen ligands at 2.17 Å. The four-coordinate nature of the complex is supported by a bond valence sum (BVS) analysis:55−57 the BVS value is in better agreement with a four-coordinate structure (0.98) compared to a three-coordinate fit (BVSs = 0.82). At pH 8.5, the best fit of the EXAFS data involves only one sulfur ligand at 2.35 Å and three nitrogen or oxygen ligands displaying one short CuI–N/O bond at 1.97 Å and two longer CuI–2N/O bonds at 2.15 Å (Table 2). Again, BVS analysis is in better agreement with a four-coordinate structure (BVSs = 1.10) relative to a three-coordinate complex (BVSs = 0.74).
Figure 3.

EXAFS spectra (A and C) and their FTs (B and D) of CuI-PrP(106–115) at pH 6.5 (A and B) and 8.5 (C and D). Experimental data are shown as solid black lines, and their best fits, using the parameters listed in Table 2, are shown as dashed red lines.
Table 2. EXAFS Fits of Spectra of CuI-PrP(106-115) Complexes at pH 6.5 and 8.5.
| pH | Fna | coord no. | path | R (Å) | σ2 (Å2) | BVS |
|---|---|---|---|---|---|---|
| 6.5 | 0.579 | 2 | Cu–O/N | 2.17 | 126 | 0.98 |
| 2 | Cu–S | 2.37 | 484 | |||
| 8.5 | 0.099 | 1 | Cu–N/O | 1.97 | 503 | 1.10 |
| 2 | Cu–N/O | 2.15 | 328 | |||
| 1 | Cu–S | 2.35 | 421 |
3.1.3. NMR Spectroscopy of CuI-PrP Complexes
In order to further explore the nature of the ligands in the CuI-PrP(106–115) complexes, 1D (1H) and 2D 1H–1H TOCSY and 1H–13C HSQC experiments were carried out. The backbone amide regions of 2D 1H–1H TOCSY and 1H–13C HSQC NMR spectra of PrP(106–115)M109A, PrP(106–115)M112A, and PrP(106–115) in the presence and absence of 0.8 equiv of CuI at pH 6.5 and 8.5 were used to assign the signals of the Hε of Met109 and Met112, the Hδ and Hε of His111, and other residues (Figures S6–S8 in the SI). Analysis of the 1H–1H TOCSY and 1H–13C HSQC NMR spectra shows that the most affected amino acids in the presence of CuI are Met109, Met112, His111, and Lys110 (Figures S6–S8 in the SI). Figure 4 shows the aromatic (6.9–8.1 1H ppm) and aliphatic (1.9–2.4 1H ppm) regions of the 1H NMR spectra of the free peptide PrP(106–115) (black line) and the CuI complex (red line) at pH 6.5 (Figure 4A) and pH 8.5 (Figure 4B). At pH 6.5, the chemical shifts associated with the Hε of Met109 and Met112 are clearly affected (Figure 4A, aliphatic region) having a broadening and downfield shift (0.15 ppm of Hε Met112 and 0.23 ppm of Hε Met109) in the presence of CuI. In agreement with XAS data, NMR data indicate that CuI is coordinated by both Met residues at low pH. The aromatic region of the spectrum displays a broadening and an upfield shift in the Hε (0.12 ppm) and Hδ (0.04 ppm) signals of His111 in the presence of CuI (Figure 4A). This result indicates that at pH 6.5 the His111 imidazole nitrogen participates in CuI binding.
Figure 4.
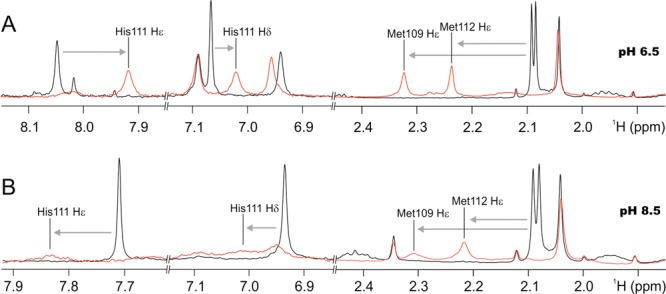
1H NMR spectra of PrP(106–115) (black line) and CuI-PrP(106–115) (red line) at pH 6.5 (A) and 8.5 (B). Black lines indicate the chemical shifts for the Hε protons of Met residues and the Hε and Hδ protons of histidine in the peptide without copper. Gray arrows show the shifts of these signals in the presence of CuI.
At pH 8.5, in the aliphatic region of the 1H NMR spectrum, the chemical shifts of Hε of Met109 and Met112 undergo a broadening and a downfield shift (0.12 ppm of Hε M112 and 0.21 ppm of Hε Met109) upon CuI binding (Figure 4B). In the aromatic region, signals of the Hε and Hδ of His111 also show broadening and downfield shift. The effect of CuI on the signals of the Hε and Hδ of His111 suggests that at pH 8.5 the His111 imidazole nitrogen participates in CuI binding. However, the line broadening observed at pH 8.5 in both regions is much more dramatic than that observed at pH 6.5. Such large signal broadening may be related to the presence of two species at pH 8.5, one with the sulfur atom from Met109 coordinating CuI and another complex with the sulfur atom from Met112. This interpretation is consistent with the behavior of the Met-to-Ala variants observed in XAS, where the only sulfur available in each variant is found to coordinate CuI. In summary, our NMR data corroborate the involvement of sulfur atoms from Met109 and Met112 of the PrP(106–115) fragment in the coordination of CuI, while they clearly indicate that the His111 imidazole nitrogen also binds to the metal ion.
3.1.4. Effect of the pH on the CuI-PrP(106–115) Complex Followed by NMR Spectroscopy
To further explore the role that Met109, Met112, and His111 play on the coordination with CuI, we tested both the PrP(106–115) peptide and the CuI-PrP(106–115) complex by 1H NMR spectroscopy at several pH values ranging from 3.5 to 9.5. Figure 5A shows the behavior of Hε (dashed black line) and Hδ (solid black line) of His111 in the free peptide and in the CuI-PrP(106–115) complex (dashed and solid red lines). The pKa associated with the free histidine is 6.2, while in the presence of CuI, the pKa of histidine shifts to ∼5 because of the coordination of His111 to CuI in the CuI-PrP(106–115) complex. This shift is consistent with that observed in CuI complexes in blue copper proteins.58−60 The signals associated with the Met residues in the CuI-PrP(106–115) complex also change drastically with the pH. Figure 5B shows in green (Hε Met109) and red (Hε Met112) dots chemical shifts of Hε of Met109 and Met112 in the presence of CuI at several pH values (3.5–9.5). The chemical shift of these protons is 2.10 ppm in the free peptide (Figure 4), and they do not change with the pH (data not shown). However, in the presence of CuI, even at a pH as low as 3.5, the signals associated with Hε of Met109 and Met112 are both shifted to 2.29 ppm, indicating coordination to CuI. Also, at higher pH values (above 8.5), both signals show a tendency to return to the chemical shift associated with the free peptide (2.10 ppm). This result indicates that, at higher pH values, both Met residues leave the coordination sphere of CuI, leading to a species without thioether ligands; such species would not be physiologically relevant and were not further characterized.
Figure 5.

(A) Chemical shifts of the Hε and Hδ protons of His111 in the free peptide (dashed and solid black lines) and in the CuI-PrP(106–115) complex (dashed and solid red lines) as a function of the pH. A shift in the pKa of His111 is observed in the presence of CuI. (B) Chemical shifts of the Hε protons of Met109 (green dots) and Met112 (red dots) in the CuI-PrP(106–115) complex as a function of the pH. The pH-independent chemical shift of these protons is 2.10 ppm in the free peptide.
These NMR data provide further insight into the role of Met109, Met112, and His111 in the coordination of CuI to the PrP(106–115) fragment as a function of the pH. At very low pH (3.5), CuI is coordinated by both Met109 and Met112. With a pKa of ∼5, the nitrogen imidazole from His111 enters the coordination sphere, while the two Met residues remain coordinated. As the pH is further increased, the Met residues play a less important role in CuI coordination, and only one Met residue (Met109 or Met112) coordinates CuI at pH >8 (Scheme 2).
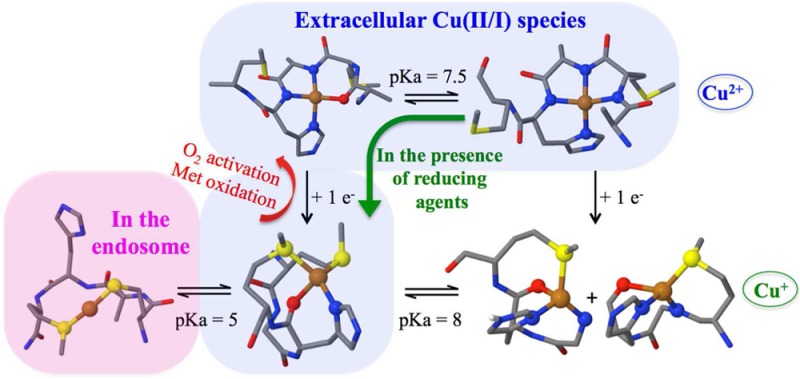
Scheme 2. Proposed Coordination Models for the CuI-PrP(106–115) at Several pH Values, Based on XAS and NMR Data.

At pH <5, CuI is anchored by both Met residues, while at a pH >5, the His111 imidazole also participates in CuI coordination. At pH >8, only one Met residue coordinates CuI. The “box” shows extracellular species.
3.1.5. DFT Modeling
The “box” in Scheme 2 shows the structures for the CuI-PrP(106–115) complexes under extracellular conditions, derived from the EXAFS and NMR data. In order to gain further insight into the nature of the unidentified N/O ligands in these CuI complexes, several models were built and RKS calculations were performed, using the CuII models previously described as starting points.33 The copper ion was reduced and three groups of ligands were evaluated (Table S3 in the SI). Group 1 included four-coordinate CuI-1N2S1x models for the complex formed at pH 6.5, with two sulfur atoms from both Met residues (Met109 and Met112), a His111 imidazole nitrogen, and a fourth ligand x, where x could be an oxygen atom from a backbone carbonyl group, a solvent H2O molecule, or a nitrogen atom from a backbone amide in a protonated (N) or deprotonated form (N–) (including those of the most affected amino acid residues in the 2D TOCSY and HSQC experiments; Figures S6–S8 in the SI). Because at pH 8.5 the EXAFS results indicate that one Met is coordinated while NMR data show that both Met residues are affected, group 2 included four-coordinate CuI-1N1S2x models, where the sulfur atom is provided by either Met109 (group 2A) or Met112 (group 2B), His111 imidazole provides a nitrogen ligand, and 2x could be any of the following combinations: 2N–, 1N1N–, 2N, 1N–1O, 1N1O, or 2O (described above). It should be noted that, because the XANES data indicate that CuI complexes are four-coordinate, only the structures that optimized as four-coordinate complexes were analyzed in each group.
3.1.5.1. CuI Structures at pH 6.5 (CuI-1N2S1x Models)
Five structures optimized as four-coordinate complexes in group 1 (Table S3 in the SI); these are shown in Figure 6, and relevant geometric parameters with and without solvent effects are listed in Table S4 in the SI. The group 1 models are divided into two subgroups, according to the total number of atoms (160 or 161), which depends on the protonation state of the backbone amides. In this group, when x is a deprotonated amide (2N2Sa and 2N2Sb), the average CuI–S bond distances (2.365 and 2.34 Å) are in good agreement with the experimental values; however, the two Cu–N bonds are quite short (2.09–2.10 Å). When x is a backbone carbonyl (1N1O2Sa and 1N1O2Sb) or a water molecule (1N1O2Sc1), the average CuI–S bond lengths (2.35, 2.34, and 2.36 Å) and CuI–N/O distances are in good agreement with the experimental data (2.17, 2.145, and 2.16 Å; Table S4 in the SI). However, the 1N1O2Sb model is discarded because it is the structure that has the highest energy in this subgroup, 13.32 kcal/mol above 1N1O2Sa.
Figure 6.
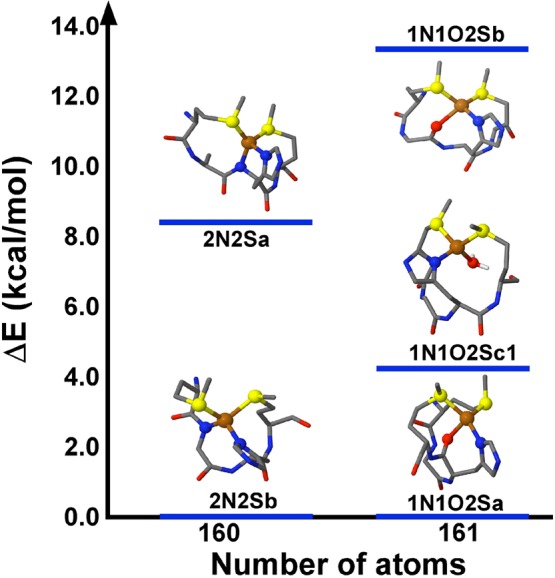
Energy diagram of optimized four-coordinate CuI-1N2S1x models for the CuI-PrP(106–115) complex at pH 6.5 (group 1), with explicit solvent. Most side chains are not shown for clarity; however, the geometry optimizations were done with the complete CuI-PrP(106–113) complex.
Thus, the best candidates for the pH 6.5 CuI structures (group 1) are the 1N1O2Sa and 1N1O2Sc1 models (Figure 6) because they display metal–ligand distances that best reproduce the experimental data in trends and values. Furthermore, using 1N1O2Sc1 to simulate an EXAFS spectrum using the EXAFSPAK program yielded a spectrum that was in good intensity agreement (but indicating an overall longer distance) with the experimental data for CuI-PrP(106–115) at pH 6.5 (Figure S9 in the SI). Therefore, at pH 6.5 the best model to represent the CuI-PrP(106–115) complex has two sulfur atoms from Met109 and Met112, a His111 imidazole nitrogen, and an oxygen atom as the fourth ligand, which could be the carbonyl oxygen atom from His111 (as in model 1N1O2Sa) or a water molecule (as in model 1N1O2Sc1).
3.1.5.2. CuI Structures at pH 8.5 (CuI-1N1S2x Models)
In these models, the sulfur atom can be provided by Met109 (group 2A) or Met112 (group 2B). The group 2 models are divided into two subgroups, according to the total number of atoms (159 or 160), which depends on the protonation state of the backbone amides. Five structures optimized as four-coordinate complexes in group 2A and only four in group 2B (Table S3 in the SI). The structures are shown in Figure 7, and their geometric parameters with and without solvent effects are listed in Tables S5 and S6 in the SI.
Figure 7.
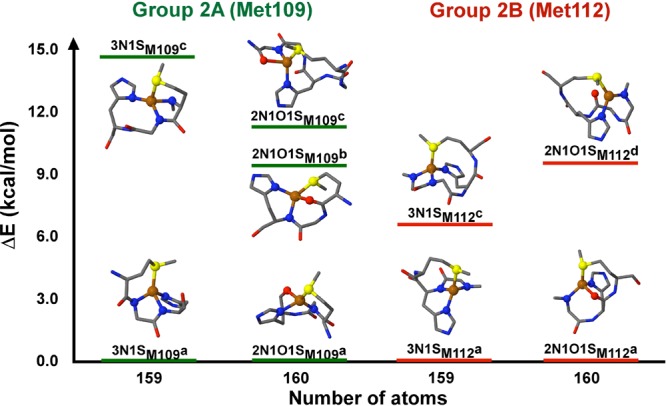
Energy diagram of optimized four-coordinate CuI-1N1S2x models for the CuI-PrP(106–115) complexes at pH 8.5, with explicit solvent and either Met109 (group 2A) or Met112 (group 2B) as the ligand. Most side chains are not shown for clarity; however, the geometry optimizations were done with the complete CuI-PrP(106–113) complex.
For the models with Met109, when 2x is a combination of two deprotonated amides (3N1SM109a and 3N1SM109c), only the 3N1SM109c model yields Cu–ligand distances that are in agreement with the experimental trend, one short Cu–N and two long Cu–N bonds (2.04 and 2.13 Å, respectively) and a CuI–S bond distance at 2.36 Å (Table S5 in the SI), although it is the highest-energy structure of the group (Figure 7). In contrast, in the models where 2x is the combination of a deprotonated amide and a backbone carbonyl (2N1O1SM109a, 2N1O1SM109b, and 2N1O1SM109c), the CuI–S bond and the trend of a short Cu–N and two long Cu–N/O bonds are reproduced in the 2N1O1SM109a model (2.32, 2.02, and 2.15 Å, respectively; Table S5 in the SI), which is the lowest-energy structure in this subgroup. Thus, we propose that the best model for the CuI-PrP(106–115) complex at pH 8.5 with a sulfur atom from Met109 is 2N1O1SM109a, with a nitrogen atom from the deprotonated amide of Lys110 and an oxygen atom from the carbonyl of His111.
The CuI-1N1S2x models with the Met112 ligand (Figure 7 and Table S6 in the SI), having 2x as a combination of two deprotonated amides (3N1SM112a and 3N1SM112c), yield a CuI–S bond at 2.28–2.30 Å and three similar Cu–N bonds (∼2.03–2.04 Å), which is not consistent with the experimental trend. On the other hand, when 2x is a combination of a deprotonated amide and a backbone carbonyl (2N1O1SM112a and 2N1O1SM112d), the 2N1O1SM112d model leads to a three-coordinate structure, while the Cu–ligand distances in the 2N1O1SM112a model show excellent agreement with the experimental data: a short Cu–N bond (2.00 Å), two long Cu–N/O bonds (2.17 Å), and a CuI–S bond at 2.34 Å (Table S6 in the SI). Therefore, the best model for the CuI-PrP(106–115) complex with Met112 at pH 8.5 is the 2N1O1SM112a structure.
Overall, the best models for the CuI-PrP(106–115) complex at pH 8.5 are 2N1O1S structures with a nitrogen from the deprotonated backbone amide of Lys110 and an oxygen from the backbone carbonyl of His111, regardless of which Met participates in the coordination shell.
Figure 8 summarizes the results from DFT modeling, showing the best models for the CuII and CuI complexes with PrP(106–115) at pH 6.5 and 8.5. It is clear that the CuII complexes (CuII-3N1O and CuII-4N) have very different geometries and coordination spheres than their reduced counterparts (CuI-1N1O2S and CuI-2N1O1S). Thus, the reduction of these complexes would involve a large reorganization energy. We calculated the inner-sphere reorganization energies associated with the reduction of the CuII species to CuI, at pH 6.5 and 8.5. For the low-pH structures CuII-3N1O and CuI-1N1O2S, the calculated λi is 1.79 eV when the oxygen ligand is the backbone carbonyl of His111 and 2.15 eV when it is a water molecule. At pH 8.5, the CuII-4N and CuI-2N1O1S structures yielded a reorganization energy of 1.60 eV when Met109 is bound and 1.66 eV for the Met112 structure (Table S7 in the SI). These are necessary for evaluating the reduction and O2 reactivity in the next section.
Figure 8.
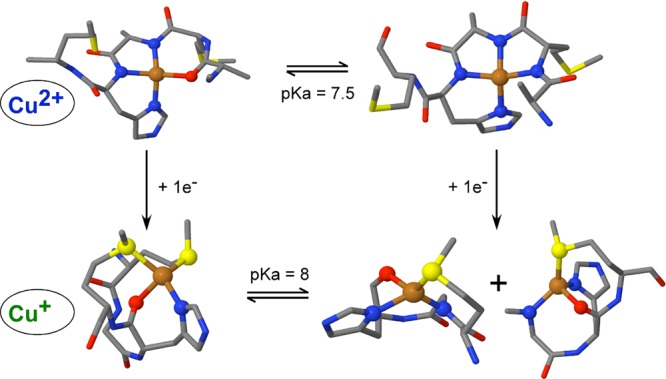
DFT-derived models for CuII and CuI complexes with the PrP(106–115) peptide at different pH values. For the CuI complex at pH 6.5, the best model has a 1N1O2S coordination sphere, where both Met residues are bound to copper and the oxygen atom can be the carbonyl oxygen from His111 (as shown here) or a water molecule. At pH 8.5, the best models are 2N1O1S structures with a deprotonated amide of Lys110, an oxygen atom from the backbone carbonyl of His111, and either Met109 or Met112 as the sulfur ligand.
3.2. Reactivity
Once the geometries and electronic structures of the CuI-PrP(106–115) complexes had been elucidated, we evaluated the role of Met109 and Met112 on the reactivity of these complexes. First, the kinetics of reduction of the CuII-PrP(106–115) complexes with ascorbate was studied, followed by reactivity studies of the resultant CuI-PrP(106–115) complexes with dioxygen.
3.2.1. Reduction of CuII-PrP(106–115) Complexes
The reduction of both complexes CuII-3N1O at pH 6.5 and CuII-4N at pH 8.5 by ascorbate was examined by stopped-flow absorption spectroscopy at 25 °C. Electronic absorption at the wavelength for the maximum intensity of the ligand-field transitions (i.e., 600 nm at pH 6.5 and 570 nm at pH 8.5) was followed over time after rapid mixing of the CuII complexes with the reductant. The reduction was followed under pseudo-first-order conditions, using at least a 20-fold excess of the reductant over the copper complex concentration. Plots of the absorption intensity at 600 and 570 nm as a function of the reaction time are shown in Figure 9, while representative data for the absorption spectra are shown in the insets; fits to the kinetic traces are summarized in Table 3. The reduction of the CuII-3N1O complex at pH 6.5 can be fitted to a single-exponential equation to obtain kobs = 0.287 ± 0.010 s–1 (Figure 9A). In contrast, the reduction of CuII-4N complex at pH 8.5 exhibited a slow biphasic behavior (Figure 9B). The kinetic trace for CuII-4N can be fitted to a double-exponential equation, yielding rates of 0.134 ± 0.023 and 0.010 ± 0.004 s–1. Thus, even the fast phase of reduction of the CuII-4N complex at pH 8.5 is 2-fold slower than reduction of the CuII-3N1O complex at pH 6.5 (Table 3). Considering the reorganization energies calculated for these two complexes, one would expect the opposite trend because λ3N1O is higher than λ4N. This could be due to a difference in the reduction potentials of the CuII-3N1O and CuII-4N species, which would result in different driving forces for the two reactions. Using anaerobic reductive titrations of the CuII complexes at pH 6.5 and 8.5 with ascorbate, we estimate a reduction potential of ∼152 mV for the CuII-3N1O complex and ∼75 mV for the CuII-4N complex. Thus, a significant difference in ΔG° for their reduction by ascorbate is expected {ΔΔG° = [ΔG°(3N1O) – ΔG°(4N)] = −3.52 kcal/mol}. Using the semiclassical Marcus equation for the rate of intermolecular electron transfer and considering the differences in the reorganization energies and reduction potentials, the ratio of the reduction rates for CuII-3N1O and CuII-4N species is calculated to be k3N1O/k4N = 2.3 (see the SI), which is consistent with the experimental values. Thus, the difference in the reduction potentials for these two complexes results in a faster reduction for the CuII-3N1O complex compared to that for the CuII-4N species.
Figure 9.

Stopped-flow absorption data for the reduction of the CuII-PrP(106–115) complex at pH 6.5 (3N1O complex) (A) and at pH 8.5 (4N complex) (B) with 20 equiv of ascorbate. Representative absorption traces, showing the loss of intensity of the d–d transitions as a function of time, are shown in each inset.
Table 3. First-Order Rate Constants for the Reduction of CuII-PrP(106–115) Complexes at pH 6.5 and 8.5 and Their Met-to-Ala Variants.
| first-order
rate constant (s–1) |
||||
|---|---|---|---|---|
| peptide | complex | pH | fast phase | slow phase |
| PrP(106–115) | CuII-4N | 8.5 | 0.134 ± 0.023 | 0.010 ± 0.004 |
| PrP(106–115) | CuII-3N1O | 6.5 | 0.287 ± 0.010 | |
| PrP(106–115)M109A | CuII-3N1O | 6.5 | 0.295 ± 0.027 | |
| PrP(106–115)M112A | CuII-3N1O | 6.5 | 0.318 ± 0.024 | |
| PrP(106–115)M109&M112A | CuII-3N1O | 6.5 | 0.327 ± 0.023 | |
In order to evaluate the effect of Met109 and Met112 on the rate of reduction of the CuII-3N1O complex, the reduction of the CuII-PrP(106–115)M109A, CuII-PrP(106–115)M112A, and CuII-PrP(106–115)M109&M112A complexes by ascorbate at pH 6.5 was also studied; representative kinetic traces are shown in Figure S10 in the SI, and the reduction rates are summarized in Table 3. Within experimental error, CuII-PrP(106–115)M109A, CuII-PrP(106–115)M112A, and CuII-PrP(106–115)M10&M112A exhibit a reduction rate constant that is the same as that of the CuII-PrP(106–115) complex. These results suggest that the Met residues are not involved in the rate-limiting step of the reduction of the CuII-PrP(106–115) complex at pH 6.5. Thus, it is likely that this involves electron transfer, followed by a slower rearrangement of the peptide, to provide the Met ligands to form the pH 6.5 CuI-1N1O2S complex.
3.2.2. Reoxidation of CuI-PrP(106–115) Complexes with Dioxygen at pH 6.5
We next investigated the first step of dioxygen activation by the CuI-PrP(106–115) complexes. Reduced complexes of PrP(106–115), PrP(106–115)M109A, PrP(106–115)M112A, and PrP(106–115)M10&M112A were mixed with an oxygenated buffer, and the reaction was followed by UV–vis absorption spectroscopy. Figure 10 shows the time traces for the appearance of the d–d transition, indicating CuII oxidation, while the reoxidation rate for each copper peptide is listed in Table 4. The end products of the reaction of CuI-PrP(106–115) complexes with oxygen were spectroscopically characterized by CD and EPR; in all cases, at least 50% of the original CuII complexes were recovered (Figure S11 in the SI). Also, analysis of the samples by matrix-assisted laser desorption/ionization time of flight/time of flight (MALDI-TOF/TOF) showed metal-catalyzed oxidation for the PrP(106–115), PrP(106–115)M109A, and PrP(106–115)M112A peptides (Figure S12 in the SI). In particular, for the PrP(106–115) peptide, two new signals with m/z peaks corresponding to the mass of the peptide, +16 and +32 Da, were observed, while single-variant peptides [PrP(106–115)M109A and PrP(106–115)M112A] only show one +16 Da peak. In contrast, the double-variant PrP(106–115)M109&M112A peptide, which contains no Met residues, suffered no modification. These results indicate that redox cycling of these copper peptide complexes results in oxidation of the Met residues into sulfoxides, while no evidence was found for their oxidation to sulfone.
Figure 10.
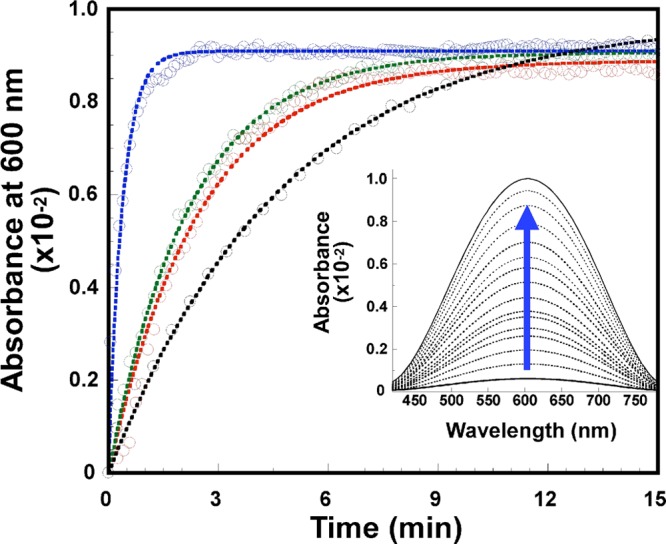
Kinetic traces for the reoxidation by dioxygen of the CuI-PrP(106–115) complex (black) and the variants CuI-PrP(106–115)M109A (green), CuI-PrP(106–115)M112A (red), and CuI-PrP(106–115)M109&M112A (blue) at pH 6.5, followed by UV–vis absorption spectroscopy at 600 nm. Inset: Representative absorption data showing the increase in the intensity of the ligand-field transitions for the CuII complex as a function of time.
Table 4. First-Order Rate Constants for Reoxidation by Dioxygen of the CuI-PrP(106–115) Complex and Its Met-to-Ala Variants at pH 6.5.
| complex | first-order rate constant (min–1) |
|---|---|
| CuI-PrP(106–115) | 0.218 ± 0.005 |
| CuI-PrP(106–115)M109A | 0.425 ± 0.060 |
| CuI-PrP(106–115)M112A | 0.421 ± 0.009 |
| CuI-PrP(106–115)M109&M112A | 2.582 ± 0.154 |
The reoxidation of CuI-PrP(106–115) by dioxygen may follow inner- or outer-sphere electron-transfer pathways. In an outer-sphere mechanism, the CuI complex would be oxidized by dioxygen in a single step to regenerate the CuII complex, while an inner-sphere reaction would involve binding of dioxygen to the CuI complex and formation of a potential copper(II) superoxide intermediate, followed by the release of superoxide to regenerate the CuII complex. The possibility of outer-sphere reoxidation of the CuI-PrP(106–115) complexes was evaluated using the Marcus equation to analyze the rates in Table 4.61,62 Using an estimated reduction potential for the copper peptide complex of 150 mV, the potential of one-electron reduction of dioxygen to O2•– (−165 mV vs NHE),63 and the calculated reorganization energy of the site, the outer-sphere electron-transfer rate from the CuI complex at pH 6.5 to the oxygen molecule is estimated to be at most ∼2.81 × 10–4 s–1 (see the SI), which is 10–102 times slower than the experimental rate of CuI reoxidation from the kinetic data (3.6 × 10–3). Thus, single-electron transfer from CuI-PrP(106–115) to dioxygen likely proceeds via an inner-sphere pathway that would involve formation of a copper(II) superoxide species. This is consistent with a previous study that showed the formation of superoxide species upon redox cycling of the Cu-PrP(106–114) complex35 and with our finding that the Met residues are oxidized to sulfoxides upon redox cycling of the CuI-PrP(106–115) complex.
It is important to note that the reoxidation rate shows an inverse correlation to the number of Met residues in the initial CuI complex, following the trend CuI-PrP(106–115) < CuI-PrP(106–115)M109A ∼ CuI-PrP(106–115)M112A < CuI-PrP(106–115)M109&M112A (Table 4). The reoxidation rate of the CuI-PrP(106–115)M109&M112A complex is 6 times faster than those of the single variants and 10 times faster than that of the CuI-PrP(106–115) complex containing both Met residues. These results suggest that the presence of Met ligands at the CuI complex affects the rate-limiting step in the reoxidation process, which likely involves formation of the copper(II) superoxide species. XANES data clearly show that, in the absence of Met residues, the CuI-PrP(106–115)M109&M112A complex is two-coordinate, while the CuI-PrP(106–115) complex is four-coordinate (vide supra). Thus, in an associative mechanism for formation of the copper(II) superoxide species, a faster reaction would be expected for the CuI-PrP(106–115)M109&M112A complex because of its coordination unsaturation, as observed. The nature of the mechanism (associative vs dissociative) for the inner-sphere oxygen reaction of the four-coordinate CuI-PrP(106–115) complex requires further computational evaluation.
4. Discussion
4.1. Copper Binding to His111
CuII-PrP(106–115) complexes at pH 6.5 and 8.5 were studied by XAS, and the role of Met109 and Met112 in CuII coordination was evaluated using peptides with Met-to-Ala variants. Our XAS results show that CuII coordination to His111 occurs through nitrogen and oxygen atoms at low pH, and all nitrogen ligation at high pH (pKa = 7.5), which is consistent with the previously proposed coordination models, as shown in Scheme 1,33 while the possibility of Met residues acting as ligands for CuII can be discarded. XAS and NMR spectroscopy, in combination with electronic structure calculations, show that CuI coordination to PrP(106–115) is also pH-dependent (Scheme 2). At pH <5, the thioether groups of Met109 and Met112 bind CuI; with a pKa of ∼5, the nitrogen imidazole from His111 enters the coordination sphere, while both Met residues remain coordinated, adopting a 1N1O2S coordination mode. At pH 8.5, two CuI species with 2N1O1S (2NOSM109 and 2NOSM112) coordination modes are formed, with a single Met residue bound to CuI (Scheme 2). Although this study has used a short fragment of PrP that only includes His111, the 2S and 1N1O2S species described here would form in longer PrP protein chains. At low pH (below 5), all His residues would be protonated and Met109 and Met112 would dominate CuI coordination to form the 2S species. At higher pH, other N-terminal His residues, such as His96, could participate in CuI binding; however, a recent study of CuI and AgI coordination to the 91–127 fragments demonstrated that a helical structure at the hydrophobic region of the 91–127 fragments causes His96 to fall out of the coordination sphere, leaving His111 as the main copper anchoring site;64 thus, the 1N1O2S complex described herein would be the dominant species.
PrPC is a synaptic glycolipid-anchored membrane protein predominantly expressed on presynaptic membranes,65 where it is exposed to CuII ions in a range of concentrations: from 15 μM at the synaptic cleft during synaptic vesicle release up to 100–300 μM during neuronal depolarization.66,67 Thus, PrPC can interact with CuII ions in the extracellular space, and at pH 7.4, there is a mixture of two species 3N1O and 4N (Scheme 1 and Figure 11A). Also, biological reductants, such as ascorbic acid, are found in concentrations of 150 μM in the cerebrospinal fluid.68,69 Thus, CuII-PrP complexes anchored at His111 in the synaptic cleft may be reduced to CuI by available reducing agents. Our results indicate that, in the presence of ascorbate, the CuII-3N1O complex would reduce faster to generate a CuI-1N1O2S complex. Therefore, at physiological pH, 3N1O and 4N complexes are the relevant CuII species, while the 1N1O2S complex is the relevant CuI coordination mode at the His111 site in PrPC (Figure 11B).
Figure 11.

Schematic representation of the main Cu-PrPC complexes under different physiological conditions. (A) At low CuII concentration (nM), CuII would be anchored at the OR (CuII-OR) and at His96 and His111 sites (CuII-3N1O and CuII-4N). (B) CuII complexes in the presence of reducing agents would be reduced. At the His111 site, the most abundant species would be CuI-1N1O2S. (C) High copper concentrations (100–500 μM) can stimulate endocytosis; in the endosomes, only the His111 site would coordinate CuI through Met109 and Met112. (D) In the extracellular space, oxygen activation by copper bound to His111 could generate superoxide, causing the subsequent oxidation of Met residues. Footnotes: a, ref (27); b, ref (28); c, ref (33); d, this work; e, ref (34); f, ref (35).
Unlike all other copper binding sites in the human PrP protein, the His111 binding site contains the MKHM motif, which provides it with unique coordination and redox properties. MX2MX2M motifs have been identified in copper-transport proteins (e.g., Yeast yCtr1) that bind CuI selectively via Met residues with affinities in each motif on the order of ∼2.5 × 10–6 M at pH 4.5.70 On the other hand, several proteins involved in copper transport that utilize His in combination with Met residues for CuI binding have emerged, such as PcoC with a CuI-(His)(Met)2 coordination mode,71 CopC [CuI-(His)(Met)3; KD ∼ 10–13 M],72,73 and CusF [CuI-(His)(Met)2(O)].74 In these cases, the presence of the His residue in the coordination sphere plays two roles: (i) to increase the affinity for CuI and (ii) to enable the coordination of CuII ions.75,76 Furthermore, it has been demonstrated that thioether ligands can be particularly effective at chelating CuI when they are separated by no more than two amino acids.70 The MKHM motif in the human PrP(106–115) sequence fulfills these conditions, such that at physiological pH CuII can be anchored via the His residue, while upon reduction, the CuI ion is bound by His and Met residues. High CuII concentrations stimulate endocytosis of the prion protein,21,23 taking the protein from the extracellular space at pH 7.4 to the endosomal space, where there is a high concentration of protons (pH 4–5) and biological reductants, such as glutathione or nicotinamide adenine dinucleotide (NADH), that are found in concentrations of 1–3 mM and ∼200 μM, respectively.77,78 Under these conditions, His111 would be protonated and CuI would be anchored only by Met residues, as illustrated in Figure 11C. Thus, this site is optimized to chelate both CuII and CuI ions and is likely to be involved in CuI transport into the cell.
4.2. Reactivity
CuII and CuI bound to PrP(106–115) form complexes with very different geometries and coordination spheres: CuII-3N1O versus CuI-1N1O2S at pH 6.5 and CuII-4N versus CuI-2N1O1S at pH 8.5, resulting in large reorganization energies of the site upon reduction of 1.79 and 1.6 eV, respectively. In spite of its higher reorganization energy, reduction of the CuII-3N1O complex is faster than that of the CuII-4N species because of its higher reduction potential. Considering that, at physiological pH, CuII-3N1O and CuII-4N complexes are present, our results indicate that, under reducing conditions, the CuII-3N1O complex would reduce faster, shifting the equilibrium between 3N1O and 4N toward the CuII-3N1O complex and favoring production of the CuI-1N1O2S complex.
Overall, the reorganization energies for the Cu-PrP(106–115) complexes are large compared to those of copper sites that are optimized to support electron transfer (blue copper T1 and CuA sites), which range from 0.5 to 0.82 eV.79−81 Despite its large reorganization energy, the CuI-PrP(106–115) complex still reacts with dioxygen. Several studies regarding the redox activity and functional implications of Cu-PrPC complexes have been put forward.35−37,82−85 Some studies have reported the generation of reactive oxygen species (H2O2 or superoxide) by Cu-PrP complexes;34−36,83,84,86 however, little is known about the mechanism of dioxygen activation by the different Cu-PrP complexes. This study investigated the kinetics of the first step of dioxygen activation by the CuI-1N1O2S complex at pH 6.5. The reoxidation rate is highly dependent on the presence of the Met residues. The estimate for the rate of outer-sphere reoxidation is 102 times slower than the experimental rate of CuI reoxidation from the kinetic data. Thus, we conclude that the CuI-1N1O2S complex at the His111 site reacts with dioxygen in an inner-sphere electron-transfer pathway that involves binding of dioxygen to the CuI complex and formation of a copper(II) superoxide intermediate, followed by the release of superoxide and partial regeneration of the CuII complex.
Our study also shows that the Met residues can be selectively oxidized to sulfoxide by the copper-catalyzed reaction (Figure 11D). It has been proposed that copper-catalyzed oxidation of Met residues may trigger a structural transition, leading to aggregation of PrP protein,87,88 or it may interfere with conversion of the prion protein into the fibrillar proteinase K-resistant conformation.89 Beyond that, Met residues in PrPC may act as an innate antioxidant defense in the protein by their ability to scavenge the produced superoxide and undergo oxidation to form methionine sulfoxide. Without the Met residues, the produced superoxide would produce cell damage. The enzymatic peptide methionine sulfoxide reductase (MsrA) reverses methionine sulfoxide back to Met, which once again is able to scavenge oxidants.90 In fact, in the Alzheimer’s disease brain, a decrease in the activity of MsrA compared to control subjects was observed,91 while the induction of MsrA activity protects neuronal cells from Amyloid β toxicity.92 It is plausible that a reversible oxidation/reduction of Met residues at the N-terminal region of the PrPC protein could be acting as an ROS sink. This would be consistent with the notion that PrPC plays a role in cellular antioxidant defense.93
5. Conclusions
In summary, our study shows that CuI binding to His111 is highly pH-dependent and that the presence of one His and two Met residues in the MKHM motif of the human PrP(106–115) fragment confers this site with unique CuI binding properties. Even upon drastic changes in the chemical environment, in particular those occurring during endocytosis, the two Met residues in the MKHM motif allow PrPC to keep CuI ions anchored, consistent with a copper-transport function for this protein. On the other hand, in the extracellular space, in the presence of reducing agents, the most populated CuI-His111 complex would be a CuI-1N1O2S species, which is capable of activating dioxygen. Our study provides further insight into the molecular mechanism of oxygen activation by this site. The CuI-1N1O2S complex reacts with dioxygen through an inner-sphere mechanism, likely involving the formation of a copper(II) superoxide intermediate, followed by the release of superoxide and partial regeneration of the CuII complex. The Met residues are oxidized to sulfoxide in this process, and their ability to scavenge superoxide may play a role in the proposed antioxidant properties of PrPC.
Acknowledgments
This research was supported by CONACYT (Grants 221134 and 193318 to L.Q., Grants 601210 and 128369 to A.V., grant 128411 to purchase stopped-flow instrument, and a graduate fellowship to T.A.-L., and by the National Institutes of Health (NIH; Grant DK31450 to E. I.S.). T.A.-L. is thankful for a Fulbright-García Robles fellowship. The authors thank Dr. Patrick Frank for technical assistance in preparing the XAS samples, Dr. Federico del Río (UNAM) for preliminary NMR tests, M. C. Emmanuel R. C. and LaNSE-Cinvestav (Unit of Genomics, Proteomics and Metabolomics) for assistance with MALDI-TOF experiments, Selena Martinez-Gomez and Atenea Villegas-Vargas for peptide synthesis, Carolina Sanchez-López and Geiser Cuellar for assistance with the acquisition of ES-MS data, and CGSTIC-Cinvestav for providing computing time for the “Xiuhcoatl Hybrid Supercomputing Cluster”. XAS data were measured at the SSRL, which is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the U.S. DOE, Office of Biological and Environmental Research, and by the NIH, National Institute of General Medical Sciences (NIGMS; Grant P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIGMS or NIH.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.5b02794.
EPR spectra, representative distribution of explicit H2O molecules, representative EXAFS and its FT data and their fits using reoptimized structures, XANES data, TOCSY 1H–1H, HSQC 1H–13C, stopped-flow absorption data, representative CD and EPR spectra at the end point of reoxidation kinetics, MALDI TOF/TOF analysis of peptides before and after a redox cycle with ascorbate and oxygen, lowest harmonic frequencies, Cu–ligand distances (Å) for selected geometry-optimized models, effects of implicit and explicit solvation, initial models of the coordination of CuI-PrP(106–113), structural and energetic parameters of the optimized geometries at OPBE/TZVP with or without six water molecules for the groups 1, 2A, and 2B, calculated reorganization energies (λi), estimation of E°(CuII/I-PrP106-115) at pH 6.5 and 8.5 by reductive titrations, calculation of kET(3N1O)/kET(4N) using the Marcus equation and calculation of the outer-sphere electron transfer rate, and Cartesian coordinates for all optimized structures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Prusiner S. B. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 13363–13383. 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer R. K.; Mckinley M. P.; Bowman K. A.; Braunfeld M. B.; Barry R.; Prusiner S. B. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 2310–2314. 10.1073/pnas.83.8.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan K. M.; Baldwin M.; Nguyen J.; Gasset M.; Serban A.; Groth D.; Mehlhorn I.; Huang Z.; Fletterick R. J.; Cohen F. E. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 10962–10966. 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner S. B. Science 1997, 278, 245–251. 10.1126/science.278.5336.245. [DOI] [PubMed] [Google Scholar]
- Gray F.; Chrétien F.; Adle-Biassette H.; Dorandeu A.; Ereau T.; Delisle M.-B.; Kopp N.; Ironside J. W.; Vital C. J. Neuropathol. Exp. Neurol. 1999, 58, 321–328. 10.1097/00005072-199904000-00002. [DOI] [PubMed] [Google Scholar]
- Prusiner S. B. Science 1982, 216, 136–144. 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- Flechsig E.; Weissmann C. Curr. Mol. Med. 2004, 4, 337–353. 10.2174/1566524043360645. [DOI] [PubMed] [Google Scholar]
- Schatzl H. M.; Da Costa M.; Taylor L.; Cohen F. E.; Prusiner S. B. J. Mol. Biol. 1995, 245, 362–374. 10.1006/jmbi.1994.0030. [DOI] [PubMed] [Google Scholar]
- Locht C.; Chesebro C.; Race R.; Keith J. M. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 6372–6376. 10.1073/pnas.83.17.6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donne D. G.; Viles J. H.; Groth D.; Mehlhorn I.; James T. L.; Cohen F. E.; Prusiner S. B.; Wright P. E.; Dyson H. J. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 13452–13457. 10.1073/pnas.94.25.13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn R.; Liu A.; Luhrs T.; Riek R.; von Schroetter C.; Lopez-Garcia F.; Billeter M.; Calzolai L.; Wider G.; Wuthrich K. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 145–150. 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouillet-Richard S. Science 2000, 289, 1925–1928. 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- Mange A.; Milhavet O.; Umlauf D.; Harris D.; Lehmann S. FEBS Lett. 2002, 514, 159–162. 10.1016/S0014-5793(02)02338-4. [DOI] [PubMed] [Google Scholar]
- Roucou X.; Gains M.; LeBlanc A. C. J. Neurosci. Res. 2004, 75, 153–161. 10.1002/jnr.10864. [DOI] [PubMed] [Google Scholar]
- Bounhar Y.; Zhang Y.; Goodyer C. G.; LeBlanc A. J. Biol. Chem. 2001, 276, 39145–39149. 10.1074/jbc.C100443200. [DOI] [PubMed] [Google Scholar]
- Linden R.; Martins V. R.; Prado M. A. M.; Cammarota M.; Izquierdo I.; Brentani R. R. Physiol. Rev. 2008, 88, 673–728. 10.1152/physrev.00007.2007. [DOI] [PubMed] [Google Scholar]
- Linden R.; Cordeiro Y.; Lima L. M. T. R. Cell. Mol. Life Sci. 2012, 69, 1105–1124. 10.1007/s00018-011-0847-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassallo N.; Herms J. J. Neurochem. 2003, 86, 538–544. 10.1046/j.1471-4159.2003.01882.x. [DOI] [PubMed] [Google Scholar]
- Kretzschmar H.; Brown D. R.; Qin K.; Herms J. W.; Madlung A.; Manson J.; Strome R.; Fraser P. E.; Kruck T.; von Bohlen A.; Schulz-Schaeffer W.; Giese A.; Westaway D. Nature 1997, 390, 684–687. 10.1038/37783. [DOI] [PubMed] [Google Scholar]
- Millhauser G. L. Acc. Chem. Res. 2004, 37, 79–85. 10.1021/ar0301678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauly P. C. J. Biol. Chem. 1998, 273, 33107–33110. 10.1074/jbc.273.50.33107. [DOI] [PubMed] [Google Scholar]
- Perera W. S. S.; Hooper N. M. Curr. Biol. 2001, 11, 519–523. 10.1016/S0960-9822(01)00147-6. [DOI] [PubMed] [Google Scholar]
- Brown L. R.; Harris D. A. J. Neurochem. 2003, 87, 353–363. 10.1046/j.1471-4159.2003.01996.x. [DOI] [PubMed] [Google Scholar]
- Aronoff-Spencer E.; Burns C. S.; Avdievich N. I.; Gerfen G. J.; Peisach J.; Antholine W. E.; Ball H. L.; Cohen F. E.; Prusiner S. B.; Millhauser G. L. Biochemistry 2000, 39, 13760–13771. 10.1021/bi001472t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns C. S.; Aronoff-Spencer E.; Legname G.; Prusiner S. B.; Antholine W. E.; Gerfen G. J.; Peisach J.; Millhauser G. L. Biochemistry 2003, 42, 6794–6803. 10.1021/bi027138+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanar L.; Rivillas-Acevedo L.; Grande-Aztatzi R.; Gómez-Castro C. Z.; Arcos-López T.; Vela A. Coord. Chem. Rev. 2013, 257, 429–444. 10.1016/j.ccr.2012.06.026. [DOI] [Google Scholar]
- Chattopadhyay M.; Walter E. D.; Newell D. J.; Jackson P. J.; Aronoff-Spencer E.; Peisach J.; Gerfen G. J.; Bennett B.; Antholine W. E.; Millhauser G. L. J. Am. Chem. Soc. 2005, 127, 12647–12656. 10.1021/ja053254z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hureau C.; Mathe C.; Faller P.; Mattioli T. A.; Dorlet P. JBIC, J. Biol. Inorg. Chem. 2008, 13, 1055–1064. 10.1007/s00775-008-0389-0. [DOI] [PubMed] [Google Scholar]
- Grande-Aztatzi R.; Rivillas-Acevedo L.; Quintanar L.; Vela A. J. Phys. Chem. B 2013, 117, 789–799. 10.1021/jp310000h. [DOI] [PubMed] [Google Scholar]
- Di Natale G.; Grasso G.; Impellizzeri G.; La Mendola D.; Micera G.; Mihala N.; Nagy Z.; Osz K.; Pappalardo G.; Rigo V.; Rizzarelli E.; Sanna D.; Sovago I. Inorg. Chem. 2005, 44, 7214–7225. 10.1021/ic050754k. [DOI] [PubMed] [Google Scholar]
- Remelli M.; Donatoni M.; Guerrini R.; Janicka A.; Pretegiani P.; Kozłowski H. Dalton Trans. 2005, 17, 2876–2885. 10.1039/b505314e. [DOI] [PubMed] [Google Scholar]
- Shearer J.; Soh P.; Lentz S. J. Inorg. Biochem. 2008, 102, 2103–2113. 10.1016/j.jinorgbio.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivillas-Acevedo L.; Grande-Aztatzi R.; Lomeli I.; Garcia J. E.; Barrios E.; Teloxa S.; Vela A.; Quintanar L. Inorg. Chem. 2011, 50, 1956–1972. 10.1021/ic102381j. [DOI] [PubMed] [Google Scholar]
- Liu L.; Jiang D.; McDonald A.; Hao Y.; Millhauser G. L.; Zhou F. J. Am. Chem. Soc. 2011, 133, 12229–12237. 10.1021/ja2045259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer J.; Soh P. Inorg. Chem. 2007, 46, 710–719. 10.1021/ic061236s. [DOI] [PubMed] [Google Scholar]
- Turnbull S.; Tabner B. J.; Brown D. R.; Allsop D. Neurosci. Lett. 2003, 336, 159–162. 10.1016/S0304-3940(02)01254-5. [DOI] [PubMed] [Google Scholar]
- Badrick A. C.; Jones C. E. J. Inorg. Biochem. 2009, 103, 1169–1175. 10.1016/j.jinorgbio.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Kates E.; Albericio F.. Solid-Phase Synthesis; Marcel Dekker Inc.: New York, 2000. [Google Scholar]
- Hood C. A.; Fuentes G.; Patel H.; Page K.; Menakuru M.; Park J. H. J. Pept. Sci. 2008, 14, 97–101. 10.1002/psc.921. [DOI] [PubMed] [Google Scholar]
- Tenderholt A.; Hedman B.; Hodgson K. O. AIP Conf. Proc. 2006, 882, 105–107. 10.1063/1.2644442. [DOI] [Google Scholar]
- George G. N.EXAFSPAK ; Stanford Synchrotron Radiation Laboratory: Menlo Park, CA, 2000.
- Rehr J. J.; Albers R. C. Rev. Mod. Phys. 2000, 72, 621–654. 10.1103/RevModPhys.72.621. [DOI] [Google Scholar]
- Koster A. M.; Geudtner G.; Calaminici P.; Casida M. E.; Dominguez V. D.; Flores-Moreno R.; Gamboa G. U.; Goursot A.; Heine T.; Ipatov A.; Janetzko F.; del Campo J. M.; Reveles J. U.; Vela A.; Zuniga-Gutierrez B.; Salahub D. R.. deMon2k, version 2; The deMon Developers, Cinvestav: Mexico City, Mexico, 2011.
- Handy N. C.; Cohen A. J. Mol. Phys. 2001, 99, 403–412. 10.1080/00268970010018431. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Godbout N.; Salahub D. R.; Andzelm J.; Wimmer E. Can. J. Chem. 1992, 70, 560–571. 10.1139/v92-079. [DOI] [Google Scholar]
- Calaminici P.; Janetzko F.; Köster A. M.; Mejia-Olvera R.; Zuniga-Gutierrez B. J. Chem. Phys. 2007, 126, 044108. 10.1063/1.2431643. [DOI] [PubMed] [Google Scholar]
- Dunlap B. I.; Connolly J. W. D.; Sabin J. R. J. Chem. Phys. 1979, 71, 3396–3402. 10.1063/1.438728. [DOI] [Google Scholar]
- Köster A. M.; del Campo J. M.; Janetzko F.; Zuniga-Gutierrez B. J. Chem. Phys. 2009, 130, 114106. 10.1063/1.3080618. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Schuurmann G. J. Chem. Soc., Perkin Trans. 2 1993, 2, 799–805. 10.1039/p29930000799. [DOI] [Google Scholar]
- Sinnecker S.; Rajendran A.; Klamt A.; Diedenhofen M.; Neese F. J. Phys. Chem. A 2006, 110, 2235–2245. 10.1021/jp056016z. [DOI] [PubMed] [Google Scholar]
- Neese F.ORCA: An ab initio density functional and semiempirical program package, version 2.6, revision 35; University of Bonn: Bonn, Germany, 2008.
- Klimkans A.; Larsson S. Chem. Phys. 1994, 189, 25–31. 10.1016/0301-0104(94)80004-9. [DOI] [Google Scholar]
- Kau L. S.; Spira-Solomon D. J.; Penner-Hahn J. E.; Hodgson K. O.; Solomon E. I. J. Am. Chem. Soc. 1987, 109, 6433–6442. 10.1021/ja00255a032. [DOI] [Google Scholar]
- Brown I. D.; Altermatt D. Acta Crystallogr., Sect. B: Struct. Sci. 1985, 41, 244–247. 10.1107/S0108768185002063. [DOI] [Google Scholar]
- Thorp H. H. Inorg. Chem. 1992, 31, 1585–1588. 10.1021/ic00035a012. [DOI] [Google Scholar]
- Liu W.; Thorp H. H. Inorg. Chem. 1993, 32, 4102–4105. 10.1021/ic00071a023. [DOI] [Google Scholar]
- Dennison C.; Kohzuma T.; McFarlane W.; Suzuki S.; Sykes A. G. Inorg. Chem. 1994, 33, 3299–3305. 10.1021/ic00093a017. [DOI] [Google Scholar]
- Yanagisawa S.; Sato K.; Kikuchi M.; Kohzuma T.; Dennison C. Biochemistry 2003, 42, 6853–6862. 10.1021/bi030030p. [DOI] [PubMed] [Google Scholar]
- Hulsker R.; Mery A.; Thomassen E. A.; Ranieri A.; Sola M.; Verbeet M. P.; Kohzuma T.; Ubbink M. J. Am. Chem. Soc. 2007, 129, 4423–4429. 10.1021/ja0690464. [DOI] [PubMed] [Google Scholar]
- Marcus R. A. Annu. Rev. Phys. Chem. 1964, 15, 155–196. 10.1146/annurev.pc.15.100164.001103. [DOI] [Google Scholar]
- Marcus R. A. Angew. Chem., Int. Ed. Engl. 1993, 32, 1111–1121. 10.1002/anie.199311113. [DOI] [Google Scholar]
- Wood P. M. Biochem. J. 1988, 253, 287–289. 10.1042/bj2530287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valensin D.; Padula E. M.; Hecel A.; Luczkowski M.; Kozlowski H. J. Inorg. Biochem. 2016, 155, 26–35. 10.1016/j.jinorgbio.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Herms J.; Tings T.; Gall S.; Madlung A.; Giese A.; Siebert H.; Schurmann P.; Windi O.; Brose B.; Kretzschmar H. J. Neurosci. 1999, 19, 8866–8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopt A.; Korte S.; Fink H.; Panne U.; Niessner R.; Jahn R.; Kretzschmar H.; Herms J. J. Neurosci. Methods 2003, 128, 159–172. 10.1016/S0165-0270(03)00173-0. [DOI] [PubMed] [Google Scholar]
- Kardos J.; Kovacs I.; Hajós F.; Kálman M.; Simonyi M. Neurosci. Lett. 1989, 103, 139–144. 10.1016/0304-3940(89)90565-X. [DOI] [PubMed] [Google Scholar]
- Shiraishi N.; Nishikimi M. FEBS Lett. 2002, 511, 118–122. 10.1016/S0014-5793(01)03324-5. [DOI] [PubMed] [Google Scholar]
- Carr R. S.; Bally M. B.; Thomas P.; Neff J. M. Anal. Chem. 1983, 55, 1229–1232. 10.1021/ac00259a012. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Nadas I. A.; Kim M. A.; Franz K. J. Inorg. Chem. 2005, 44, 9787–9794. 10.1021/ic051180m. [DOI] [PubMed] [Google Scholar]
- Peariso K.; Huffman D. L.; Penner-Hahn J. E.; O’Halloran T. V. J. Am. Chem. Soc. 2003, 125, 342–343. 10.1021/ja028935y. [DOI] [PubMed] [Google Scholar]
- Arnesano F.; Banci L.; Bertini I.; Mangani S.; Thompsett A. R. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3814–3819. 10.1073/pnas.0636904100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koay M.; Zhang L.; Yang B.; Maher M. J.; Xiao Z.; Wedd A. G. Inorg. Chem. 2005, 44, 5203–5205. 10.1021/ic0506198. [DOI] [PubMed] [Google Scholar]
- Loftin I. R.; Franke S.; Roberts S. A.; Weichsel A.; Heroux A.; Montfort W. R.; Rensing C.; McEvoy M. M. Biochemistry 2005, 44, 10533–10540. 10.1021/bi050827b. [DOI] [PubMed] [Google Scholar]
- Haas K. L.; Putterman A. B.; White D. R.; Thiele D. J.; Franz K. J. J. Am. Chem. Soc. 2011, 133, 4427–4437. 10.1021/ja108890c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino J. T.; Chenkin M. P.; Keller M.; Riggs-Gelasco P.; Franz K. J. Metallomics: integrated biometal science 2011, 3, 61–73. 10.1039/C0MT00044B. [DOI] [PubMed] [Google Scholar]
- Lin S.; Guarente L. Curr. Opin. Cell Biol. 2003, 15, 241–246. 10.1016/S0955-0674(03)00006-1. [DOI] [PubMed] [Google Scholar]
- Handbook of Neurochemistry and Molecular Neurobiology. Amino Acids and Peptides in the Nervous System, 3rd ed.; Springer: Berlin, 2007; p 352. [Google Scholar]
- Di Bilio A. J.; Hill M. G.; Bonander N.; Karlsson G.; Villahermosa R. M.; Malmstrom B. G.; Winkler J. R.; Gray H. B. J. Am. Chem. Soc. 1997, 119, 9921–9922. 10.1021/ja971518e. [DOI] [Google Scholar]
- Olsson M. H. M.; Ryde U. J. Am. Chem. Soc. 2001, 123, 7866–7876. 10.1021/ja010315u. [DOI] [PubMed] [Google Scholar]
- Farver O.; Hwang H. J.; Lu Y.; Pecht I. J. Phys. Chem. B 2007, 111, 6690–6694. 10.1021/jp0672555. [DOI] [PubMed] [Google Scholar]
- Brown D. R.; Wong B.; Hafiz F.; Clive C.; Haswell S. J.; Jones I. M. Biochem. J. 1999, 344, 1–5. 10.1042/bj3440001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano T. Int. J. Biol. Sci. 2007, 3, 57–63. 10.7150/ijbs.3.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal R. C.; Abdelraheim S. R.; Brazier M. W.; Rigby S. E.; Brown D. R.; Viles J. H. Free Radical Biol. Med. 2007, 42, 79–89. 10.1016/j.freeradbiomed.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Zhou F.; Millhauser G. L. Coord. Chem. Rev. 2012, 256, 2285–2296. 10.1016/j.ccr.2012.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull S.; Tabner B. J.; Brown D. R.; Allsop D. Biochemistry 2003, 42, 7675–7681. 10.1021/bi030036e. [DOI] [PubMed] [Google Scholar]
- Requena J. R.; Groth D.; Legname G.; Stadtman E. R.; Prusiner S. B.; Levine R. L. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 7170–7175. 10.1073/pnas.121190898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younan N. D.; Nadal R. C.; Davies P.; Brown D. R.; Viles J. H. J. Biol. Chem. 2012, 287, 28263–28275. 10.1074/jbc.M112.354779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breydo L.; Bocharova O. V.; Makarava N.; Salnikov V. V.; Anderson M.; Baskakov I. V. Biochemistry 2005, 44, 15534–15543. 10.1021/bi051369+. [DOI] [PubMed] [Google Scholar]
- Lowther W. T.; Brot N.; Weissbach H.; Matthews B. W. Biochemistry 2000, 39, 13307–13312. 10.1021/bi0020269. [DOI] [PubMed] [Google Scholar]
- Gabbita S. P.; Aksenov M. Y.; Lovell M. A.; Markesbery W. R. J. Neurochem. 1999, 73, 1660–1666. 10.1046/j.1471-4159.1999.0731660.x. [DOI] [PubMed] [Google Scholar]
- Moskovitz J.; Maiti P.; Lopes D. H.; Oien D. B.; Attar A.; Liu T.; Mittal S.; Hayes J.; Bitan G. Biochemistry 2011, 50, 10687–10697. 10.1021/bi201426b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klamt F.; Dal-Pizzol F.; Conte da Frota M. L. Jr.; Walz R.; Andrades M. E.; Da Silva E. G.; Brentani R. R.; Izquierdo I.; Moreira J. C. F. Free Radical Biol. Med. 2001, 30, 1137–1144. 10.1016/S0891-5849(01)00512-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.