

Abstract
Background
Dexmedetomidine is a highly selective α2-adrenoreceptor agonist with sedative, analgesic and sympatholytic properties. Its cardiac protective effect cannot be ignored, notwithstanding its associated adverse drug reactions. This study aimed to investigate the effects of dexmedetomidine on L-type calcium current (ICa-L) in adult rat ventricular myocytes, and to clarify the electrophysiological mechanism of its effect on cardiomyocytes.
Methods
Single rat ventricular myocytes were obtained by enzymatic dissociation method. Myocytes were perfused with external solutions containing various concentrations of dexmedetomidine at a flow rate of 2-3 ml/min for 5 min. Whole-cell current recordings were performed using the conventional whole-cell patch-clamp technique. Besides, the effects of 1 μM yohimbine, an alpha-2 adrenergic antagonist, were given alone or in combination with 10 ng/ml dexmedetomidine.
Results
Dexmedetomidine inhibited the amplitude of ICa-L in a concentration-dependent manner. The current voltage curve was shifted upwards. The steady activated curves were shifted to the right and the V1/2 activation of the ICa-L were increased by dexmedetomidine at the high concentration (10 and 200 ng/ml). Dexmedetomidine did not affect the ICa-L steady-state inactivation curve, but shifted down the recovery curve. Yohimbine did not have influence on ICa-L. However, inhibition of ICa-L by dexmedetomidine at the concentration of 10 ng/ml was partially reversed by yohimbine.
Conclusions
Dexmedetomidine can attenuate ICa-L in adult rat ventricular myocytes, which may contribute to its negative effects on myocardia contractility and cardiac electrophysiology. Its inhibitory effect on ICa-L is partially associated with alpha-2 adrenergic receptors.
Keywords: Cardiology, Dexmedetomidine, L-type calcium current, Ventricular myocytes, Whole-cell patch clamp
INTRODUCTION
Dexmedetomidine (DEX) is a highly specific and selective α2-adrenoreceptor agonist that possesses sedative, analgesic and sympatholytic properties. DEX is currently widely used in the intensive care setting and in clinical anesthesia.1,2 It has been noted that DEX offers remarkable “awake sedation” properties with the unique characteristic of causing no respiratory depression. In addition, it depresses the stress reaction caused by the tracheal intubation and surgical stimulation, which allows hemodynamic stability. It decreases the frequency of use of propofol, fentanyl, sevoflurane and other anesthetics, and reduces agitation during anesthesia recovery. It can be applied in postoperative analgesia as well. However, hypotension, bradycardia and nausea were the most frequently observed adverse drug reactions. In animals, DEX has marked cardiovascular effects, such as bradycardia, and decreases in stroke volume, cardiac output and myocardial oxygen consumption. Although it can have negative effects on the cardiovascular system, it offers remarkable pharmacological properties such as a cardioprotective effect in a wide range of circumstances.3,4 Results observed in animal experiments indicate that DEX can have a moderating effect on myocardial ischemia and reperfusion injury. Pre-clinical application also showed that DEX can reduce the incidence of myocardial ischemia and myocardial infarction and other cardiovascular events, and decrease the perioperative mortality of patients with non-cardiac surgery. ICa-L, which are the bridges of communicating myocardial electrical activity and mechanical activity, are important ion channels in cardiomyocytes.5,6 This study was conducted to test the following hypo-theses: its negative effects on cardiac electric and mechanical activity is related to the ICa-L, and can be attributed to a direct effect of DEX on the myocardium. The whole-cell patch-clamp technique was used to assess the effects of DEX on ICa-L in isolated rat ventricular myocytes so as to reveal its associated electrophysiological mechanism of its inhibitory and cardioprotective effect on cardiomyocytes.
MATERIALS AND METHODS
Cardiomyocyte isolation
Ventricular myocytes were isolated from the heart of male Sprague-Dawley rats (150-250 g body weight). The rats were purchased from the Experiment Animal Center of Tongji Medical College, Huazhong University of Science and Technology. Room temperature was maintained at 23 celcius with constant (55%) humidity, and lights were maintained on a 12-hour light (8:00 am to 8:00 pm)/dark cycle. The heart was mounted on a Langendorff apparatus and perfused with a nominally Ca2+-free Tyrode’s solution for 10 min and then with an enzymatic solution (Ca2+-free Tyrode’s solution containing 0.15 mg/ml. collagenase I, and 0.5 mg/ml bovine serum albumin, Sigma) for 25-30 min. When the heart was soft, it was sequentially washed with 50 ml 0.2 mM Ca2+ Tyrode’s solution plus 1 mg/ml bovine serum albumin. The ventricles were cut off, chunked and gently stirred, ventricular myocytes were collected in Ca2+-free Tyrone’s solution plus 1 mg/ml bovine serum albumin, and used on the same day. All steps were performed at 37 celcius in solutions gassed with 95% O2 + 5% CO2.
Drugs and solutions
Tyrone’s solution contained (in mM): NaCl 135, KCl 5.4, CaCl2 1.8, MgCl2 1.0, NaH2PO4 0.33, HEPES 10, Glucose 10, with the pH adjusted to 7.35 with NaOH. The external solution for recording ICa-L was Tyrode’s solution containing different concentrations of DEX (0.6, 1.8, 5.4, 10, 200 ng/ml) or 1 μM yohimbine. The electrode internal solution for recording the ICa-L contained (in mM): CsCl 120, CaCl2 1.0, MgCl2 5, Na2ATP 5.0, EGTA 11, HEPES 10, Glucose 11, with the pH adjusted to 7.35 with CsOH. Collagenase I, yohimbine hydrochloride (Y3125-1G), HEPES, Na2ATP, CsCl, EGTA, Na2ATP and CsOH were purchased from Sigma. Dexmedetomidine (production batch number 09081232) were purchased from Jiangsu Hengrui Medicine Company Ltd. NaCl, KCl, CaCl2, NaH2PO4, MgCl2, Glucose, NaOH, KOH are domestic products with the analytical grade.
Electrophysiological recordings
The whole patch-clamp technique was performed to record currents in ventricular myocardial cells of healthy rats as previous studies have described.7,8 Isolated ventricular myocytes were placed in the experimental chamber (1.5 ml) mounted on the stage of an inverted microscope (IX70, Olympus, Japan). After settling down to the bottom of the chamber, cells were perfused with external solution included different concentrations of DEX (0.6, 1.8, 5.4, 10, 200 ng/ml) for 5 min at a rate of 2-3 ml/min. Glass microelectrodes were made using microelectrodes (PB-7, Narishige, Japan) by two-stage pulling and they had a resistance of 3.0 to 5.0 MΩ when filled with electrode internal solution. Mean capacitance of the cells was 109.20 ± 28.12 pF. Series resistances were less than 20 MΩ. All currents were digitally sampled at 10 kHz, low pass filtered at 1 kHz and saved on a hard drive for post hoc measurement. The measurements were performed at room temperature (20-25 celcius). Currents recordings were obtained and analyzed with an EPC-9 patch clamp amplifier (HEKA, Lambrecht, Germany) in the whole-cell mode and Pulse/Pulsefit software program (HEKA Elektronik, Lambrecht, Germany). In order to estimate the spontaneous decline of ICa-L with time (run-down) occurring during the first five minutes of recording, 5 mmol/L Mg-ATP was added to the pipette solution and commenced data acquisition after 5-15 min of equilibration between pipette solution and intracellular contents.
Date analysis
All data were analyzed by the use of Origin 5.0 software (Microcal Software, Northampton, MA, USA). For activation, current at each test potential was converted to conductance (G) using the following formula: G = I/(V – V rev), (Vrev is reversal potential). The peak conductance value for each test potential was normalized to Gmax and plotted against the test potential to produce a voltage - conductance relationship curve, which were fitted well with a Boltzmann equation G/Gmax = 1/{1 + exp[-(V – Vh)/k]}, where Vh is the potential of half-maximal activation and k is the slope factor. For inactivation, the current value was normalized by dividing the test current by the maximal current. The following form of Boltzmann function was used for data fitting: I/Imax = 1/{1 + exp[(V – Vh)/k]}, where Vh is the potential of half-maximal inactivation and k is slope factor. For recovery from inactivation, the data were fitted by a double exponential function. For time course of inactivation, fast and slow inactivation time constants were calculated by fitting a two exponential function to the extrapolated decay phase. All values were presented as means ± SEM. Statistical significance was evaluated by the two-tailed Student’s t-test, or if more than two conditions were compared by One-way analysis of variance (ANOVA), with p < 0.05 were considered significant.
RESULTS
Effect on Ica-L by different concentrations of DEX
ICa-L in rat ventricular myocytes was elicited by a 200 ms depolarization step pulse from the holding potential of -40 to +60 mV with step 10 mV. The acute effect of DEX on ICa-L was determined by monitoring the amplitude of ICa-L elicited by the step pulse mentioned above. The results showed that ICa-L activated around -30 mV, and then peaked around +10 mV. The inhibition of peak current amplitude was significantly enhanced with the increase of DEX concentration (0.6, 1.8, 5.4, 10, 200 ng/ml). DEX decreased the peak amplitude of ICa-L by 8.8 ± 2.4%, 14.6 ± 3.6%, 21.4 ± 8.4%, 25.2 ± 6.4%, 32.1 ± 6.6% (n = 6, p < 0.05) at 0.6, 1.8, 5.4, 10 and 200 ng/ml, respectively (Figure 1A). During perfusion with the extracellular fluid containing 10 ng/ml DEX, the peak current decreased by 25.2%, followed by perfusion for 5 minutes with the extracellular fluid containing 1 μM yohimbine, where the peak current increased by about 15% (Figure 1B). However, no significant changes had been shown before and after perfusion with the extracellular fluid containing 1 μM yohimbine alone.
Figure 1.
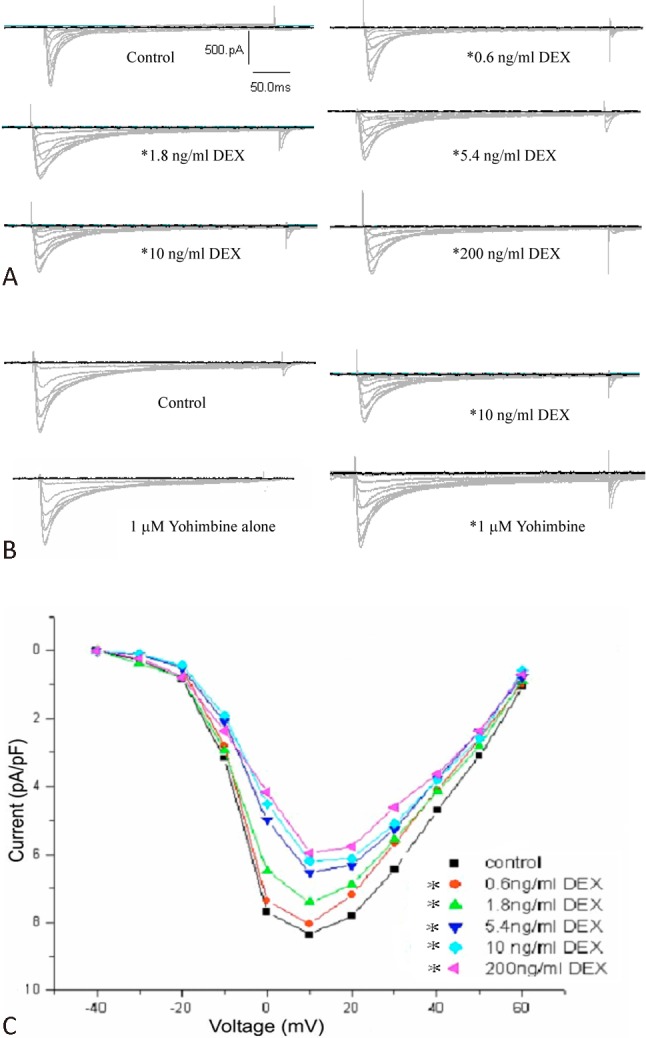
Dexmedetomedine (DEX) inhibited voltage-gated L-type calcium current (ICa-L) in rat ventricular myocytes. (A) Traces of ICa-L evoked in the absence and presence of DEX. Current was elicited by depolarization from a holding potential of -40 mV to +60 mV. (B) Effect on ICa,L by 10 ng/ml DEX and 1 μM yohimbine. (C) DEX decreased ICa-L at the test potential and made current-voltage (IV) curves upwards. (n = 6, *p < 0.05)
Current-voltage (I-V) curves were constructed by plotting the current amplitudes as a function of teat potentials. As shown in Figure 1C, DEX shifted current-voltage (IV) curves upwards, and markedly decreased the am-plitude of ICa-L at all test potentials. This inhibition efficiency increased as an increase in DEX concentration. Its shape, activation potential, peak potential and the reversal potential activation remained virtually unchanged.
Effect of DEX on steady-state activation and inactivation of ICa-L
Currents were evoked by a series of 10 mV voltage step to potential -40 mV and +60 mV from the holding potential of -40 mV to test the effect of DEX on ICa-L activation. In order to quantify the effects of DEX on the channel activation, conductance-voltage (G-V) curves were constructed (Figure 2A). Figure 2 shows the voltage dependence of steady-state activation and inactivation in the absence and presence of DEX. Effects of 0.6, 1.8, 5.4 ng/ml DEX on ICa-L activation were not significantly different, while 10, 200 ng/ml DEX shifted activation curves to the right, and slowed down the activation process. The values at Vh of the normalized activation conductance curves were -2.41 ± 1.21 mV with a slope factor (K) of 4.97 ± 0.34 for control and 5.93 ± 3.02 mV (n = 6, p < 0.05) with a K value of 9.00 ± 1.67 (n = 6, p < 0.05) for 10 ng/ml DEX, and 12.99 mV (n = 6, p < 0.05) with a K value of 13.05 (n = 6, p < 0.05) for 200 ng/ml (Figure 2A).
Figure 2.
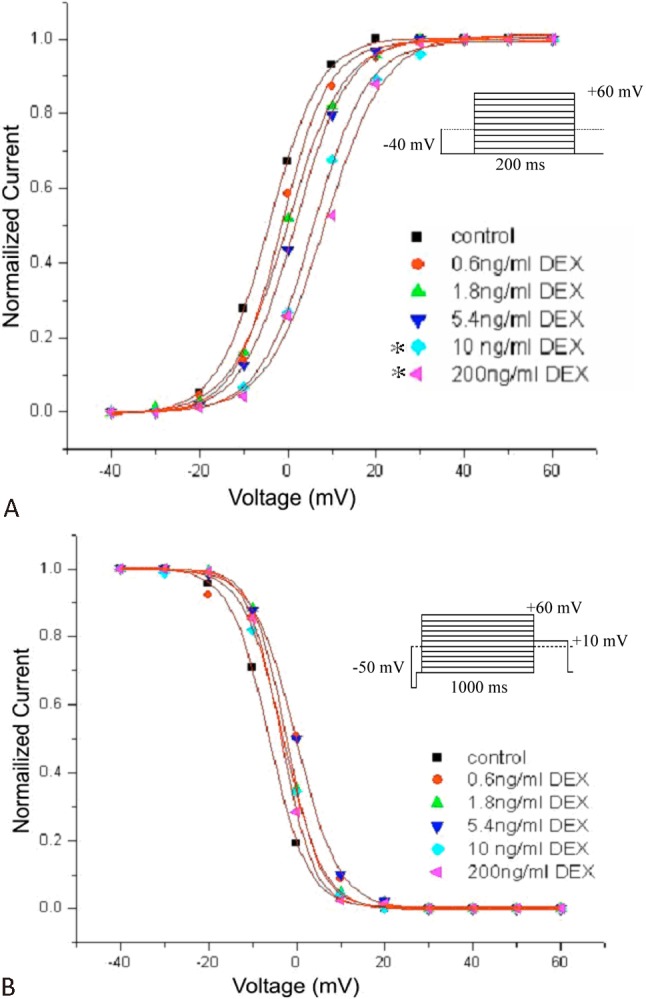
Effects of Dexmedetomedine (DEX) on steady-state activation (A) and inactivation (B) of L-type calcium current (ICa-L) in the absence and presence of different concentrations of DEX (0.6, 1.8, 5.4, 10 and 200 ng/ml). The values of activation and inactivation were fitted well with a Boltzmann equation. (n = 6, *p < 0.05)
The steady-state inactivation was determined using a double-pulse protocol: a 1000 ms prepulse to potentials between -50 and +60 mV with 10 mV increments, followed by a fixed 400 ms test pulse to 10 mV. The inactivation curves shown in Figure 2B were obtained by plotting the normalized ICa-L against the prepulse voltages. The inactivation values were not significantly different between the absence and presence of DEX. These results suggested that DEX altered the activation gating property of the cardiac ICa-L, but not the inactivation gating property.
Effect of DEX on the recovery of ICa-L from inactivation
The recovery of ICa-L was determined by using double-pulse protocol consisting of two identical pulses (holding potential from -50 mV to +10 mV for 300 ms) in variable intervals from 50 to 500 ms in 50 ms increment. The data were fitted with a double exponential function. DEX shifted the curve to the right (Figure 3). The recovery time of ICa-L from inactivation was 98.80 ± 16.88 ms for control, 102.34 ± 13.01 ms, 139.82 ± 15.52 ms, 175.11 ± 19.65 ms, 205.44 ± 22.63 ms, 239.54 ± 28.43 ms for 0.6, 1.8, 5.4, 10 and 200 ng/ml, respectively (n = 6, p < 0.05).
Figure 3.
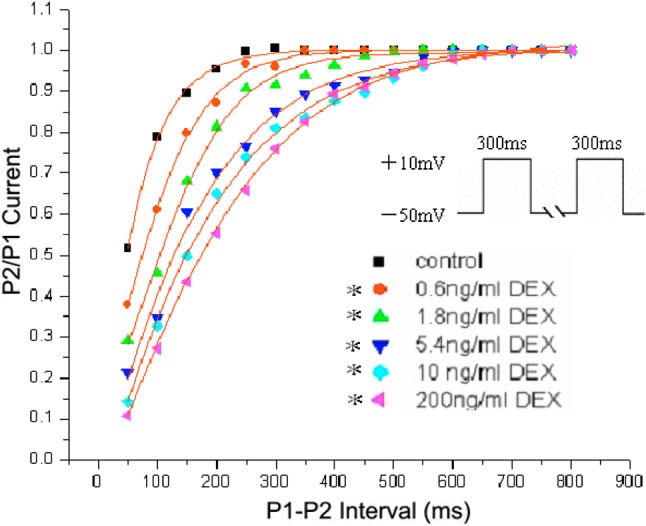
Effect of Dexmedetomedine (DEX) on the recovery of L-type calcium current (ICa-L) from inactivation. The pulse was composed of two pulses, one 300 ms prepulse from -50 mV to +10 mV, followed by a repolarizing pulse to -50 mV which progressively prolonged durations from 50 to 500 ms with 50 in 50 ms increment, and then a 200 ms test pulse to +10 mV. DEX (0.6, 1.8, 5.4, 10 and 200 ng/ml) shifted the curve to the right. (n = 6, *p < 0.05)
DISCUSSION
The most common manifestations of the negative effects of DEX are sinus bradycardia and hypotension.9-11 The trend of effective refractory period (ERP) prolongation (p = 0.045) of atrial and ventricular muscles also can be observed.12 There have been reported cases of asystole, aggravated atrioventricular block or refractory cardiogenic shock after administration of DEX.13,14 Additionally, DEX can depress the cardiac output (CO) in a dose-dependent manner in the plasma concentration of 0.5-10 ng/ml, and it can significantly reduce the cardiac stroke volume when the target level of 5.1 ng/ml was achieved. Animal experiments indicate that DEX can be effective against myocardial ischemia/reperfusion injury.9,15 Moreover, DEX may have a potential therapeutic role in the acute phase of perioperative atrial and junc-tional tachyarrhythmias for congenital cardiac surgery.16
Some researchers hold that DEX has a negative effect on the cardiovascular system due to the reduction in sympathetic tone. However, norepinephrine and adrenaline concentrations showed no significantly greater decreases from the value at the 1.9 ng/ml concentration of DEX.9 Additionally, the effects of DEX are not limited to its interactions with alpha 2-adrenergic receptors.17 DEX suppressed the amplitude of delayed rectifier K+ current [IK(DR)] in a concentration-dependent manner in NG108-15 cells. It depressed the peak amplitude of Na+ current (INa). In isolated cerebellar granule cells, DEX also effectively suppressed IK(DR). Therefore, its negative effect on the cardiac contractile function cannot be simply attributed to its depression on the sympathetic nervous system. It suggests that DEX may have direct myocardial effects.
ICa-L, plays an important role in maintaining the normal electric activity and mechanical force of cardiomyocytes. During contraction of the cardiac muscle, an action potential is conducted from the non-contractile cardiac myocytes to contractile cells through gap junction. When a myocyte is depolarized by an action potential which travels along T-tubules among Z-discs, calcium ions enter the cell during the plateau phase of the cardiac action potential through ICa-L located on the sarcolemma. This calcium triggers a subsequent release of calcium that is stored in the sarcoplasmic reticulum (SR) through calcium-release channels (“ryanodine receptors”). The cyoplasmic calcium binds to Troponin C, causing the contraction of the myocyte. The excitability of myocyte depends on free intracellular calcium concentration regulating the myocardial contractility. In the period of relaxation, intracellular calcium is taken up by the sarco/endoplasmic reticulum ATPase pump into the sarcoplasm, ejected from the cell by the sodium-calcium exchanger or the plasma membrane calcium ATPase or taken up by the mitochondrial calcium tubulin uniporter.18 Intracellular calcium concentration significantly drops causing the relaxation of myocyte. The rapid decline of calcium concentration is the determinant to relaxation. ICa-L, which is the bridge for communicating myocardial electrical activity and mechanical activity, the important ion channel in cardiomyocytes. It is the glycosylated polypeptide complex consisted of subunits α1, α2, β, γ and δ. Subunit α that contains sites of calcium activation and deactivation, voltage feeling and ion selection is the main functional unit of the ion channel. It is the binding site of calcium channel activators and blockers as well. The channel would be activated by the powerful depolarization (up to -40 mv~-30 mv), and its conductance is large (25 ps). The whole cell patch clamp technique was used to investigate the effects of DEX on ICa-L. The results suggested that DEX can attenuate ICa-L, slow the activity of calcium channels and extend the recovery time from inactivation of channels so as to reduce calcium inward flow. However, it may contribute to its negative effects on myocardia contractility and cardiac electric activity.
The result also shows that the inhibition on ICa-L of DEX can be weakened by Yohimbine. This also indicates that the inhibition on ICa-L of DEX may be mediated by alpha 2-adrenergic receptor (alpha 2AR). Alpha 2ARs belong to the G-protein-coupled receptor family,1 which are widely distributed in the central and peripheral nervous system, kidney and other organs and tissues. Alpha 2ARs are also detected in rat and adult ventricular myocytes. The effects of DEX including sedation, analgesia and the sympatholytic properties are mediated by Protein (Gi/Go) – adenylate cyclase (AC) – protein kinase A (PKA). However, this needs further research to determine whether the effect on myocardial ICa-L of DEX is relevant to this signal pathway.
CONCLUSIONS
In conclusion, DEX can attenuate ICa-L in a dose-dependent manner, which contributes to its negative effects on myocardia contractility and cardiac electric activity. Therefore, the patients with cardiac insufficiency, bradycardia or cardiac conduction system abnormalities need be used with caution in the clinical application.
Acknowledgments
We are grateful to Prof.Xi Wang (Department of Cardiology, Renmin Hospital of Wuhan University) for assistance in researching.
REFERENCES
- 1.Mantz J, Josserand J, Hamada S. Dexmedetomidine: new insights. Eur J Anaesthesiol. 2011;28:3–6. doi: 10.1097/EJA.0b013e32833e266d. [DOI] [PubMed] [Google Scholar]
- 2.Carollo DS, Nossaman BD, Ramadhyani U. Dexmedetomidine: a review of clinical applications. J Curr Opin Anaesthesiol. 2008;21:457–461. doi: 10.1097/ACO.0b013e328305e3ef. [DOI] [PubMed] [Google Scholar]
- 3.Wijeysundera DN, Bender JS, Beattie WS. Alpha-2 adrenergic agonists for the prevention of cardiac complications among patients undergoing surgery. J Cochrane Database Syst Rev. 2009;7:CD004126. doi: 10.1002/14651858.CD004126.pub2. [DOI] [PubMed] [Google Scholar]
- 4.Biccard BM, Goga S, de Beurs J. Dexmedetomidine and cardiac protection for non-cardiac surgery: a meta-analysis of randomised controlled trials. J Anaesthesia. 2008;63:4–14. doi: 10.1111/j.1365-2044.2007.05306.x. [DOI] [PubMed] [Google Scholar]
- 5.Benitah JP, Alvarez JL, Gomez AM. L-type Ca2+ current in ventricular cardiomyocytes. Journal of Molecular and Cellular Cardiology. 2010;48:26–36. doi: 10.1016/j.yjmcc.2009.07.026. [DOI] [PubMed] [Google Scholar]
- 6.Lacinová L. Voltage-dependent calcium channels. J Gen Physiol Biophys. 2005;24:1–78. [PubMed] [Google Scholar]
- 7.Wang X, Wang X, Tang YH, et al. Effects of minocycline on L-type calcium current in rat ventricular myocytes. Chinese Journal of Cardical Arrhythmias. 2011;14:457–459. [Google Scholar]
- 8.Chen YJ, Chen YC, Chen SA, et al. Cardiac cellular electrophysiology, voltage clamp, and patch clamp. Acta Cardiol Sin. 2009;25:59–63. [Google Scholar]
- 9.Ebert TJ, Hall JE, Barney JA, et al. The effects of increasing plasma concentrations of dexmedetomidine in humans. J Anesthesiology. 2000;93:382–394. doi: 10.1097/00000542-200008000-00016. [DOI] [PubMed] [Google Scholar]
- 10.Gertler R, Brown HC, Mitchell DH, et al. Dexmedetomidine: a novel sedative-analgesic agent. J Proc (Bayl Univ Med Cent) 2001;14:13–21. doi: 10.1080/08998280.2001.11927725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bloor BC, Ward DS, Belleville JP, et al. Effects of intravenous dexmedetomidine in humans II. Hemodynamic changes. J Anesthesiology. 1992;77:1134–1142. doi: 10.1097/00000542-199212000-00014. [DOI] [PubMed] [Google Scholar]
- 12.Hammer GB, Drover DR, Cao H, et al. The effects of dexmedetomidine on cardiac electrophysiology in children. J Anesth Analg. 2008;106:79–83. doi: 10.1213/01.ane.0000297421.92857.4e. [DOI] [PubMed] [Google Scholar]
- 13.Sichrovsky TC, Mittal S, Steinberg JS. Dexmedetomidine sedation leading to refractory cardiogenic shock. J Anesth Analg. 2008;106:1784–1786. doi: 10.1213/ane.0b013e318172fafc. [DOI] [PubMed] [Google Scholar]
- 14.Hutchens MP, Thorborg P. Dexmedetomidine sedation (and cardiac perforation, pericardial tamponade, cardiac arrest, and cardiopulmonary resuscitation) leading to refractory cardiogenic shock. J Anesth Analg. 2009;108:379–380. doi: 10.1213/ane.0b013e31818c0d06. [DOI] [PubMed] [Google Scholar]
- 15.Talke P, Li J, Jain U, et al. Effects of perioperative dexmedetomidine infusion in patients undergoing vascular surgery. The Study of Perioperative Ischemia Research Group. J Anesthesiology. 1995;82:620–633. doi: 10.1097/00000542-199503000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Chrysostomou C, Beerman L, Shiderly D, et al. Dexmedetomidine: a novel drug for the treatment of atrial and junctional tachyarrhythmias during the perioperative period for congenital cardiac surgery: a preliminary study. Pediatric anesthesiology. J Anesthesia and Analgesia. 2008;107:1514–1522. doi: 10.1213/ane.0b013e318186499c. [DOI] [PubMed] [Google Scholar]
- 17.Chen BS, Peng H, Wu SN. Dexmedetomidine, an alpha2-adrenergic agonist, inhibits neuronal delayed-rectifier potassium current and sodium current. Br J Anaesth. 2009;103:244–254. doi: 10.1093/bja/aep107. [DOI] [PubMed] [Google Scholar]
- 18.Chan YH, Wu CT, Yeh YH, et al. Reappraisal of Luo-Rudy dynamic cell model. Acta Cardiol Sin. 2010;26:69–80. [Google Scholar]