

Abstract
It is well-known that aldosterone plays an important role in reabsorption of sodium and fluid, and in potassium excretion in kidneys via epithelial mineralocorticoid receptor (MR) activation. Recent studies have shown that aldosterone causes cardiovascular remodeling not only in a blood pressure-dependent manner, but also in a blood pressure-independent manner by decreasing nitric oxide bioavailability and modulating oxidative stress, leading to vascular inflammation. In addition, MR blockade does provide beneficial effects associated with cardiovascular protection, resulting in a reduction of cardiovascular morbidity and mortality. A growing body of evidence suggests that MR blockade is a promising therapeutic target to help prevent cardiovascular events.
Keywords: Aldosterone, Mineralocorticoid receptor, Nitrix oxide, Renin-angiotensin-aldosterone system
INTRODUCTION
Aldosterone plays important roles in the reabsorption of sodium and fluid, and in potassium excretion in epithelial cells of the collecting ducts of the kidney via cytosolic mineralocorticoid receptor (MR) activation. Previously, it was believed that the main role of aldosterone was to expand extracellular volume after sodium absorption in renal distal tubules, resulting in elevated blood pressure, as a terminal effecter of the renin-angiotensin-aldosterone system (RAAS) cascade. However, recent studies have revealed that aldosterone also has additional physiological and/or pathophysiological effects, such as decreasing nitric oxide (NO) bioavailability and modulating oxidative stress, leading to vascular inflammation.1 Aldosterone therefore causes organ damage and remodeling not only in a blood pressure-dependent manner, but also in a blood pressure-independent manner. In this review, we focus on the effects of mineralocorticoid receptor blockade on cardiovascular systems and the mechanisms by which aldosterone evokes cardiovascular inflammation through imbalance of NO bioavailability and oxidative stress.2
PHYSIOLOGICAL MINERALOCORTICOIDS ARE ESSENTIAL FOR HUMANS
Mineralocorticoids including aldosterone are steroid hormones that are produced in the zona glomerulosa of the adrenal gland. The RAAS first appeared in bony fishes and has been gradually strengthened through the evolutionary process. It is apparent that aldosterone has an essential role in maintaining body fluid and sodium, and therefore represented a major leap in RAAS evolution in early organisms that transitioned out of the water and onto dry land. If mineralocorticoids had not been secreted appropriately, they would have suffered from salt and water wasting. Thus, aldosterone is an essential hormone for maintaining body fluid homeostasis; however, in this age of relative affluence, humans usually have more salt than is necessary. Therefore, alterations in lifestyle behavior that impact excessive salt intake have made aldosterone a strong risk factor for cardiovascular disease (CVD).
ALDOSTERONE SYNTHESIS IS STIMULATED AS A RESULT OF THE RAAS ACTIVATION
RAAS activation is initiated by release of angiotensinogen into the circulation by renin. Renin is secreted from the juxtaglomerular apparatus of the kidney to form angiotensin (Ang) I. Ang I is then converted to Ang II by angiotensin-converting enzyme (ACE), which is expressed on pulmonary endothelial cells. Subsequently, Ang II activates Ang II type 1 receptors (AT1R) on vascular smooth muscle cells to induce vasoconstriction. Ang II then stimulates the synthesis and release of aldosterone in the adrenal cortex. In this way, activation of the RAAS enhances the production of aldosterone. Building evidence has shown that aldosterone enhances Ang II expression in neonatal rat cardiomyocytes through up-regulation of ACE messenger ribonucleic acid (mRNA) expression.3 Aldosterone was also shown to up-regulate Ang II receptors.4 Therefore, even when the expression of ACE or Ang II receptor is suppressed by ACE inhibitors or Ang II receptor blockers, the RAAS could be enhanced by aldosterone per se, forming a vicious cycle of the RAAS5 (Figure 1). In addition, the local RAAS also plays a pivotal role in cardiovascular organ damage. Components of the RAAS have been detected in multiple organs, such as the heart and kidney, and in the vasculature. The local RASS is activated independently of the systemic RAAS, indicating that organ damage may occur although the systemic RAAS is not activated. Moreover, inflammatory cells, including monocytes and macrophages, express angiotensinogen, renin, ACE, and AT1R, and activation of the RAAS in inflammatory cells promotes monocyte migration into the vascular wall; this leads to differentiation of monocytes into macrophages, resulting in further activation of the local RAAS.6
Figure 1.
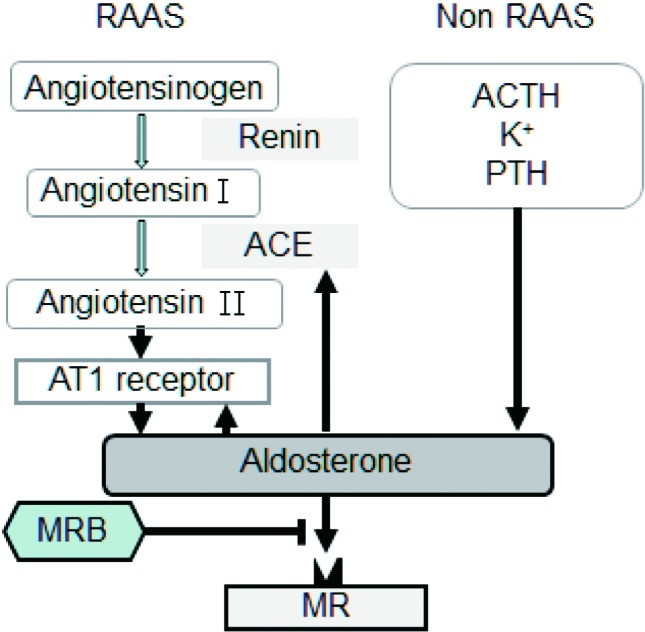
Aldosterone synthesis is activated by renin-angiotensin-aldosterone system (RAAS)/nonRAAS stimuli, which was blocked by mineralocorticoid receptor blocker (MRB). ACE, angiotensin-converting enzyme; ACTH, adrenocorticotropic hormone; MR, mineralocorticoid receptors; PTH, parathyroid hormone.
OTHER REGULATORS OF ALDOSTERONE SYNTHESIS
In addition to Ang II, recent studies have revealed several stimuli for aldosterone synthesis and secretion: adrenocorticotropic hormone (ACTH) and potassium ion can increase aldosterone secretion acutely by activating the G-protein coupled receptor/cyclic adenosine monophosphate/P450 pathway, as well as increasing aldosterone secretion chronically by increasing gene expression of CYP11B2.7 Furthermore, it has been shown that aldosterone is involved in biological aging, a pivotal risk factor for CVD, because plasma aldosterone concentration was found to be inversely associated with telomere length.8 In addition, age-dependent inactivation of 11β hydroxysteroid dehydrogenase type 2 (11β-HSD2), leading to cortisol-mediated MR activation, was seen in elderly hypertensive patients, and age-dependent inactivation of dehydroepiandrosterone sulfate (DHEAS), which inhibits Ang II-mediated release of aldosterone, was seen in the elderly.9 We previously showed that age-dependent reduction of DHEAS was an independent negative factor for intima-media thickness in carotid arteries of males.10 Inactivation of 11β-HSD and DHEAS might therefore be involved in age-dependent MR activation leading to CVD. Moreover, the results of recent studies have supported an interaction between aldosterone and parathyroid hormone (PTH): PTH increases the secretion of aldosterone from the adrenal glands directly and/or indirectly by activating the RAAS, suggesting that up-regulation of aldosterone synthesis in patients with primary parahyperthyroidism contributes to the development of hypertension and CVD.11 It has also been shown that 3-beta-hydroxyl-steroid dehydrogenase is involved in aldosterone production as well as aldosterone synthase, the main enzyme for aldosterone production.12
ALDOSTERONE IS A RISK FACTOR FOR CVD
It is known that aldosterone is a risk factor for CVD in both blood pressure-dependent and independent manners. Patients presenting with primary aldosteronism experienced more cardiovascular events than did essential hypertension patients independent of blood pressure.13 Aldosterone levels were shown to be correlated with the degree of vascular stiffness and the degree of left ventricular hypertrophy.14 The Framingham and SUVIMAX studies showed that elevated aldosterone level was positively associated with the incidence of new onset of hypertension.15,16 In addition, the CONSENSUS trial showed a relationship between plasma level of aldosterone and mortality in patients with heart failure (HF).17 Thus, aldosterone and activation of the MR play important roles in the occurrence of CVD and HF.
MR ACTIVATION BY ALDOSTERONE AND CORTISOL
Aldosterone binds to the MR, a nuclear receptor located in the cell cytosol. The activated receptor/hormone complex binds to hormone-responsive elements in the 5’ untranslated region of aldosterone-responsive genes that activate gene transcription leading to protein synthesis, which is called genomic action of aldosterone. Rapid action of aldosterone is known as a non-genomic action of aldosterone. The MR also binds cortisol, a glucocorticoid. Cortisol has a greater affinity than aldosterone to the MR; however, the enzyme 11βHSD2, detected in epithelial cells in the renal tubule, lung, placenta and vasculature, allows aldosterone to selectively activate the MR by converting cortisol to cortisone, an inactive metabolite. In the myocardium, 11βHSD2 is not abundant; however, under the condition of RAAS activation with increased oxidative stress, the MR could be activated by cortisol, leading to enhanced redox signaling.18
VASCULAR INFLAMMATION MEDIATED BY MR ACTIVATION
Aldosterone, as a terminal effector in the RAAS, plays a pivotal role in inflammation, leading to fibrosis of cardiovascular tissue. In fact, treatment with eplerenone or other aldosterone antagonists has been shown to reduce vascular inflammation and remodeling.1
Aldosterone exacerbates vascular injury through local inflammation in the heart, aorta, and kidney in an MR-dependent manner. Rocha et al. reported that treatment of uninephrectomized rats with aldosterone and salt caused the development of extensive coronary inflammatory lesions with perivascular macrophage infiltration. They assumed that excessive aldosterone and salt increased the expression levels of cyclooxygenase-2, osteopontin, and macrophage chemoattractant protein-1 (MCP-1), and that these effects were attenuated by MR blockade.19 MR antagonism also decreased aortic inflammation, fibrosis, and hypertrophy in hypertensive rats and decreased oxidative stress and expression levels of inflammatory cytokines and chemokines such as TNF-α and MCP-1, in apolipoprotein E-deficient mice fed a high-cholesterol diet.20,21 Interestingly, aldosterone triggers the exocytosis of endothelial cells, the first step in leucocyte trafficking, resulting in the adherence of leucocytes to endothelial cells in a P-selectin-dependent manner. Conversely, aldosterone-induced endothelial exocytosis was inhibited by MR antagonism and knockdown, whereas actinomycin D had no effect, suggesting that the MR activates vascular inflammation through non-genomic pathways.22
The MR antagonist potassium canrenoate was shown to block markers of vascular inflammation, including osteopontin, cyclooxygenase-2 and ED-1, in uninephrectomized rats treated with deoxycorticosterone and a high salt diet.23 In addition, coronary vascular inflammation and fibrosis caused by mineralocorticoid administration was reversed by eplerenone but not by deoxycorticosterone withdrawal.23 Thus, these findings indicate that MR activation plays a crucial role in the pathogenesis of vascular inflammation and fibrosis.
REDUCED NO BIOAVAILABILITY IN MR-MEDIATED VASCULAR INFLAMMATION
RAAS activation increases oxidative stress and decreases NO bioavailability, leading to tissue inflammation: aldosterone breaks the balance between NO bio-availability and oxidative stress leading to inflammation1 (Figure 2). NO bioavailability is decreased in atherosclerosis due to an increase in NO inactivation by reactive oxygen species (ROS) and reduced endothelial NO synthase (eNOS) expression. MR activation also reduces NO bioavailability in endothelial cells through an increase in ROS production. Endothelial-dependent vasorelaxation was improved by eplere-none treatment through suppressed ACE activity and decreased serum MCP-1 levels in hypertensive or atherosclerotic animal models. MR antagonists, together with an ACE inhibitor, normalized NO-mediated relaxation in rats with congestive HF by beneficially modulating the balance between NO bio-availability and super-oxide generation.24 In addition, in the presence of aldosterone, a small increase in plasma sodium concentration per se decreased NO release and increased the stiffness of endothelial cells, suggesting that salt intake plays a key role in the decrease in NO bioavailability.25 Nagata et al. showed that aldosterone inhibits NO synthase in human endothelial cells by inducing oxidation of its co-factor 5,6,7,8-tetrahydrobiopterin as well as by activating protein phosphatase 2A.26 Leopold et al. showed that aldosterone decreased the expression and activity of glucose-6-phosphate dehydrogenase (G6PD), which modulates vascular function by reducing oxidant stress to preserve bioavailable NO. Aldosterone antagonism or gene transfer of G6PD improved vascular reactivity by restoring G6PD activity.27 It is therefore likely that eNOS uncoupling and/or G6PD activity mediate the aldosterone-induced decease in NO bioavailability leading to vascular inflammation.
Figure 2.
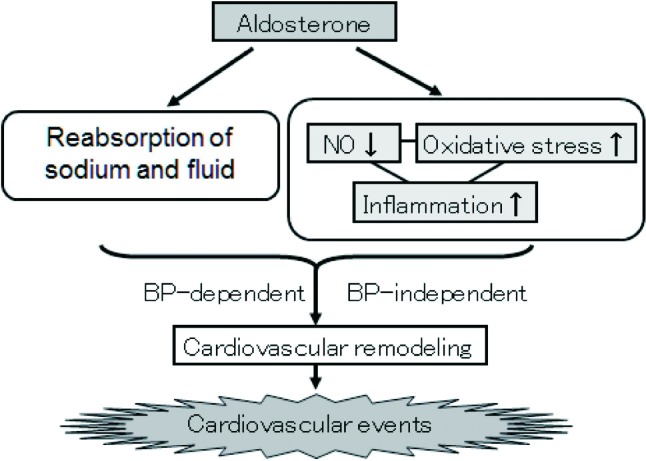
Aldosterone decreases NO bioavailability and increases oxidative stress. Imbalance between NO bioavailability and oxidative stress evokes vascular inflammation, leading to cardiovascular events due to cardiovascular remodeling in blood pressure dependent/independent manners. BP, blood pressure; NO, nitric oxide.
INCREASED OXIDATIVE STRESS IN MR-MEDIATED VASCULAR INFLAMMATION
Systemic administration of aldosterone increases oxidative stress in the heart, vasculature, and kidneys.28 Aldosterone up-regulates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in macrophages and vascular smooth muscle cells through genomic and/or non-genomic pathways, and the up-regulation of NADPH oxidase was attenuated by eplerenone.29,30 MR activation also contributes to Ang II-mediated activation of NADPH oxidase in the heart and aorta:31 aldosterone stimulates aortic expression of p22 phox and NOX2 (gp91 phox ) through an MR-dependent mechanism, and up-regulates p47 phox mRNA through both AT1 receptor and MR-dependent mechanisms. It has been reported that superoxide production is also induced by aldosterone through MR-mediated activation of NADPH-oxidase and Rac1 in endothelial cells, contributing to the development of aldosterone-induced vascular injury.32,33
Increased oxidative stress stimulates activation of pro-inflammatory transcriptional factors such as activator protein-1 and nuclear transcription factor-κ, resulting in up-regulation of adhesion molecules, chemokines, and inflammatory cytokines that cause vascular inflammation due to the activation of redox signaling. In addition, administration of antioxidant drugs including radical scavengers and NADPH oxidase inhibitors decreased inflammation and injury in aldosterone-treated rodents.34,35 Oxidative stress, therefore, is one of the key components of vascular inflammation induced by MR activation as a counterpart to NO.
ADVANTAGEOUS EFFECTS OF MR BLOCKADE IN CLINICAL PRACTICE
Blockade of the MR has advantageous effects on hypertension and the cardiovascular system. Administration of spironolactone to resistant hypertensive patients was effective in decreasing blood pressure along with attenuating cardiovascular damage such as carotid intima-media thickening.36,37 In addition, eplelenone was effective as well as enalapril for reducing left ventricular mass.38
Two large clinical trials, RALES and EPHESUS, revealed that MR blockade improves morbidity and mortality in patients with moderate to severe HF with left ventricular dysfunction after myocardial infarction.39,40 The EMPHASIS-HF trial showed that MR blockade also has advantageous effects in patients with mild symptoms of NYHA class II.41 In addition, it has been reported that MR blockade with spirono-lactone improves the echocardiographic indices of diastolic function in patients with diastolic HF.42 The TOPCAT trial is currently being carried out to clarify whether MR blockade of spironolactone reduces cardiovascular mortality and hospitalization in patients with HF of preserved systolic function (http://clinical-trials.gov/). In addition, REMINDER and ALBATROSS are also ongoing trials to clarify whether early MR blockade improves clinical outcomes in patients with acute myocardial infarction (http://clinicaltrials.gov/). Taken together, emerging evidence is unveiling the therapeutic value of aldosterone blockade in patients with CVD or HF.
NEW TARGETS FOR MR BLOCKADE TO REVERSE VASCULAR INFLAMMATION
We showed that perivascular adipose tissue plays a crucial role in vascular inflammation and lesion formation.43 Recent evidence suggests that MR activation in adipose tissue is involved in the pathogenesis of metabolic syndrome and insulin resistance. Serum aldosterone levels were elevated in spontaneously hypertensive rats/NDmcr-cp rats, a model of metabolic syndrome, and high MR expression level was detected in adipocytes from obese mice.44 Conversely, MR blockade decreased the production of ROS and inflammatory cytokine gene expression in adipose tissue of these mice and improved their insulin resistance.45 Thus, MR blockade in adipose tissue, particularly perivascular adipose tissue, may reduce vascular inflammation. We also showed that bone marrow-derived vascular progenitor cells contribute to vascular repair and remodeling,46 and that a local RAAS exists in bone marrow and may play a crucial role in vascular inflammation.47 In addition, it has been shown that aldosterone inhibits the formation of bone marrow-derived endothelial progenitor cells, suggesting that MR blockade and/or co-treatment with antioxidants may enhance vascular regeneration.48 Thus, blockade of the MR in bone marrow may also have the potential to promote vascular repair and reverse vascular inflammation.
NEW DRUGS FOR MR BLOCKADE
New, non-steroidal mineralocorticoid receptor antagonists with greater selectivity than spironolactone and stronger mineralocorticoid receptor binding affinity than eplerenone are currently being tested in clinical trials (http://clinicaltrials.gov/). Conventional MR blockers have several disadvantages: spironolactone has side effects such as gynecomastia or irregular menstruation, and eplerenone has a half life of only 4 hours. These new drugs therefore are expected to have a strong effect on cardiovascular protection, with fewer side effects.
CONCLUSIONS
Recent experimental evidence has shown the benefits of MR blockade for cardiovascular protection by the mechanism of MR blockade reducing vascular inflammation through improvement of the imbalance of NO bioavailability and oxidative stress. Large clinical trials have also indicated the benefits of MR antagonism in patients with HF. Mounting evidence suggests that MR blockade is a promising therapeutic target for preventing CVD or HF.
REFERENCES
- 1.Gilbert KC, Brown NJ. Aldosterone and inflammation. Curr Opin Endocrinol Diabetes Obes. 2010;17:199–204. doi: 10.1097/med.0b013e3283391989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yagi S, Sata M. Pre-clinical data on the role of mineralocorticoid receptor antagonists in reversing vascular inflammation. Eur Heart J Suppl. 2011;13:B15–B20. [Google Scholar]
- 3. Harada E, Yoshimura M, Yasue H, et al. Aldosterone induces angiotensin-converting-enzyme gene expression in cultured neonatal rat cardiocytes. Circulation. 2001;104:137–139. doi: 10.1161/01.cir.104.2.137. [DOI] [PubMed] [Google Scholar]
- 4.Ullian ME, Schelling JR, Linas SL. Aldosterone enhances angio-tensin ii receptor binding and inositol phosphate responses. Hypertension. 1992;20:67–73. doi: 10.1161/01.hyp.20.1.67. [DOI] [PubMed] [Google Scholar]
- 5. Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006;86:747–803. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
- 6.Yabu M, Senda T, Nonaka Y, et al. Localization of the gene transcripts of 11 beta-hydroxylase and aldosterone synthase in the rat adrenal cortex by in situ hybridization. Histochemistry. 1991;96:391–394. doi: 10.1007/BF00315995. [DOI] [PubMed] [Google Scholar]
- 7.Hattangady NG, Olala LO, Bollag WB, Rainey WE. Acute and chronic regulation of aldosterone production. Molecular and Cellular Endocrinology. 2012;350:151–162. doi: 10.1016/j.mce.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benetos A, Gardner JP, Kimura M, et al. Aldosterone and telomere length in white blood cells. J Gerontol A Biol Sci Med Sci. 2005;60:1593–1596. doi: 10.1093/gerona/60.12.1593. [DOI] [PubMed] [Google Scholar]
- 9.Chang LL, Wun WSA, Wang PS. Effects of dehydro-epiandro-sterone on aldosterone release in rat zona glomerulosa cells. J Biomed Sci. 2008;15:463–470. doi: 10.1007/s11373-008-9241-3. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida S, Aihara K, Azuma H, et al. Dehydroepiandrosterone sulfate is inversely associated with sex-dependent diverse carotid atherosclerosis regardless of endothelial function. Atherosclerosis. 2010;212:310–315. doi: 10.1016/j.atherosclerosis.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 11.Tomaschitz A, Ritz E, Pieske B, et al. Aldosterone and parathyroid hormone: a precarious couple for cardiovascular disease. Cardiovasc Res. 2012;94:10–19. doi: 10.1093/cvr/cvs092. [DOI] [PubMed] [Google Scholar]
- 12.Doi M, Takahashi Y, Komatsu R, et al. Salt-sensitive hypertension in circadian clock-deficient cry-null mice involves dysregulated adrenal hsd3b6. Nat Med. 2010;16:67–74. doi: 10.1038/nm.2061. [DOI] [PubMed] [Google Scholar]
- 13.Milliez P, Girerd X, Plouin PF, et al. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol. 2005;45:1243–1248. doi: 10.1016/j.jacc.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 14.Safar ME, Levy BI, Struijker-Boudier H. Current perspectives on arterial stiffness and pulse pressure in hypertension and cardiovascular diseases. Circulation. 2003;107:2864–2869. doi: 10.1161/01.CIR.0000069826.36125.B4. [DOI] [PubMed] [Google Scholar]
- 15.Vasan RS, Evans JC, Larson MG, et al. Serum aldosterone and the incidence of hypertension in nonhypertensive persons. N Engl J Med. 2004;351:33–41. doi: 10.1056/NEJMoa033263. [DOI] [PubMed] [Google Scholar]
- 16. Meneton P, Galan P, Bertrais S, et al. High plasma aldosterone and low renin predict blood pressure increase and hypertension in middle-aged caucasian populations. J Hum Hypertens. 2008;22:550–558. doi: 10.1038/jhh.2008.27. [DOI] [PubMed] [Google Scholar]
- 17.Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. Consensus trial study group. Circulation. 1990;82:1730–1736. doi: 10.1161/01.cir.82.5.1730. [DOI] [PubMed] [Google Scholar]
- 18.Pitt B. The role of mineralocorticoid receptor antagonists (mras) in very old patients with heart failure. Heart Fail Rev. 17:573–579. doi: 10.1007/s10741-011-9286-7. [DOI] [PubMed] [Google Scholar]
- 19.Rocha R, Rudolph AE, Frierdich GE, et al. Aldosterone induces a vascular inflammatory phenotype in the rat heart. Am J Physiol Heart Circ Physiol. 2002;283:H1802–H1810. doi: 10.1152/ajpheart.01096.2001. [DOI] [PubMed] [Google Scholar]
- 20.Lacolley P, Labat C, Pujol A, et al. Increased carotid wall elastic modulus and fibronectin in aldosterone-salt-treated rats: effects of eplerenone. Circulation. 2002;106:2848–2853. doi: 10.1161/01.cir.0000039328.33137.6c. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki J, Iwai M, Mogi M, et al. Eplerenone with valsartan effectively reduces atherosclerotic lesion by attenuation of oxidative stress and inflammation. Arterioscler Thromb Vasc Biol. 2006;26:917–921. doi: 10.1161/01.ATV.0000204635.75748.0f. [DOI] [PubMed] [Google Scholar]
- 22.Jeong Y, Chaupin DF, Matsushita K, et al. Aldosterone activates endothelial exocytosis. Proc Natl Acad Sci U S A. 2009;106:3782–3787. doi: 10.1073/pnas.0804037106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young M. Eplerenone, but not steroid withdrawal, reverses cardiac fibrosis in doc/salt rats. Endocrinology. 2004;145:3153–3157. doi: 10.1210/en.2004-0005. [DOI] [PubMed] [Google Scholar]
- 24.Bauersachs J, Heck M, Fraccarollo D, et al. Addition of spironolactone to angiotensin-converting enzyme inhibition in heart failure improves endothelial vasomotor dysfunction: role of vascular superoxide anion formation and endothelial nitric oxide synthase expression. J Am Coll Cardiol. 2002;39:351–358. doi: 10.1016/s0735-1097(01)01729-6. [DOI] [PubMed] [Google Scholar]
- 25.Oberleithner H, Riethmuller C, Schillers H, et al. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc Natl Acad Sci U S A. 2007;104:16281–16286. doi: 10.1073/pnas.0707791104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagata D, Takahashi M, Sawai K, et al. Molecular mechanism of the inhibitory effect of aldosterone on endothelial no synthase activity. Hypertension. 2006;48:165–171. doi: 10.1161/01.HYP.0000226054.53527.bb. [DOI] [PubMed] [Google Scholar]
- 27.Leopold JA, Dam A, Maron BA, et al. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13:189–197. doi: 10.1038/nm1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun Y, Zhang J, Lu L, et al. Aldosterone-induced inflammation in the rat heart: role of oxidative stress. Am J Pathol. 2002;161:1773–1781. doi: 10.1016/S0002-9440(10)64454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Keidar S, Kaplan M, Pavlotzky E, et al. Aldosterone administration to mice stimulates macrophage nadph oxidase and increases atherosclerosis development: a possible role for angiotensin-converting enzyme and the receptors for angiotensin ii and aldosterone. Circulation. 2004;109:2213–2220. doi: 10.1161/01.CIR.0000127949.05756.9D. [DOI] [PubMed] [Google Scholar]
- 30.Callera GE, Touyz RM, Tostes RC, et al. Aldosterone activates vascular p38map kinase and nadph oxidase via c-src. Hypertension. 2005;45:773–779. doi: 10.1161/01.HYP.0000154365.30593.d3. [DOI] [PubMed] [Google Scholar]
- 31.Johar S, Cave AC, Narayanapanicker A, et al. Aldosterone mediates angiotensin ii-induced interstitial cardiac fibrosis via a nox2-containing nadph oxidase. FASEB J. 2006;20:1546–1548. doi: 10.1096/fj.05-4642fje. [DOI] [PubMed] [Google Scholar]
- 32.Iwashima F, Yoshimoto T, Minami I, et al. Aldosterone induces superoxide generation via rac1 activation in endothelial cells. Endocrinology. 2008;149:1009–1014. doi: 10.1210/en.2007-0864. [DOI] [PubMed] [Google Scholar]
- 33. Shibata S, Nagase M, Yoshida S, et al. Modification of mineralocorticoid receptor function by rac1 gtpase: implication in proteinuric kidney disease. Nat Med. 2008;14:1370–1376. doi: 10.1038/nm.1879. [DOI] [PubMed] [Google Scholar]
- 34.Hirono Y, Yoshimoto T, Suzuki N, et al. Angiotensin ii receptor type 1-mediated vascular oxidative stress and proinflammatory gene expression in aldosterone-induced hypertension: the possible role of local renin-angiotensin system. Endocrinology. 2007;148:1688–1696. doi: 10.1210/en.2006-1157. [DOI] [PubMed] [Google Scholar]
- 35.Iglarz M, Touyz RM, Viel EC, et al. Involvement of oxidative stress in the profibrotic action of aldosterone. Interaction wtih the renin-angiotension system. Am J Hypertens. 2004;17:597–603. [PubMed] [Google Scholar]
- 36.de Souza F, Muxfeldt E, Fiszman R, Salles G. Efficacy of spirono-lactone therapy in patients with true resistant hypertension. Hypertension. 2010;55:147–152. doi: 10.1161/HYPERTENSIONAHA.109.140988. [DOI] [PubMed] [Google Scholar]
- 37. Vukusich A, Kunstmann S, Varela C, et al. A randomized, double-blind, placebo-controlled trial of spironolactone on carotid intima-media thickness in nondiabetic hemodialysis patients. Clin J Am Soc Nephrol. 2010;5:1380–1387. doi: 10.2215/CJN.09421209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pitt B, Reichek N, Willenbrock R, et al. Effects of eplerenone, enalapril, and eplerenone/enalapril in patients with essential hypertension and left ventricular hypertrophy - the 4e-left ventricular hypertrophy study. Circulation. 2003;108:1831–1838. doi: 10.1161/01.CIR.0000091405.00772.6E. [DOI] [PubMed] [Google Scholar]
- 39.Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized aldactone evaluation study investigators. N Engl J Med. 1999;341:709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 40.Pitt B, Remme W, Zannad F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–1321. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- 41.Zannad F, McMurray JJV, Krum H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med . 2011;364:11–21. doi: 10.1056/NEJMoa1009492. [DOI] [PubMed] [Google Scholar]
- 42.Mottram PM, Haluska B, Leano R, et al. Effect of aldosterone antagonism on myocardial dysfunction in hypertensive patients with diastolic heart failure. Circulation. 2004;110:558–565. doi: 10.1161/01.CIR.0000138680.89536.A9. [DOI] [PubMed] [Google Scholar]
- 43.Takaoka M, Suzuki H, Shioda S, et al. Endovascular injury induces rapid phenotypic changes in perivascular adipose tissue. Arterioscler Thromb Vasc Biol. 2009 doi: 10.1161/ATVBAHA.110.207175. [DOI] [PubMed] [Google Scholar]
- 44.Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol. 2006;17:3438–3446. doi: 10.1681/ASN.2006080944. [DOI] [PubMed] [Google Scholar]
- 45. Hirata A, Maeda N, Hiuge A, et al. Blockade of mineralocorticoid receptor reverses adipocyte dysfunction and insulin resistance in obese mice. Cardiovasc Res. 2009;84:164–172. doi: 10.1093/cvr/cvp191. [DOI] [PubMed] [Google Scholar]
- 46.Sata M, Saiura A, Kunisato A, et al. Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat Med. 2002;8:403–409. doi: 10.1038/nm0402-403. [DOI] [PubMed] [Google Scholar]
- 47.Fukuda D, Sata M. Role of bone marrow renin-angiotensin system in the pathogenesis of atherosclerosis. Pharmacol Ther. 2008;118:268–276. doi: 10.1016/j.pharmthera.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 48.Marumo T, Uchimura H, Hayashi M, et al. Aldosterone impairs bone marrow-derived progenitor cell formation. Hypertension. 2006;48:490–496. doi: 10.1161/01.HYP.0000235681.25685.cf. [DOI] [PubMed] [Google Scholar]