

Abstract
Background
Cardiac cellular injury as a consequence of ischemia and reperfusion involves nuclear factor-κB (NF-κ B), amongst other factors, and NF-κ B inhibitors could substantially reduce myocardial infarct size. Parthenolide, a sesquiterpene lactone compound which could inhibit NF-κ B, has been shown to ameliorate myocardial reperfusion injury but may also produce toxic effects in cardiomyocytes at high concentrations. The aim of this study was to examine the cytotoxic effects of this drug on H9c2 cardiomyoblasts, which are precursor cells of cardiomyocytes.
Methods
Cell viability and apoptosis were examined by MTT and TUNEL assay, respectively, and protein expression was analyzed by western blot. Reactive oxygen species (ROS) production was measured using DCFH-DA as dye. Cytosolic Ca2+ concentration and mitochondrial membrane potential were measured microfluorimetrically using, respectively, fura 2 and rhodamine 123 as dyes.
Results
Parthenolide caused apoptosis at 30 μ M, as judged by TUNEL assay and Bax and cytochrome c translocation. It also caused collapse of mitochondrial membrane potential and endoplasmic reticulum stress. Parthenolide triggered ROS formation, and vitamin C (antioxidant) partially alleviated parthenolide-induced cell death.
Conclusions
The results suggested that parthenolide at high concentrations caused cytotoxicity in cardiomyoblasts in part by inducing oxidative stress, and demonstrated the imperative for cautious and appropriate use of this agent in cardioprotection.
Keywords: Cardiomyoblast, Endoplasmic reticulum stress, Oxidative stress, Parthenolide, Reperfusion injury
INTRODUCTION
Ischemia and reperfusion to the heart pose the threats of energetic stress and oxidative stress, respectively; these threats lead to both apoptotic and necrotic cell death.1,2 What follows the post-reperfusion healing process may be long-term damage, where inflammatory cells infiltrate and produce cytokines and toxic reactive oxygen species (ROS). One of the key signals in reperfusion injury is nuclear factor-κB (NF-κB), a family of transcription factors which regulate the expression of genes involved in inflammation and apoptosis.3,4 Antagonizing the activities of NF-κB could substantially reduce myocardial infarct size by up to 60%.5
A number of compounds isolated from plants, such as lignans, sesquiterpenes, diterpenes and triterpenes, have been known to be NF-κB inhibitors.6 One of the sesquiterpenes, parthenolide, has attracted particular attention in the research community. This sesquiterpene lactone compound is isolated from the leaves and flowerheads of the plant feverfew (Tanacetum parthenium).7 For instance, parthenolide produces ameliorative effects on myocardial reperfusion injury in rats, evidenced by a decrease in infarct size, reduction in inflammatory cell infiltration and consequent oxidative damage.8 Furthermore, parthenolide has been shown to enhance the protective effect of sevoflurane anesthetic preconditioning on ischemia/reperfusion injury in rats.9
However, intermediate to high concentrations of parthenolide produce toxic effects in cardiomyocytes due to the release of ROS and the collapse of mitochondrial membrane potential.10 Therefore, caution needs to be exercised in using this compound in cardioprotection. The effects of parthenolide on cardiomyoblasts, the precursor cells of cardiomyocytes, remain unexplored. In the present work we showed that parthenolide triggered ROS generation, endoplasmic reticulum (ER) stress, mitochondrial membrane potential decrease and eventually caused apoptotic death in rat H9c2 cardiomyoblasts.
MATERIALS AND METHODS
Materials
Dulbecco’s Modified Eagle’s Medium (DMEM), fetal calf serum, and tissue culture reagents were purchased from the Invitrogen Corporation (Carlsbad, CA, USA). Parthenolide and cyclopiazonic acid were obtained from Sigma-Aldrich (St Louis, MO, USA). Fura-2 AM and rhodamine-123 were purchased from Calbiochem-Millipore (Billerica, MA, USA).
Cell culture
H9c2 cells were cultured at 37 °C in 5% CO2 in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Invitrogen).
Assay of cell viability and apoptosis
Cell viability was examined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenltetrazolium bromide (MTT) method. Cells were cultured in a 96-well plate at a density of 1.5 × 104/well, and were then treated with or without parthenolide and/or other drugs for 15 h. MTT (final concentration at 0.5 mg/ml) was subsequently added to each well and then further incubated for 4 h. The culture medium was then removed and 100 μ l of dimethyl sulfoxide was added to each well for 15 min (with shaking) to dissolve the cells. The absorbance at 595 nm was measured using an ELISA reader and was used as an indicator of cell viability.
Apoptosis was assayed by the terminal deoxynucleotidyltransferase dUTP nick end labeling (TUNEL) method using a kit from Roche Applied Science (Branford, CT, USA) according to the manufacturer’s instructions.
Microfluorimetric measurement of cytosolic Ca2+
Microfluorimetric measurement of cytosolic Ca2+ concentration was performed using fura-2 as the Ca2+-sensitive fluorescent dye as described previously.11 Briefly, cells were incubated with 5 μ M fura-2 AM for 1 hr at 37 °C and then washed in extracellular bath solution which contained (mM): 140 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES (pH 7.4 adjusted with NaOH). When intracellular Ca2+ release was assayed, Ca2+-free solution was used. This Ca2+-free solution was the same as the extracellular bath solution mentioned above except that Ca2+ was omitted and 20 μ M EGTA was supplemented. Cells were alternately excited with 340 nm and 380 nm using an optical filter changer (Lambda 10-2, Sutter Instruments, Novato, CA, USA). Emission was collected at 500 nm and images were captured using a CCD camera (CoolSnap HQ2, Photometrics, Tucson, AZ, USA) linked to an inverted Nikon TE 2000-U microscope. Images were analysed with an MAG Biosystems Software (Sante Fe, MN, USA). All imaging experiments were performed at room temperature (25 °C).
Western blot
Western blotting was performed as described previously.12 In case of separation of cytosolic and mitochondrial fractions, a kit from BioVision (Milpitas, CA, USA) was used. Briefly, cells were washed in cold phosphate buffered saline (PBS), and then lysed for 30 min on ice using a radioimmunoprecipitation assay (RIPA) buffer. Protein samples containing 30 μg protein were separated on 10% sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). The membranes were incubated for 1 h with 5% nonfat milk in PBS buffer to block nonspecific binding. The membranes were incubated with various primary antibodies such as anti-actin (1:10000), anti-COX4 (1: 2000), anti-Bax (1:1000), anti-cyt c (1:1000) and anti-CHOP (1:1000) (Cell Signaling Technology, Danvers, MA, USA). Subsequently, the membranes were incubated with goat anti-rabbit or goat anti-mouse peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 1 h. The blots were visualized by enhanced chemiluminescence (Millipore, Billerica, MA, USA) using Kodak X-OMAT LS film (Eastman Kodak, Rochester, NY, USA).
ROS assay
H9c2 cells were incubated in serum-free DMEM containing 20 μM 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA, Sigma) for 30 min at 37 °C in the dark. Cells were subsequently treated with or without 30 μM parthenolide for 1 h and then gently scraped. Then, the cells were analyzed using a FACS Canto flow cytometer system (BD Biosciences, San Jose, CA, USA).
Microfluorimetric measurement of mitochondrial membrane potential
Cells were loaded with 5 μM rhodamine-123 for 40 min and then treated in the absence or presence of parthenolide (2 h). Excitation and emission wavelengths were set at 500 and 542 nm, respectively. Authentic mitochondrial signals were confirmed by addition of 2 μM carbonyl cyanide 4-trifluoromethoxyphenylhydrazone (FCCP; Sigma-Aldrich, MO, USA) to observe an increase in fluorescence intensity (depolarization).
Statistical analysis
Data are presented as means ± standard error of the mean. The unpaired or paired Student’s t test was used where appropriate to compare two groups. ANOVA was used to compare multiple groups, followed by the Tukey’s HSD post-hoc test. A value of p < 0.05 was considered to represent a significant difference.
RESULTS
Cytotoxic effects of parthenolide
Using the MTT assay, we found that treatment of H9c2 myoblasts with parthenolide for 15 h resulted in a concentration-dependent cell death (Figure 1). Cell death was revealed to be apoptotic by the TUNEL assay after a 15-h treatment with parthenolide (Figure 2). Note that after parthenolide and hydrogen peroxide treatment, there was nuclear shrinkage or condensation typical of apoptosis (DAPI staining). Furthermore, we confirmed that parthenolide treatment (4 h) caused translocation of Bax from cytosol to mitochondria and translocation of cytochrome c from mitochondria to cytosol (Figure 3A; quantitative results in Figure 3B). Therefore, the data suggested that parthenolide caused apoptotic death in H9c2 myoblasts.
Figure 1.
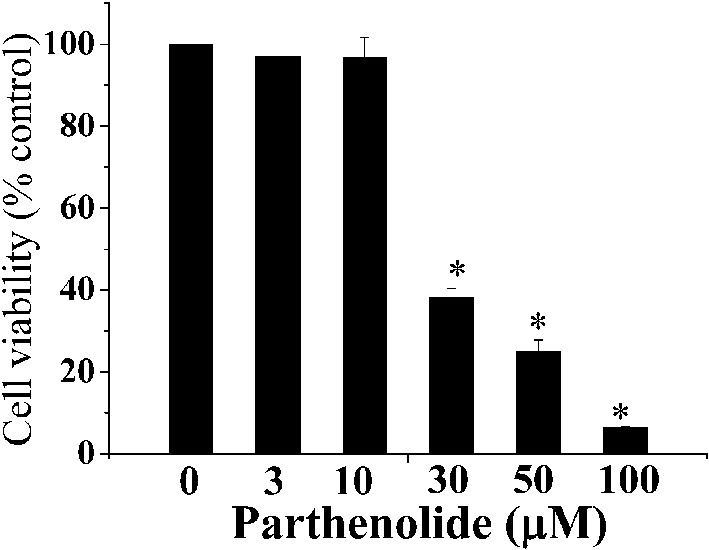
Parthenolide caused H9c2 cell lethality. H9c2 cells were treated with different concentrations of parthenolide for 15 h and then assayed for cell viability using the MTT assay. Results are mean ± standard error of the mean of 3 experiments. * indicates significant difference (p < 0.05) from the control.
Figure 2.
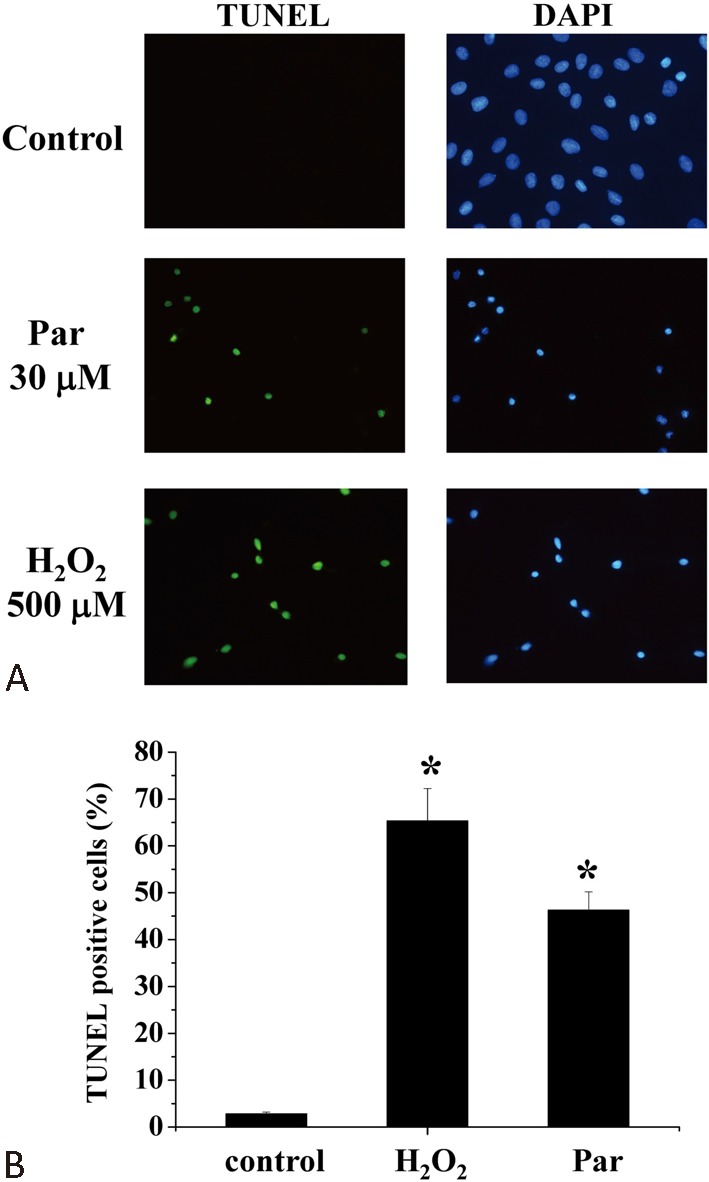
Parthenolide caused apoptosis in H9c2 myoblasts. (A) Cells were treated with either parthenolide or hydrogen peroxide for 15 h and then assayed for apoptosis using the TUNEL assay. Apoptotic cells were stained green while nuclei were stained blue using DAPI as dye. Magnification: 200X. (B) Quantification of percentage of TUNEL-positive cells. Results are mean ± standard error of the mean of 3 experiments. * indicates significant difference (p < 0.05) from the control. DAPI, 4’,6-diamidino-2-phenylindole; Par, parthenolide; TUNEL, terminal deoxynucleotidyltransferase dUTP nick end labeling.
Figure 3.
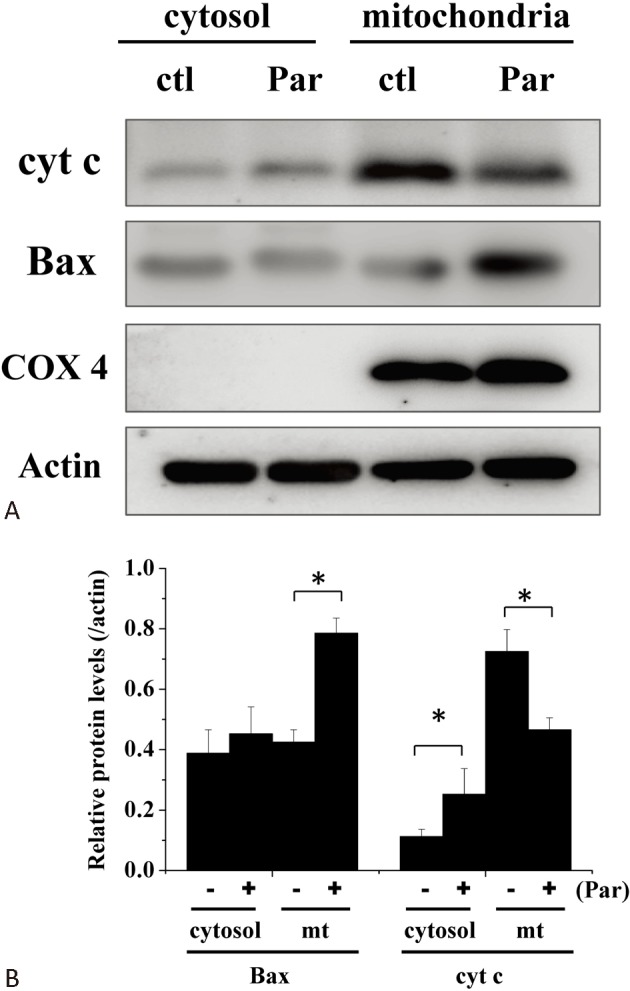
Parthenolide induced translocation of Bax and cytochrome c. (A) Protein levels of Bax and cytochrome c in the cytosol and mitochondria in the absence and presence of parthenolide (30 mM) treatment (4 h) were analysed by western blot. COX4 was used as a mitochondrial marker. (B) Quantification of results from (A). Results are mean ± standard error of the mean of 6 experiments. * indicates significant difference (p < 0.05). Ctl, control; mt, mitochondria; Par, parthenolide.
Parthenolide caused ER stress and oxidative stress
Whether parthenolide caused ER stress in H9c2 cells was then examined. Cyclopiazonic acid (CPA), which could cause depletion of intracellular Ca2+ store and thus ER stress, was used as a positive control. Both CPA and parthenolide treatment (4 h) caused the appearance of an ER stress marker, CCAAT/-enhancer-binding protein homologous protein (CHOP) (Figure 4). It is remarkable that while CPA increased the level of activating transcription factor 6 (ATF6), parthenolide increased the level of phospho-JNK, suggesting that these two agents triggered different pathways leading to CHOP expression. Both CPA and parthenolide did not increase the levels of eIF2-α and phospho-eIF2-α.
Figure 4.
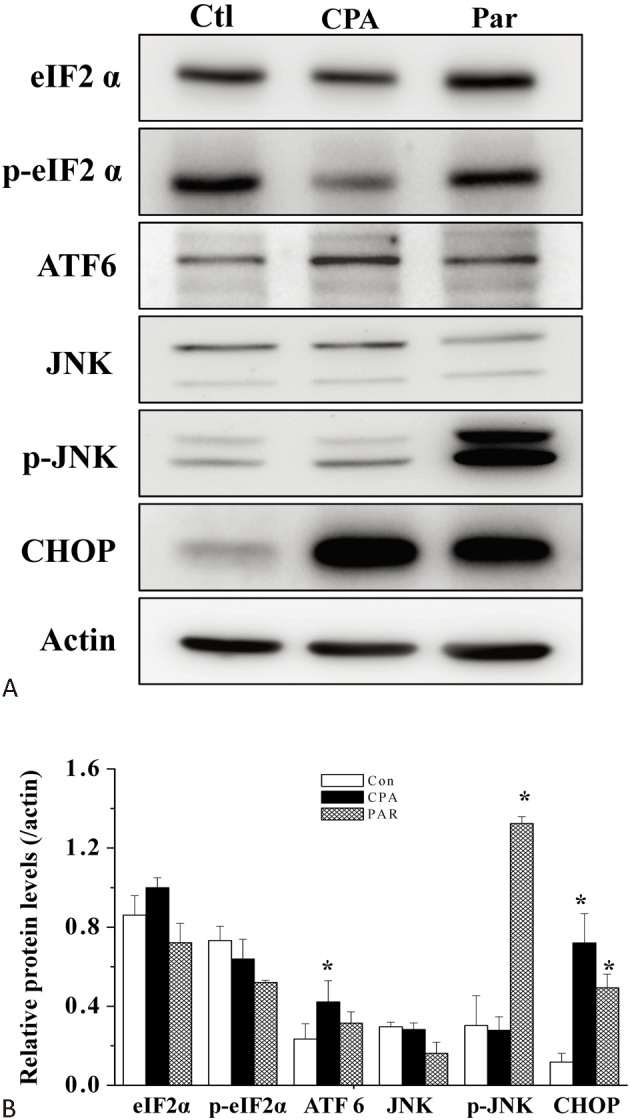
Parthenolide induced ER stress. (A) Protein levels of various ER stress markers in the absence or presence of CPA (50 μM) or parthenolide (30 μM) treatments (4 h) were analysed by western blot. (B) Quantification of results from (A). Results are mean ± standard error of the mean of 6 experiments. * indicates significant difference (p < 0.05) from the control. Con, control; CPA, cyclopiazonic acid; Ctl, control;Par, parthenolide.
We also investigated whether parthenolide triggered ROS formation in H9c2 cells. Data revealed that treatment with parthenolide for 1 h resulted in significant generation of ROS, as assayed by flow cytometry (Figure 5).
Figure 5.
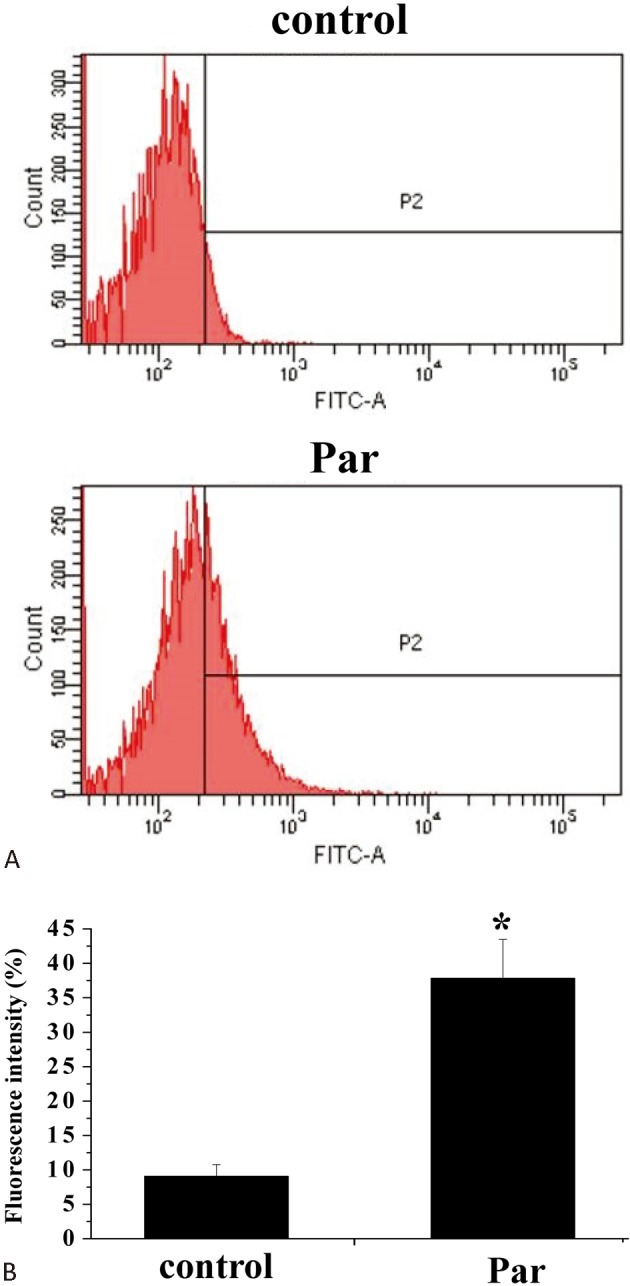
Parthenolide elicited ROS formation. (A) H9c2 cells were treated with DCFH-DA and then incubated in the absence or presence of parthenolide (30 μM) for 1 h and ROS generation was assayed using flow cytometry. (B) Quantification of results in (A). Results are mean ± standard error of the mean of 4 experiments. * indicates significant difference (p < 0.05) from the control. DCFH-DA, 2,7-dichlorodihydrofluorescein diacetate; FITC, Fluorescein isothiocyanate; Par, parthenolide.
Parthenolide did not trigger a Ca2+ signal or reduce Ca2+ pool size
Perturbations in Ca2+ homeostasis, such as cytosolic Ca2+ overload and depletion of intracellular Ca2+ stores, could be a mediator of apoptotic cell death. We therefore examined whether such perturbations were involved in parthenolide cytotoxic actions. Addition of parthenolide did not cause an acute elevation of Ca2+ concentration (Figure 6A). When cells were treated with parthenolide for 4 h and then examined for CPA-induced Ca2+ release (as a measure of Ca2+ pool size), it was found that parthenolide did not decrease Ca2+ pool size but instead slightly enhanced it when compared to the Ca2+ pool size of the control group (p < 0.05; Figure 6B, C).
Figure 6.
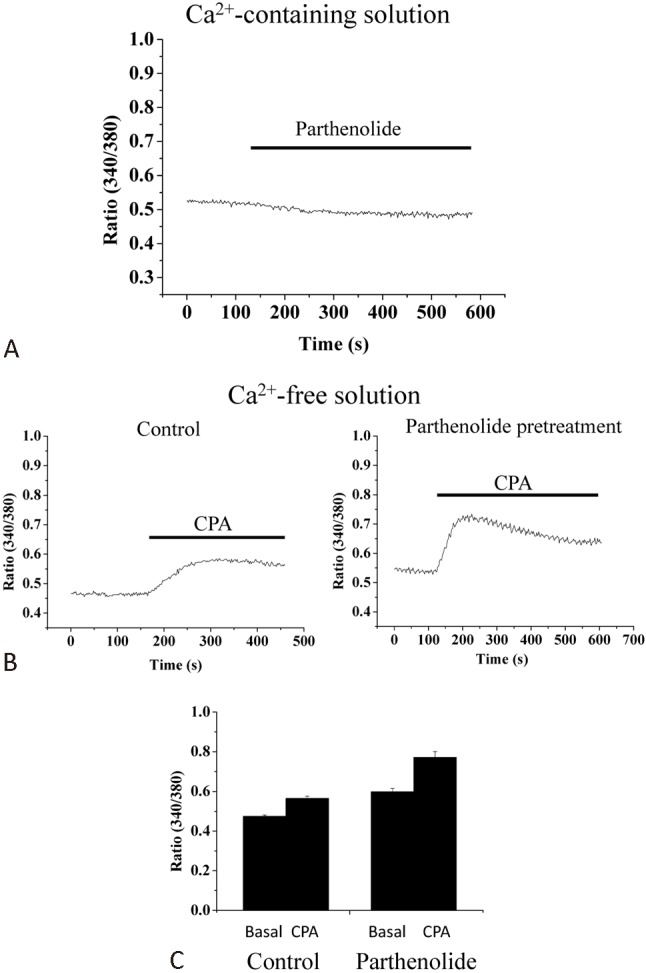
Effects of parthenolide on Ca2+ homeostasis in H9c2 cells. (A) Parthenolide (30 μM) did not cause any acute change in cytosolic Ca2+ concentration in cells bathed in Ca2+-containing bath solution. Results shown are representative of 6 experiments. (B) Cells were pretreated with (right panel) or without (left panel) parthenolide (30 μM) for 4 h and then bathed in Ca2+-free bath solution and were subsequently challenged with 50 μM cyclopiazonic acid (CPA; endoplasmic reticulum Ca2+ pump inhibitor) to examine Ca2+ release. (C) Quantification of results from (B). Results are mean ± standard error of the mean of 6 experiments.
Parthenolide caused a collapse in mitochondrial membrane potential
Collapse of the mitochondrial membrane potential is often involved in apoptotic cell death. We examined whether parthenolide affected mitochondrial membrane potential. Parthenolide did not cause an acute change in mitochondrial membrane potential (not shown), but treatment of parthenolide for 2 h resulted in reduction in mitochondrial membrane potential (thus depolarization, as indicated in an increase in rhodamine 123 fluorescence) (Figure 7). FCCP was used to collapse mitochondrial membrane potential in order to confirm the authenticity of mitochondrial signals.
Figure 7.
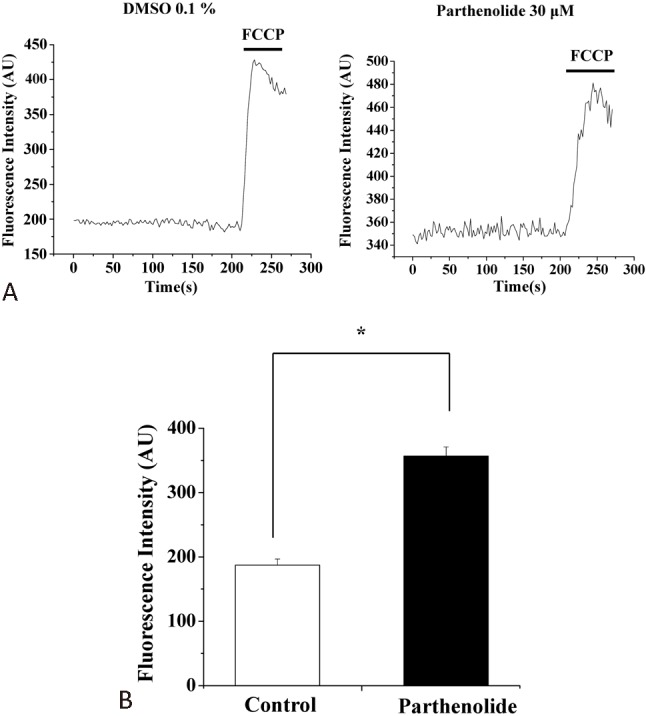
Parthenolide depolarized mitochondrial membrane potential in H9c2 cells. (A) Cells were pretreated with (right panel) or without (left panel) parthenolide (30 μM) for 2 h and then assayed for mitochondrial membrane potential using rhodamine 123 as dye. For authentication of the mitochondrial signals, FCCP (2 μM) was used to collapse mitochondrial membrane potential. (B) Quantification of results from (A). Results are mean ± standard error of the mean of 37-47 cells from 4 experiments. * indicates significant difference (p < 0.05) from the control. DMSO, dimethyl sulfoxide; FCCP, carbonyl cyanide4-trifluoromethoxyphenylhydrazone.
Cytotoxic mechanism of parthenolide in part involves oxidative stress
Since parthenolide induced both ER stress and oxidative stress in H9c2 myoblasts (Figure 4, Figure 5), we then proceeded to determine the relative contribution of these stresses to cell death. We employed vitamin C and salubrinol as an antioxidant and an inhibitor of ER stress, respectively. As shown in Figure 8, parthenolide-induced cell death could be partially prevented by optimal concentrations of vitamin C and salubrinol; a complete prevention of cell death required a combination of both compounds. To test the specificity of actions of vitamin C and salubrinal, the effects of these two compounds on ER stress and ROS generation were examined. Vitamin C did not affect parthenolide-induced CHOP expression; unexpectedly, the latter was also not modulated by salubrinal (Figure 9A, B), casting doubt on the proposal that salubrinal rescued cell death by suppressing ER stress. We also examined the effects of vitamin C and salubrinal on ROS generation. These two compounds neither elicited ROS production, nor did they suppress parthenolide-triggered ROS formation. The observation that vitamin C did not affect parthenolide-triggered ROS formation could be explained by the fact that vitamin C is able to scavenge, but not inhibit the formation of oxygen free radicals.
Figure 8.
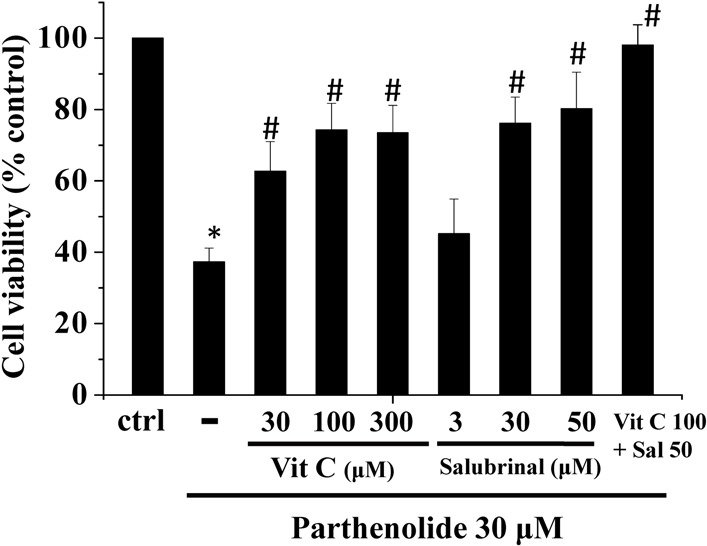
Parthenolide-induced cell death could be rescued by vitamin C and salubrinal. H9c2 cells were treated with parthenolide and/or different combinations of vitamin C and salubrinol for 15 h and then assayed for cell viability using the MTT assay. Results are mean ± standard error of the mean of 3 experiments. * indicates significant difference (p < 0.05) from the control. # indicates significant difference (p < 0.05) from parthenolide alone. Ctrl, control; Vit C; vitamin C.
Figure 9.
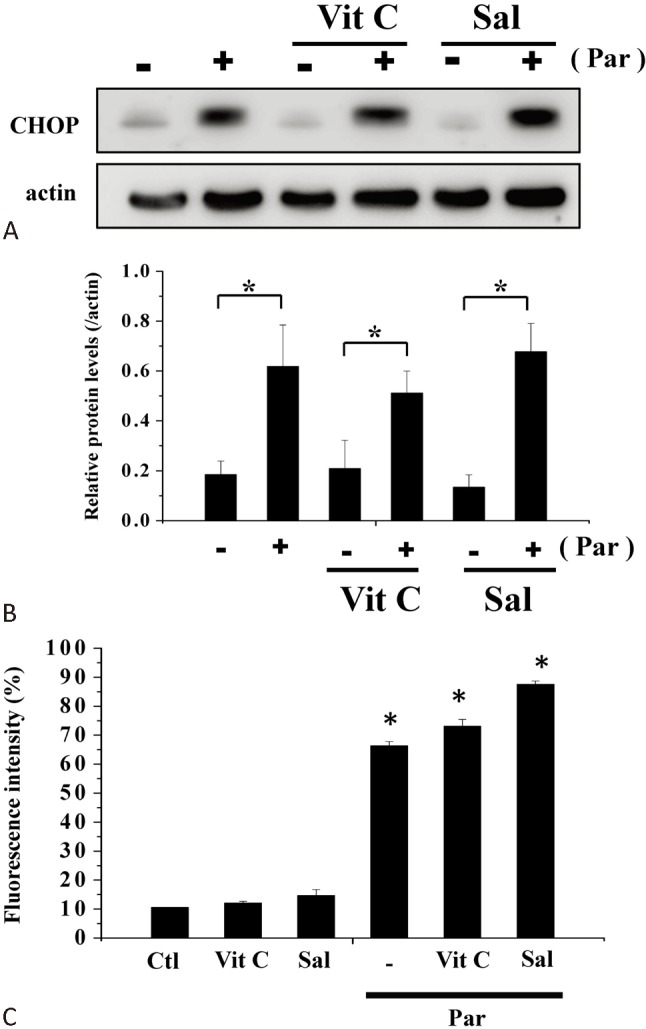
Effects of vitamin C and salubrinal on ER stress and ROS formation. (A) Protein levels of CHOP in the absence or presence of parthenolide (30 μM), vitamin C (100 μM) or salubrinal (50 μM) treatments (4 h) were analyzed by western blot. (B) Quantification of results from (A). Results are mean ± standard error of the mean of 4 experiments. * indicates significant difference (p < 0.05). (C) H9c2 cells were treated with DCFH-DA and then incubated in the absence or presence of parthenolide (30 μM), vitamin C (100 μM) or salubrinal (50 μM) for 1 h and ROS generation was assayed using flow cytometry. Results are mean ± standard error of the mean of 4 experiments. * indicates significant difference (p < 0.05) from the control. Ctl, control; ER; endoplasmic reticulum; Par, parthenolide; ROS, reactiveoxygen species; Sal, salubrinal; Vit C; vitamin C.
DISCUSSION
Parthenolide has been shown to be a cardioprotective agent in myocardial reperfusion injury because of its ability to inhibit NF-κB.8,9 However, because of its reported toxic effects in cardiac myocytes,10 caution is needed in using this drug. Furthermore, whether parthenolide elicits toxic effects in cardiac myoblasts, the progenitor cells of cardiomyocytes, is still unknown. We here reported the toxic actions of parthenolide in rat H9c2 myoblasts.
Our data supported earlier findings that parthenolide induced oxidative stress and mitochondrial membrane potential collapse in cardiac myocytes (Figure 5, Figure 7).10 We further confirmed that cell death was apoptotic, as evidenced by the TUNEL assay and translocation of Bax from cytosol to mitochondria and translocation of cytochrome c from mitochondria to cytosol (Figure 2, Figure 3). Our findings seemed to rule out the involvement of Ca2+ overload and Ca2+ store depletion in parthenolide-induced cell death (Figure 6).
Interestingly, we observed that parthenolide not only caused oxidative stress but also ER stress (Figure 4). The latter is presumably unrelated to Ca2+ store depletion since parthenolide-treated H9c2 cells had functionally intact Ca2+ stores (Figure 6B, C). Further, Ca2+ store depletion by CPA increased the levels of ATF6 and CHOP, while parthenolide treatment raised the levels of phospho-JNK and CHOP (Figure 4). While data (triggering of ROS formation by parthenolide and partial inhibition of cell death by vitamin C, Figure 5, Figure 8) suggest oxidative stress contributed to parthenolide-induced cell death, the role of ER stress is uncertain, since salubrinal failed to suppress CHOP expression (Figure 9A,B). It is thus possible that the rescue effect of salubrinal (Figure 8) is attributable to the effect(s) other than ER stress suppression. However, a definitive understanding of this mechanism awaits future investigation. We did not deploy another ER stress inhibitor, tauroursodeoxycholic acid (TUDCA), since the latter has been reported to be non-specific and show antioxidant activities.13
Parthenolide has been shown to be an anti-inflammatory drug. For instance, it inhibits inflammatory responses on macrophages by suppressing the protease activity of caspase-1.14 It attenuates lipopolysaccharide-inflicted fever in rats by reducing circulating levels of IL-6 and TNF-α.15 In addition, parthenolide prevents down-regulation of PPARβ/ δ-target gene pyruvate dehydrogenase kinase 4 and PPARγ coactivator 1 gene expression in lipopolysaccharide-treated H9c2 cells.16 In line with this, parthenolide has been shown to suppress cytokine-activated STAT3 and JAK1 activation in cardiomyocytes.17 Its cardioprotective effect is further demonstrated by its prevention of pressure overload-induced left ventricular hypertrophy by inhibiting STAT3 signaling and fibroblast activation.18 Parthenolide has also been shown to be an effective anticancer agent in many tumor cell and models and is currently under clinical trials.19-25 Hitherto, there are no clinical trials yet to investigate the cardioprotective effects of parthenolide. Given that both its beneficial and toxic effects in cardiac tissues may happen at close concentration ranges (see next paragraph), caution must be exercised in future clinical trials to monitor the concentration of this drug to prevent overdose.
In animal studies demonstrating beneficial cardiac effects of parthenolide,8,9,18 the effective concentrations of parthenolide were not known. In in vitro studies using H9c2 cells and cardiomyocytes,16,17 parthenolide shows protective effects at ≥ 10 μM. Our data here showed that parthenolide at concentrations ≥ 30 μM triggered ROS formation, caused mitochondrial membrane potential collapse and cell death. In rat cardiomyocytes, parthenolide at ≥ 10 μM causes mitochondrial membrane potential collapse and cell death; at ≥ 5 μM it decreases the levels of reduced glutathione, and it triggers ROS formation at a concentration as low as 0.5 μM.10 Therefore, beneficial and detrimental effects of parthenolide may co-exist at overlapping micromolar ranges. One of the possible strategies to render parthenolide a safer and more effective cardioprotective agent may be to co-administer anti-oxidants as supplements.
CONCLUSIONS
In conclusion, this is the first report to show that parthenolide caused oxidative stress and apoptotic death in cardiomyoblasts. This new information adds to our understanding of the potential toxic effects of this agent in cardiomyoblasts and thus in cardiac muscle tissue development.
Acknowledgments
Y.M.L. and K.L.W. would like to thank China Medical University, Taiwan, and the Taiwan National Science Council for providing funding (NSC 100-2320-B-039-006-; DMR-99-097).
REFERENCES
- 1.Baines CP. How and when do myocytes die during ischemia and reperfusion: the late phase. J Cardiovasc Pharmacol Ther. 2011;16:239–243. doi: 10.1177/1074248411407769. [DOI] [PubMed] [Google Scholar]
- 2.Gottlieb RA. Cell death pathways in acute ischemia/reperfusion injury. J Cardiovasc Pharmacol Ther. 2011;16:233–238. doi: 10.1177/1074248411409581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nichols TC. NF-kappaB and reperfusion injury. Drug News Perspect. 2004;17:99–104. doi: 10.1358/dnp.2004.17.2.829042. [DOI] [PubMed] [Google Scholar]
- 4.Van der Heiden K, Cuhlmann S, Luong le A, et al. Role of nuclear factor kappaB in cardiovascular health and disease. Clin Sci (Lond) 2010;118:593–605. doi: 10.1042/CS20090557. [DOI] [PubMed] [Google Scholar]
- 5.Latanich CA, Toledo-Pereyra LH. Searching for NF-kappaB-based treatments of ischemia reperfusion injury. J Invest Surg. 2009;22:301–315. doi: 10.1080/08941930903040155. [DOI] [PubMed] [Google Scholar]
- 6.Nam NH. Naturally occurring NF-kappaB inhibitors. Mini Rev Med Chem. 2006;6:945–951. doi: 10.2174/138955706777934937. [DOI] [PubMed] [Google Scholar]
- 7.Hendricks H, Anderson-Wildeboer Y, Engels G, et al. The content of parthenolide and its yield per plant during the growth of Tanacetum parthenium. Planta Med. 1997;63:356–359. doi: 10.1055/s-2006-957700. [DOI] [PubMed] [Google Scholar]
- 8.Zingarelli B, Hake PW, Denenberg A, et al. Sesquiterpene lactone parthenolide,an inhibitor of IkappaB kinase complex and nuclear factor-kappaB,exerts beneficial effects in myocardial reperfusion injury. Shock. 2002;17:127–134. doi: 10.1097/00024382-200202000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Konia MR, Schaefer S, Liu H. Nuclear factor-[kappa]B inhibition provides additional protection against ischaemia/reperfusion injury in delayed sevoflurane preconditioning. Eur J Anaesthesiol. 2009;26:496–503. doi: 10.1097/eja.0b013e328324ed2e. [DOI] [PubMed] [Google Scholar]
- 10.Kurdi M, Bowers MC, Dado J, et al. Parthenolide induces a distinct pattern of oxidative stress in cardiac myocytes. Free Radic Biol Med. 2007;42:474–481. doi: 10.1016/j.freeradbiomed.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 11.Leung YM, Huang CF, Chao CC, et al. Voltage-gated K+ channels play a role in cAMP-stimulated neuritogenesis in mouse Neuro-2A cells. J Cell Physiol. 2011;226:1090–1098. doi: 10.1002/jcp.22430. [DOI] [PubMed] [Google Scholar]
- 12.Lu DY, Yu WH, Yeh WL, et al. Hypoxia-induced matrix metalloproteinase-13 expression in astrocytes enhances permeability of brain endothelial cells. J Cell Physiol. 2009;220:163–173. doi: 10.1002/jcp.21746. [DOI] [PubMed] [Google Scholar]
- 13.Oveson BC, Iwase T, Hackett SF, et al. Constituents of bile,bilirubin and TUDCA,protect against oxidative stress-induced retinal degeneration. J Neurochem. 2011;116:144–153. doi: 10.1111/j.1471-4159.2010.07092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Juliana C, Fernandes-Alnemri T, Wu J, et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem. 2010;285:9792–10502. doi: 10.1074/jbc.M109.082305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rummel C, Gerstberger R, Roth J, et al. Parthenolide attenuates LPS-induced fever, circulating cytokines and markers of brain inflammation in rats. Cytokine. 2011;3:739–748. doi: 10.1016/j.cyto.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 16.Planavila A, Sánchez RM, Merlos M, et al. Atorvastatin prevents peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) downregulation in lipopolysaccharide-stimulated H9c2 cells. Biochim Biophys Acta. 2005;1736:120–127. doi: 10.1016/j.bbalip.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 17.Kurdi M, Booz GW. Evidence that IL-6-type cytokine signaling in cardiomyocytes is inhibited by oxidative stress:parthenolide targets JAK1 activation by generating ROS. J Cell Physiol. 2007;212:424–431. doi: 10.1002/jcp.21033. [DOI] [PubMed] [Google Scholar]
- 18.Skoumal R, Tóth M, Serpi R, et al. Parthenolide inhibits STAT3 signaling and attenuates angiotensin II-induced left ventricular hypertrophy via modulation of fibroblast activity. J Mol Cell Cardiol. 2011;50:634–641. doi: 10.1016/j.yjmcc.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Cheng G, Xie L. Parthenolide induces apoptosis and cell cycle arrest of human 5637 bladder cancer cells in vitro. Molecules. 2011;16:6758–6768. doi: 10.3390/molecules16086758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuch D, Giang AH, Shapovalov Y, et al. Targeting radioresistant osteosarcoma cells with parthenolide. J Cell Biochem. 2012;113:1282–1291. doi: 10.1002/jcb.24002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim SL, Trang KT, Kim SH, et al. Parthenolide suppresses tumor growth in a xenograft model of colorectal cancer cells by inducing mitochondrial dysfunction and apoptosis. Int J Oncol. 2012;41:1547–1553. doi: 10.3892/ijo.2012.1587. [DOI] [PubMed] [Google Scholar]
- 22.Nakabayashi H, Shimizu K. Involvement of Akt/NF-κB pathway in antitumor effects of parthenolide on glioblastoma cells in vitro and in vivo. BMC Cancer. 2012;12:453. doi: 10.1186/1471-2407-12-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quanjun Y, Lili W, Zhiyong Z, et al. Parthenolide from parthenium integrifolium reduces tumor burden and alleviate cachexia symptoms in the murine CT-26 model of colorectal carcinoma. Phytomedicine. 2013;20:992–998. doi: 10.1016/j.phymed.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 24.Ghantous A, Gali-Muhtasib H, Vuorela H, et al. What made sesquiterpene lactones reach cancer clinical trials? Drug Discov Today. 2010;15:668–678. doi: 10.1016/j.drudis.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Ghantous A, Sinjab A, Herceg Z, et al. Parthenolide:from plant shoots to cancer roots. Drug Discov Today. 2013;18:894–905. doi: 10.1016/j.drudis.2013.05.005. [DOI] [PubMed] [Google Scholar]