

Abstract
The etiology of Alzheimer’s disease (AD) remains unclear. Epidemiologic studies suggest hypertension plays a contributing role to AD. Recently, several experimental and observational studies showed interaction between the renin-angiotensin system and amyloid-β, a key pathologic feature of AD, with diverse results. This article reviews molecular, genetic, experimental and clinical data to clarify the impact on an AD patient with angiotensin converting enzyme inhibitor and angiotensin II receptor blocker therapy, with some guidance for the direction of possible future research.
Keywords: Alzheimer’s disease, Amyloid-β, Renin angiotensin system
INTRODUCTION
Alzheimer’s disease (AD) is well-known to be the most common form of dementia in developed countries.1 It is a complex and heterogeneous disease, characterized by progressive episodic memory deficit and personality changes, accompanied by specific structural abnormalities in the brain.2 The main pathological features of AD are extracellular deposits of amyloid-β (Aβ) in the form of senile plaques, and intracellular inclusions of hyperphosphorylated tau in the form of neurofibrillary tangles (NFT). Importantly, there is growing evidence indicating an association between vascular risk factors, for example, hypertension, and AD. Several epidemiological studies have shown that hypertension is related to the development of AD.3 Since the renin-angiotensin system (RAS) plays a crucial role in the pathogenesis of hypertension, it is not surprising that RAS is also related to the development of AD.4 However, the exact impact and mechanisms involved remain largely unknown. On the other hand, clinical differentiation between AD and vascular dementia (VaD), the two most common forms of dementia, can often be confusing. Unlike memory impairment as the earliest symptom of AD, the main problem of VaD was executive function, speed of information processing and attention. Besides, the course of AD was slowly progressive but stepwise in VaD. To provide greater focus, this review only summarizes the updated knowledge of the influence of RAS on the pathogenesis of AD in cell culture systems, animals, and humans.
EPIDEMIOLOGY AND CLINICAL STUDIES
Previous studies revealed that high midlife blood pressure is a risk factor for dementia5 and plays a role in AD progression.6 A report from Taiwan also showed that hypertension, especially diastolic blood pressure (DBP), is a significant risk for AD.7 However, some adverse results have indicated that low DBP (≤ 70 mm Hg) in older adults is related to an increased dementia risk.8 Since blood pressure, especially hypertension, plays a role in AD, clinical anti-hypertensive therapy trials have addressed the issue. Two large studies covering stroke incidence and antihypertensive therapy all mentioned dementia. The Systolic Hypertension in Europe (SYST-EUR) trial9 was a double-blind placebo-controlled trial that was early terminated in just two years due to a significant reduction of stroke. In two years of follow-up, the study showed a 50% reduced incidence of AD and VaD, regardless of stroke. Similarly, results from The Perindopril Protection Against Recurrent Stroke (PROGRESS) Study, a randomised, double-blind, placebo-controlled trial with previous cerebrovascular accident (CVA) patients10 suggested angiotensin converting enzyme inhibitor (ACEI) reduced the risk of dementia in stroke patients, and calcium channel blocker (CCB) also had some benefit. Khachaturian et al.11 found that diuretics provided a benefit for AD but ACEI had no significant influence (hazard ratio 1.08, 95% CI 0.53-1.99). Ohrui et al.12 stated a new concept that “brain penetrating ACEI”, not mentioned in the Khachaturian study, was more effective for slowing cognitive decline in AD patient compared with non-brain penetrating ACEI and CCB. The finding suggested that ACEI may have some potential effect for slowing AD progression beyond a blood pressure-lowing effect. Recently, two large cohort studies13,14 and one small randomized clinical trial (RCT)15 also found that angiotensin II receptor blocker (ARB) has protective effect for AD (Table 1). However, another two RCT studies revealed no difference in AD incidence between ARB treatment group and control.16,17 One study based on the Taiwan National Health Insurance database also found no protective effect of ARB.18
Table 1. Clinical trials evaluating the effects of antihypertensive drugs in AD .
| Source | Drugs | Study size and members | Length of follow up | outcome |
| SYST-EUR trial (1998) | Placebo vs. nitrendipine (10-40 mg/day) ± enalapril (5-20 mg/day) ± hydrochlorothiazide | Age > 60 years HTN p’t,n = 2148 | 2 years | ACEI reduced AD and VaD |
| PROGRESS trial (2003) | Placebo vs perindopril ± indapamide | Prior stroke HRN p’t, n = 6105 | 3.9 years | ACEI reduced dementia |
| Ohrui et al. (2004) | Perindopril 2 mg/day vs. enalapril 5 mg/day vs. nifedipine 20 mg/day | Mild to moderate AD p’t n = 162 | 1 years | Brain penetrating ACEI slowed cognitive decline |
| Khachaturian et al. (2006) | ACEI vs. β-blocker vs. CCB vs. diuretics | Age > 65 years HTN p’t, n = 3308 | 5 years | ACEI had no influence on AD risk |
| Rozzini (2006) | ACEI vs. β-blocker vs. CCB | MCI p’t, n = 74 | 1 year | ACEI have protect effect for MCI |
| Kazumasa (2012) | Telmisartan 40-80 mg/day vs. amlodipine 5-10 mg/day | AD p’t, n = 20 | 6 months | ARB had protective effect for AD |
ACEI, angiotensin converting enzyme inhibitor; AD,Alzheimer’s disease; ARB, angiotensin II receptor blocker; HTN, hypertension; MCI, mild cognitive impairment; p’t, patient.
In Summary, although low blood pressure in later life may be associated with dementia, almost all studies showed that anti-hypertensive therapy can reduce the incidence and progression of AD. However, the effect of ACEI and ARB was inconclusive and controversial. To date, large-scale clinical trials of ARB and ACEI in AD are lacking and previous studies were too small, having short follow-up periods with some confounding factors such as stroke or other uncontrolled metabolic factors. In addition, the dementia type of AD and VaD was not clearly separated. Further trials addressed a head-to-head comparison of ARB and ACEI, and other antihypertensive drugs were needed.
FINDING OF CELL-BASED EXPERIMENTS AND ANIMAL MODELS
Aβ is a key pathological feature of Alzheimer’s disease which consists of 40-42 amino acid peptides.19 It is commonly known that Aβ accumulation in AD patients reflects imbalance between Aβ production and removal. Transmembrane amyloid precursor protein (APP) was cleavaged by β- and γ-secretases (the amyloidogenic pathway)20 and formed Aβ. Aβ may aggregate and be deposited in the brain, leading to neurotoxicity.21
Besides, reduced cholinergic fibers in the brain were another pathological abnormality contributing to the development of AD.22 Currently, primarily cholinesterase inhibitors such as Donepezil and Rivastigmine have been approved by the Food and Drug Administration for treatment of AD.
There are several in vivo and in vitro studies that showed interaction regarding Aβ and angiotensin-converting enzyme (ACE). Hu et al.23 first described that human affinity-purified ACE had an inhibitory effect for synthetic Aβ aggregation in vitro by cleavage at the site Asp7-Ser8 of Aβ. Compared with Aβ, the degraded product had reduced aggregation ability and cytotoxicity. They also demonstrated ACE had reduced Aβ toxic effects on rat cell line, and was nullified if the ACEI lisinopril was added to the cultures. Besides, Hemming and Selkoe24 used cloned ACE from human neuroblastoma cells to demonstrate both the N- and C-domains of ACE were able to degrade Aβ and the properties were inhibited by captopril. According to these in vivo studies, ACE seems to have some measure of protective effects while drugs inhibiting ACE may be harmful.
A review of the prior literature also indicates that there have been varied results in animal studies (Table 2). One study showed that in ACE knockout mice and wild type mice treated with perindopril, the Aβ level was not altered.25 Similarly, another study demonstrated that in APP transgenic mice, treatment with captopril failed to change the Aβ level.26 Drugs such as ARB were also tested in these animal studies. Valsartan27 and telmisartan28 were tested and showed reduced Aβ level and improvement of mice cognitive function. Since telmisartan also possesses peroxisome proliferator-activated receptor (PPAR-γ) agonist properties, PPAR-γ antagonist was also tested and it reduced the protective effect of telmisartan.28
Table 2. Effects of ACEI and ARB on animal serum Aβ level .
| Animal models | Tested drugs | Results |
| Tg 2576 (3-4 weeks) | Perindopril (0.2 mg/kg/ day) | ACEI not affect Aβ level25 |
| 3*TG (3 months) | Captopril (2 g/l-28 days) | ACEI not affect Aβ level26 |
| Tg2576 (6 months) | Valsartan (10 mg/kg-5 months) | ARB reduced serum Aβ level and improved cognitive function27 |
| ddY (8 weeks) | Telmisartan (100 mg/kg-28 days) | ARB reduced serum Aβ level and improved cognitive function28 |
Aβ, amyloid-β; ACEI,angiotensin converting enzyme inhibitor; ARB,angiotensin II receptor blocker.
To explore the possible explanations for the conflicting results of ACE and AD in vivo and in vitro studies, we need to review the renin-angiotensin system pathway. Renin acts on angiotensinogen to produce angiotensin I, which in turn is cleaved by ACE to form the active angiotensin II, a potent vasoconstrictor exerting the hypertensive effects by its action on two receptors (AT1 and AT2).29 Some evidence has showed that angiotensin 2 inhibits potassium-mediated release of acetylcholine,30 which was also involved in pathophysiology in AD as mentioned previously. Therefore, ACE inhibitors, by reducing the level of both ACE and angiotensin II, caused both beneficial and negative effects for AD. Such an explanation also provides clues as to why the effect can only be seen in vivo but not in vitro (Figure 1). Another possible explanation for the beneficial effects of ACE inhibitors is that they increase brain substance P,31 which is normally degraded by ACE. Substance P was reported to augment the activity of neprilysin, a recognized Aβ degrading enzyme.32
Figure 1.
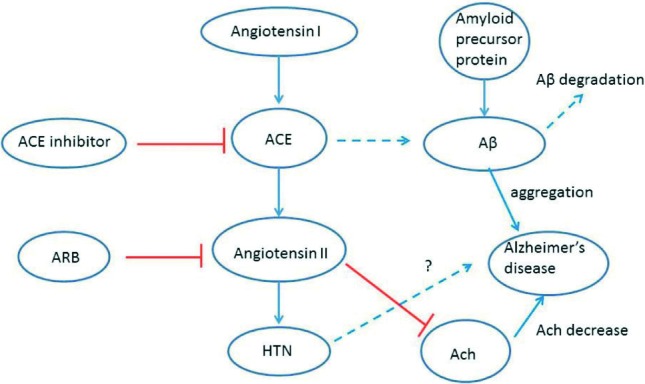
Potential interaction of renin-angiotensinsystem (RAS) system and amyloid-β (Aβ) metabolism. Angiotensin converting enzyme (ACE) has positive effects for Aβ degradation (dotted line) and angiotensin II has inhibitory effects on acetylcholine release (red line). Therefore, ACE inhibitors, by reducing the level of both ACE and angiotensin II, generate both beneficial and negative effects for AD. Ach, acetylcholine; ARB, angiotensin II receptor blocker; HTN, hypertension.
Besides, it is important to clarify that the data derived from the animal models might not apply to its human-equivalent disease. Moreover, the animals used in AD studies were too young (only 3 and 4 weeks of age) such that age-related metabolic abnormalities as seen in older humans might not exist. Finally, It is not clear whether mouse ACE acted on Aβ in the same way as that seen in humans. Consequently, additional studies focusing on these issues are needed.
THE HUMAN GENE AND BRAIN TISSUE FINDINGS
One post-mortem brain tissue study from AD patients33 showed elevated ACE activity in the medial hippocampus and parahippocampal gyrus, frontal cortex and caudate nucleus correlated with Aβ plaque load. Another study34 using ACE radioligand [3H] ceranapril to assess the binding density in brain tissue showed increased ACE in the temporal cortex in patients with AD. On the other hand, studies of ACE level and activity in the CSF have yielded equivocal results, with reports of reduced,35 no difference36 or elevated37 ACE level. However, these studies were small in sample size and therefore required further evaluation. It is not clear why ACE activity is increased in the brain and how alternation of brain ACE contributes to blood flow change in the AD patients.
In human genetic studies, Kehoe et al.38 first found AD was associated with insertion (I)/deletion (D) polymorphism within intron 16 of the ACE gene. Later, multiple case-control studies were published with variable results. A meta-analysis39 supports a modest contribution (OR: 1.27, 95% CI, 1.10 to 1.47; p < 0.001) of ACE gene variation to the risk of developing late onset AD. In recent years, genome-wide association studies (GWASs) of single nucleotide polymorphisms (SNPs) found evidence to support the involvement of ACE in AD,40,41 which possibly work through interaction with LRRTM3 gene42 and A2M gene.43 However, it is still a question how genetic variations in ACE affect risk of AD.
CONCLUSION
There are several controversial results concerning the connection between RAS and AD. Fortunately, no human clinical data showed harmful effects of using an ACE inhibitor in AD patients. Besides, whether angiotensin-receptor blockers are appropriate drugs for hypertension control in AD patients remain unclear. To answer these questions, future prospective studies monitoring the effects of ACEI and ARB on cognitive performance and blood pressure as well as post-mortem assessment of pathology in the AD patients are required.
REFERENCES
- 1.Aguero-Torres H, Fratiglioni L, Guo Z, et al. Dementia is the major cause of functional dependence in the elderly:3-year follow-up data from a population-based study. Am J Public Health. 1998; 88:1452–1456. doi: 10.2105/ajph.88.10.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Markowitsch HJ, Staniloiu A. Amnesic disorders. Lancet. 2012; 380:1429–1440. doi: 10.1016/S0140-6736(11)61304-4. [DOI] [PubMed] [Google Scholar]
- 3.Skoog I, Gustafson D. Hypertension,hypertension-clustering factors and Alzheimer’s disease. Neurol Res. 2003;25:675–680. doi: 10.1179/016164103101201986. [DOI] [PubMed] [Google Scholar]
- 4.Trenkwalder P. Potential for antihypertensive treatment with an AT(1)-receptor blocker to reduce dementia in the elderly. J Hum Hyperte. 2002;16:S71–S75. doi: 10.1038/sj.jhh.1001443. [DOI] [PubMed] [Google Scholar]
- 5.Launer LJ, Masaki K, Petrovitch H, et al. The association between midlife blood pressure levels and late-life cognitive function. The Honolulu-Asia Aging Study. . JAMA. 1995;274:1846–1851. [PubMed] [Google Scholar]
- 6.Mielke MM, Rosenberg PB, Tschanz J, et al. Vascular factors predict rate of progression in Alzheimer disease. Neurology. 2007;69:1850–1858. doi: 10.1212/01.wnl.0000279520.59792.fe. [DOI] [PubMed] [Google Scholar]
- 7.Yang YH, Roe CM, Morris JC. Relationship between late-life hypertension,blood pressure,and Alzheimer’s disease. Am J Alzheimers Dis Othe. 2011;26:457–462. doi: 10.1177/1533317511421779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verghese J, Lipton RB, Hall CB, et al. Low blood pressure and the risk of dementia in very old individuals. Neurology. 2003;61:1667–1672. doi: 10.1212/01.wnl.0000098934.18300.be. [DOI] [PubMed] [Google Scholar]
- 9.Forette F, Seux ML, Staessen JA, et al. Prevention of dementia in randomised double-blind placebo-controlled Systolic Hypertension in Europe (Syst-Eur) trial. Lancet. 1998;352:1347–1351. doi: 10.1016/s0140-6736(98)03086-4. [DOI] [PubMed] [Google Scholar]
- 10.Tzourio C, Anderson C, Chapman N, et al. Effects of blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in patients with cerebrovascular disease. Arch Intern Med. 2003;163:1069–1075. doi: 10.1001/archinte.163.9.1069. [DOI] [PubMed] [Google Scholar]
- 11.Khachaturian AS, Zandi PP, Lyketsos CG , et al. Antihypertensive medication use and incident Alzheimer disease:the Cache County Study. Arch Neurol. 2006;63:686–692. doi: 10.1001/archneur.63.5.noc60013. [DOI] [PubMed] [Google Scholar]
- 12.Ohrui T, Matsui T, Yamaya M , et al. Angiotensin-converting enzyme inhibitors and incidence of Alzheimer’s disease in Japan. J Am Geriatr Soc. 2004;52:649–650. doi: 10.1111/j.1532-5415.2004.52178_7.x. [DOI] [PubMed] [Google Scholar]
- 13.Li NC, Lee A, Whitmer RA, et al. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population:prospective cohort analysis. BMJ. 2010;340:b5465–b5465. doi: 10.1136/bmj.b5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies NM, Kehoe PG, Ben-Shlomo Y, et al. Associations of anti-hypertensive treatments with Alzheimer’s disease,vascular dementia,and other dementias. J Alzheimers Dis. 2011;26:699–708. doi: 10.3233/JAD-2011-110347. [DOI] [PubMed] [Google Scholar]
- 15.Kume K, Hanyu H, Sakurai H, et al. Effects of telmisartan on cognition and regional cerebral blood flow in hypertensive patients with Alzheimer’s disease. Geriatr Gerontol Int. 2012;12:207–214. doi: 10.1111/j.1447-0594.2011.00746.x. [DOI] [PubMed] [Google Scholar]
- 16.Diener HC, Sacco RL, Yusuf S, et al. Effects of aspirin plus extended-release dipyridamole versus clopidogrel and telmisartan on disability and cognitive function after recurrent stroke in patients with ischaemic stroke in the Prevention Regimen for Effectively Avoiding Second Strokes (PRoFESS) trial:a double-blind,active and placebo-controlled study. Lancet Neurol. 2008; 7:875–884. doi: 10.1016/S1474-4422(08)70198-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lithell H, Hansson L, Skoog I, et al. The Study on Cognition and Prognosis in the Elderly (SCOPE):principal results of a randomized double-blind intervention trial. J Hypertens. 2003;21:875–886. doi: 10.1097/00004872-200305000-00011. [DOI] [PubMed] [Google Scholar]
- 18.Hsu CY, Huang CC, Chan WL, et al. Angiotensin-receptor blockers and risk of Alzheimer’s disease in hypertension population - a nationwide cohort study. Circ J. 2013;77:405–410. doi: 10.1253/circj.cj-12-0658. [DOI] [PubMed] [Google Scholar]
- 19.Glenner GG, Wong CW. Alzheimer’s disease:initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Re. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 20.Vassar R, Bennett BD, Babu-Khan S, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 21.Bayer TA, Wirths O, Majtenyi K, et al. Key factors in Alzheimer’s disease:beta-amyloid precursor protein processing,metabolism and intraneuronal transport. Brain Pathol. 2001;11:1–11. doi: 10.1111/j.1750-3639.2001.tb00376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pakaski M, Kalman J. Interactions between the amyloid and cholinergic mechanisms in Alzheimer’s disease. Neurochem Int. 2008;53:103–111. doi: 10.1016/j.neuint.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Hu J, Igarashi A, Kamata M, et al. Angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide (A beta); retards A beta aggregation,deposition,fibril formation; and inhibits cytotoxicity. J Biol Chem. 2001;276:47863–47868. doi: 10.1074/jbc.M104068200. [DOI] [PubMed] [Google Scholar]
- 24.Hemming ML, Selkoe DJ. Amyloid beta-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem. 2005;280:37644–37650. doi: 10.1074/jbc.M508460200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eckman EA, Adams SK, Troendle FJ, et al. Regulation of steady-?state beta-amyloid levels in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme. J Biol Chem. 2006;281:30471–30478. doi: 10.1074/jbc.M605827200. [DOI] [PubMed] [Google Scholar]
- 26.Hemming ML, Selkoe DJ, Farris W. Effects of prolonged angiotensin-converting enzyme inhibitor treatment on amyloid beta-protein metabolism in mouse models of Alzheimer disease. Neurobiol Dis. 2007;26:273–281. doi: 10.1016/j.nbd.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Ho L, Chen L, et al. Valsartan lowers brain beta-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Invest. 2007;117:3393–3702. doi: 10.1172/JCI31547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mogi M, Li JM, Tsukuda K, et al. Telmisartan prevented cognitive decline partly due to PPAR-gamma activation. Biochem Biophys Res Commun. 2008;375:446–449. doi: 10.1016/j.bbrc.2008.08.032. [DOI] [PubMed] [Google Scholar]
- 29.Chiu AT, Herblin WF, McCall DE, et al. Identification of angio?tensin II receptor subtypes. Biochem Biophys Res Commun. 1989; 165:196–203. doi: 10.1016/0006-291x(89)91054-1. [DOI] [PubMed] [Google Scholar]
- 30.Barnes JM, Barnes NM, Costall B, et al. Angiotensin-converting enzyme inhibition,angiotensin,and cognition. J Cardiovasc Pharmacol. 1992;S63:S71. doi: 10.1097/00005344-199219006-00011. [DOI] [PubMed] [Google Scholar]
- 31.Emanueli C, Grady EF, Madeddu P, et al. Acute ACE inhibition causes plasma extravasation in mice that is mediated by bradykinin and substance P. Hypertension. 1998;31:1299–1504. doi: 10.1161/01.hyp.31.6.1299. [DOI] [PubMed] [Google Scholar]
- 32.Carson JA, Turner AJ. Beta-amyloid catabolism: roles for ne?prilysin (NEP) and other metallopeptidases? J Neurochem. 2002;81:1–8. doi: 10.1046/j.1471-4159.2002.00855.x. [DOI] [PubMed] [Google Scholar]
- 33.Arregui A, Perry EK, Rossor M, et al. Angiotensin converting enzyme in Alzheimer’s disease increased activity in caudate nucleus and cortical areas. J Neurochem. 1982;38:1490–1492. doi: 10.1111/j.1471-4159.1982.tb07930.x. [DOI] [PubMed] [Google Scholar]
- 34.Barnes NM, Cheng CH, Costall B, et al. Angiotensin converting enzyme density is increased in temporal cortex from patients with Alzheimer’s disease. Eur J Pharmacol. 1991;200:289–292. doi: 10.1016/0014-2999(91)90584-d. [DOI] [PubMed] [Google Scholar]
- 35.Zubenko GS, Volicer L, Direnfeld LK, et al. Cerebrospinal fluid levels of angiotensin-converting enzyme in Alzheimer’s disease,Parkinson’s disease and progressive supranuclear palsy. Brain Res. 1985;328:215–221. doi: 10.1016/0006-8993(85)91032-7. [DOI] [PubMed] [Google Scholar]
- 36.Konings CH, Kuiper MA, Scheltens P, et al. Re-evaluation of cerebrospinal fluid angiotensin-converting enzyme activity in patients with ‘probable’ Alzheimer’s disease. Eur J Clin Chem Clin Biochem. 1993;31:495–497. [PubMed] [Google Scholar]
- 37.Miners S, Ashby E, Baig S, et al. Angiotensin-converting enzyme levels and activity in Alzheimer’s disease:differences in brain and CSF ACE and association with ACE1 genotypes. Am J Transl Res. 2009;1:163–177. [PMC free article] [PubMed] [Google Scholar]
- 38.Kehoe PG, Russ C, McIlory S, et al. Variation in DCP1,encoding ACE,is associated with susceptibility to Alzheimer disease. Nat Genet. 1999;21:71–72. doi: 10.1038/5009. [DOI] [PubMed] [Google Scholar]
- 39.Elkins JS, Douglas VC, Johnston SC. Alzheimer disease risk and genetic variation in ACE:a meta-analysis. Neurology. 2004;62:363–368. doi: 10.1212/01.wnl.0000106823.72493.ff. [DOI] [PubMed] [Google Scholar]
- 40.Li H, Wetten S, Li L, et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol. 2008;65:45–53. doi: 10.1001/archneurol.2007.3. [DOI] [PubMed] [Google Scholar]
- 41.Feulner TM, Laws SM, Friedrich P, et al. Examination of the current top candidate genes for AD in a genome-wide association study. Mol Psychiatry. 2010;15:756–766. doi: 10.1038/mp.2008.141. [DOI] [PubMed] [Google Scholar]
- 42.Thornton-Wells TA, Moore JH, Martin ER, et al. Confronting complexity in late-onset Alzheimer disease:application of two-stage analysis approach addressing heterogeneity and epistasis. Genet Epidemiol. 2008;32:187–203. doi: 10.1002/gepi.20294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edwards TL, Pericak-Vance M, Gilbert JR, et al. An association analysis of Alzheimer disease candidate genes detects an ancestral risk haplotype clade in ACE and putative multilocus association between ACE,A2M,and LRRTM3. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:721–735. doi: 10.1002/ajmg.b.30899. [DOI] [PMC free article] [PubMed] [Google Scholar]