

Abstract
Metal–organic frameworks (MOFs) are usually synthesized using a single type of metal ion, and MOFs containing mixtures of different metal ions are of great interest and represent a methodology to enhance and tune materials properties. We report the synthesis of [Ga2(OH)2(L)] (H4L = biphenyl-3,3′,5,5′-tetracarboxylic acid), designated as MFM-300(Ga2), (MFM = Manchester Framework Material replacing NOTT designation), by solvothermal reaction of Ga(NO3)3 and H4L in a mixture of DMF, THF, and water containing HCl for 3 days. MFM-300(Ga2) crystallizes in the tetragonal space group I4122, a = b = 15.0174(7) Å and c = 11.9111(11) Å and is isostructural with the Al(III) analogue MFM-300(Al2) with pores decorated with −OH groups bridging Ga(III) centers. The isostructural Fe-doped material [Ga1.87Fe0.13(OH)2(L)], MFM-300(Ga1.87Fe0.13), can be prepared under similar conditions to MFM-300(Ga2) via reaction of a homogeneous mixture of Fe(NO3)3 and Ga(NO3)3 with biphenyl-3,3′,5,5′-tetracarboxylic acid. An Fe(III)-based material [Fe3O1.5(OH)(HL)(L)0.5(H2O)3.5], MFM-310(Fe), was synthesized with Fe(NO3)3 and the same ligand via hydrothermal methods. [MFM-310(Fe)] crystallizes in the orthorhombic space group Pmn21 with a = 10.560(4) Å, b = 19.451(8) Å, and c = 11.773(5) Å and incorporates μ3-oxo-centered trinuclear iron cluster nodes connected by ligands to give a 3D nonporous framework that has a different structure to the MFM-300 series. Thus, Fe-doping can be used to monitor the effects of the heteroatom center within a parent Ga(III) framework without the requirement of synthesizing the isostructural Fe(III) analogue [Fe2(OH)2(L)], MFM-300(Fe2), which we have thus far been unable to prepare. Fe-doping of MFM-300(Ga2) affords positive effects on gas adsorption capacities, particularly for CO2 adsorption, whereby MFM-300(Ga1.87Fe0.13) shows a 49% enhancement of CO2 adsorption capacity in comparison to the homometallic parent material. We thus report herein the highest CO2 uptake (2.86 mmol g–1 at 273 K at 1 bar) for a Ga-based MOF. The single-crystal X-ray structures of MFM-300(Ga2)-solv, MFM-300(Ga2), MFM-300(Ga2)·2.35CO2, MFM-300(Ga1.87Fe0.13)-solv, MFM-300(Ga1.87Fe0.13), and MFM-300(Ga1.87Fe0.13)·2.0CO2 have been determined. Most notably, in situ single-crystal diffraction studies of gas-loaded materials have revealed that Fe-doping has a significant impact on the molecular details for CO2 binding in the pore, with the bridging M–OH hydroxyl groups being preferred binding sites for CO2 within these framework materials. In situ synchrotron IR spectroscopic measurements on CO2 binding with respect to the −OH groups in the pore are consistent with the above structural analyses. In addition, we found that, compared to MFM-300(Ga2), Fe-doped MFM-300(Ga1.87Fe0.13) shows improved catalytic properties for the ring-opening reaction of styrene oxide, but similar activity for the room-temperature acetylation of benzaldehyde by methanol. The role of Fe-doping in these systems is discussed as a mechanism for enhancing porosity and the structural integrity of the parent material.
Short abstract
Key structural insights were obtained for CO2-loaded MFM-300(Ga2) and mixed GaFe analogues by combining in situ single-crystal X-ray diffraction and in situ polarized IR spectroscopic studies. These static and dynamic experiments revealed that the adsorbed CO2 molecules in MFM-300(Ga2) form hydrogen bonds with the free −OH groups on the surface of the pores, and a previously unobserved pattern of intermolecular dipole interactions was found to stabilize two CO2 molecules within the pores.
Introduction
Porous metal–organic frameworks (MOFs) have attracted a great deal of interest because of their potential applications in gas adsorption and separation, catalysis, and drug delivery.1−3 The assembly of MOF materials from various metal ions and organic linkers, usually via solvothermal reactions, allows the fine-tuning of their crystal structures and the incorporation of designed functional groups for specific applications. Indeed, the properties, size, and functionality of the cavity of these porous materials can be optimized by using different metal centers or organic ligands.4,5 Amine (−NH2) groups can bind selectively to CO2 due to the formation of strong electrostatic interactions between the electronegative N center of the −NH2 group and the electropositive C center of the CO2 molecule.6 For this reason, a number of amine-functionalized MOFs have been designed and synthesized to capture CO2 from flue gases.7,8 However, the effect of the different metal ions in MOFs on CO2 adsorption properties has been rarely studied. While frameworks based on divalent transition metal centers have been extensively explored and are now well-developed,9 MOFs with trivalent metal centers have generally attracted less attention.10 Furthermore, few examples of gallium-carboxylate MOFs are described in the literature.11 MOFs are usually constructed from a single type of metal cation and organic linker, but there are increasing examples of MOFs which contain two different types of ligand linkers or metal cations with a homogeneous distribution, as found in solid solutions.12−23 Kitagawa et al.17 and Cheetham18 et al. have reported the syntheses and properties of some binary and ternary MOF solid solutions which contain a mixture of similar organic linkers. In addition, Serre et al. have reported mixed-metal MIL-53(FeCr), which exhibits a new type of framework breathing behavior that is distinct from that in either pure MIL-53(Fe) or MIL-53(Cr).19 Hill et al. reported that CO2 uptake could be increased through metal ion exchange in UiO-66.20 Thus, postsynthetic modification of UiO-66 by stirring UiO-66 in a solution of TiCl4(THF)2 for 1, 5, or 15 days gave UiO-66 (Ti32), UiO-66 (Ti44), and UiO-66 (Ti56), respectively. The BET surface area of these materials was found to increase as the proportion of Ti within the framework increased, and an increase in Ti content from 0–56% gave an increase in CO2 uptake by up to 81% compared to UiO-66 (Zr100).20 Similar replacement of heavier metal ions with lighter ones has also been shown to increase surface area.21,22 Long et al. have recently described23 a magnesium-diluted [Fe0.1Mg0.9(dobdc)] [dobdc4– = 2,5-dioxido-1,4-benzenedicarboxylate] analogue of [Fe2(dobdc)], also known as Fe-MOF-74. [Fe0.1Mg1.9(dobdc)] was synthesized by mixing MgCl2, FeCl2, and H4(dobdc) in a mixture of DMF (N,N′-dimethylformamide) and MeOH in a Schlenk flask at 120 °C. [Fe0.1Mg1.9(dobdc)] converts ethane into EtOH and acetaldehyde using N2O as the terminal oxidant, in contrast to the complex mixture of products formed by its analogue Fe-MOF-74.23
We report herein the synthesis and structural characterization of a Ga(III)-based metal–organic material [Ga2(OH)2(L)]·1.6DMF·4H2O [denoted as MFM-300(Ga2)-solv; MFM = Manchester Framework Material replacing NOTT designation], which is found to be isostructural with the previously reported MFM-300(Al2),24 and, to the best of our knowledge, shows the highest CO2 uptake (20.8 wt % at 195 K, 1 bar) for Ga-based MOFs.11 In order to investigate the effect of metal doping on CO2 adsorption and catalytic properties, we synthesized the isostructural mixed-metal solid solution material, [Ga1.87Fe0.13(OH)2(L)] [denoted as MFM-300(Ga1.87Fe0.13)], by including Fe(NO3)3 in the synthesis of MFM-300(Ga2). Gas adsorption studies have confirmed a significant enhancement (49%) of CO2 adsorption upon controlled Fe-doping. Single-crystal X-ray diffraction studies on both solvated and desolvated materials confirm the structural integrity of these materials, and notably, in situ single-crystal diffraction studies under a CO2 atmosphere have been used to determine the preferred binding sites of CO2 within these MOFs and to explain the changes in the molecular guest binding mechanism upon Fe-doping. In situ synchrotron IR spectroscopy has also been used to study the details of CO2 binding with respect to the −OH groups in the pore. Furthermore, catalytic experiments have also confirmed that MFM-300(Ga1.87Fe0.13) shows higher conversion rates than MFM-300(Ga2) for the ring-opening reaction of styrene oxide.
Materials and Methods
Chemicals
Gallium nitrate (Ga(NO3)3, 99%, Aldrich), iron nitrate (Fe(NO3)3·9H2O, 99%, Sigma), hydrochloric acid (HCl, >99%, Fisher Scientific), nitric acid (HNO3, >99%, Fisher Scientific), piperazine (99%, Sigma-Aldrich), DMF (>99%, Fisher Scientific), and THF (99.9%, Fisher Scientific) were used as purchased. Biphenyl-3,3′,5,5′-tetracarboxylic acid (H4L) (Scheme 1) was synthesized by a literature method.25
Scheme 1. View of Biphenyl-3,3′,5,5′-tetracarboxylic Acid (H4L).

Synthesis of MFM-300(Ga2)-solv
Biphenyl-3,3′,5,5′-tetracarboxylic acid (H4L, 21.8 mg, 0.067 mmol) and Ga(NO3)3 (38.0 mg, 0.148 mmol) in a 2:5:1 mixture of DMF, THF, and water (8 mL) slightly acidified with 37% HCl (2 drops) were reacted in a pressure tube at 348 K for 3 days. The white crystalline product was separated by filtration as single crystals (∼0.05 mm cubes), washed several times with warm DMF, and dried in air. Yield: 85%. Elemental analysis (% calc/found) for [Ga2(OH)2(L)]·5H2O·1DMF (C19H25Ga2NO16): C, 34.43/34.21; H, 3.80/3.14; N, 2.11/1.70. Rapid loss of free solvent molecules and hydration during the sample transfer are responsible for the discrepancy and the variability observed in the elemental analytical data for this material. Selected IR: ν/cm–1: 3648(m), 3236(w), 1667(vs), 1541(vs), 1458(s), 1425(s), 1376(m), 1318(m), 1095(m), 915(w), 775(m), 664(m).
Synthesis of MFM-310(Fe)
Fe(NO3)3·9H2O (290 mg, 0.72 mmol), H4L (60 mg, 0.18 mmol), and piperazine (0.10 g, 1.26 mmol) were mixed and dispersed in water (10.0 mL) containing nitric acid (2.0 mL, 2.7M) in a Teflon autoclave and heated at 483 K for 3 days. The yellow crystalline product was separated by filtration, washed several times with water, and dried in air. Yield: 10%. These hydrothermal conditions afforded two phases which were light yellow and dark purple in color. Single-crystal X-ray diffraction identified the light yellow phase as MFM-310(Fe), while the other phase (Fe3O4) was isolated only as a fine powder. Since we have found no method as yet for their separation as pure phases, no further characterization has been undertaken.
Synthesis of MFM-300(Ga1.87Fe0.13)-solv
A mixture of Fe(NO3)3·9H2O (3.16 mg, 0.00782 mmol), Ga(NO3)3 (38 mg, 0.1486 mmol), and H4L (22.0 mg, 0.067 mmol) in a 2:5:1 mixture of DMF, THF, and water (8 mL) slightly acidified with 37% HCl (5 drops) was reacted at 348 K for 3 days. The microcrystalline pale orange precipitate was separated by filtration, washed several times with warm DMF, and dried in air. Yield: 84%. Elemental analysis (% calc/found) for [Ga1.87Fe0.13(OH)2(L)]·1.5H2O (C16Fe0.13Ga1.87H11O11.5): C, 36.61/36.80; H, 2.11/2.11; N, 0/0. Selected IR: ν/cm–1: 3648(m), 3232(w), 1662(vs), 1539(vs), 1455(s), 1418(s), 1374(m), 1318(m), 1096(m), 913(w), 803(w), 775(m), 659(m). The presence and quantity of Fe within the single crystals was determined by SEM and EDX, which also confirmed the even distribution of Fe throughout the material (Figure S16), and ICP analysis confirmed the overall metal content of the bulk product. The Ga:Fe stoichiometry in the final product was consistent over a range of syntheses using the above conditions.
Growth of MOF Crystals on SiO2 Surfaces
Silicon (100) wafers with a 300 nm SiO2 thin oxide layer were cut mechanically and then washed in methanol, acetone, and isopropanol for 10 min each. The wafers were dried under a stream of N2 and placed into the above reaction solutions, which were then heated in a pressure tube. Single crystals of MOF products grew on these surfaces over 3 days under the same solvothermal conditions. We found this methodology to be highly effective in growing good quality single crystals for subsequent structural characterization.
SEM, EDX, and ICP Measurements
MOF crystals grown on SiO2 substrates were coated with carbon using an Agar turbo carbon coater to improve conductivity. Scanning electron microscopic (SEM) analysis was performed on a FEI Quanta 3D 200 dual beam focused ion beam scanning electron microscope (FIB-SEM). The images were acquired using secondary electron imaging at an accelerating voltage of 5 kV. An Oxford Instruments integrated INCA Energy 250 Microanalysis System was used for EDX elemental spectra and mapping, and ICP-OES measurements were made on a PerkinElmer Optima 2000.
Growth of Single Crystals of MFM-300(Ga1.87Fe0.13)-solv
Single crystals of MFM-300(Ga2)-solv were obtained under solvothermal conditions as detailed above, and we also attempted to grow crystals of both MFM-300(Ga2)-solv and MFM-300(Ga1.87Fe0.13)-solv on a SiO2 surface. Immersion of the prepared SiO2 substrate into a reaction solution of MFM-300(Ga1.87Fe0.13) leads to the formation of uniform cubic single crystals of 30–50 μm in length. The growth of MOF crystallites on surfaces has been observed previously26 but has been used rarely for the growth of single crystals. We were unable to grow single crystals of MFM-300(Ga1.87Fe0.13)-solv in the absence of the SiO2 substrate (Figure 1).
Figure 1.

SEM images for (a, b) [MFM-300(Ga1.87Fe0.13)]-solv and (c, d) [MFM-300(Ga2)]-solv grown on SiO2 surfaces.
Structure Determination and Refinement for MFM-300(Ga2–xFex)-solv (x = 0, 0.13)
High-resolution powder diffraction data for the as-synthesized MOFs were collected on Beamline I11 at Diamond Light Source using multi-analyzing-crystal (MAC) detectors.27 The powder pattern was first indexed on a body-centered tetragonal lattice, and the independent unit cell parameters were refined using TOPAS.28 The body centring and the reflection condition 00l: l = 4n indicates that the space group is one of the enantiomeric pair I4122 and I4322. In the absence of any component capable of directing chirality, the product is expected to be a 1:1 racemic mixture. The structure solution was initially established in space group I4122 starting from the structural model of MFM-300(Ga2) and further developed by subsequent difference Fourier analysis using TOPAS.28 The final structure refinement was carried out using the Rietveld method with isotropic displacement parameters for all atoms. The highly disordered DMF molecules in the pores could not be located and modeled and, therefore, were treated as discrete water molecules in the refinement. A total of 40 disordered water molecules per unit cell were found within the channels and included in the final structure refinement for MFM-300(Ga2–xFex)-solv (x = 0, 0.13). The final stage of the Rietveld refinement involved soft restraints to the C–C bond lengths within the benzene rings.
Structure Determination and Refinement for MFM-300(Ga2)-solv, MFM-300(Ga2), MFM-300(Ga2)·2.35CO2, MFM-300(Ga1.87Fe0.13)-solv, MFM-300(Ga1.87Fe0.13), MFM-300(Ga1.87Fe0.13)·2.0CO2
Single-crystal diffraction data of MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13) were collected on Beamline I19 at Diamond Light Source within a gas cell system. A single crystal of MFM-300(Ga2)-solv or MFM-300(Ga1.87Fe0.13)-solv was loaded into the gas cell, which was then flushed with dry N2. The diffraction data were collected at room temperature. The gas cell was then evacuated at 10–4 mbar and heated at 393 K using an Oxford Cryosystems open-flow nitrogen cryostat for 4 h to generate the desolvated material MFM-300(Ga2) or MFM-300(Ga1.87Fe0.13). Diffraction data were collected under vacuum after cooling to room temperature. The gas cell was then cooled to 195 K and exposed to dry CO2 for 2 h. The diffraction data were then collected at 195 K under 1.0 bar pressure of CO2. The structures were solved by direct methods and developed by difference Fourier techniques using the SHELXTL software package.29 A total of 6.4 DMF and 16 water molecules, and 10.8 water molecules per unit cell were found within the pores, and these were included in the refinement of MFM-300(Ga2)·1.6DMF·4H2O and MFM-300(Ga1.87Fe0.13)·2.7H2O, respectively. Importantly, no residual electron density was apparent within the pores of desolvated MFM-300(Ga2) and desolvated MFM-300(Ga1.87Fe0.13), confirming the complete removal of guest solvent molecules. A total of 9.4 and 8.0 CO2 molecules per unit cell were found to occupy two independent sites within the pore and were included in the refinement of MFM-300(Ga2)·2.35CO2 and MFM-300(Ga1.87Fe013.)·2.0CO2, respectively. The hydrogen atoms on the ligands and DMF molecules were placed geometrically and refined using a riding model. The hydrogen atoms of free water molecules could not be located but are included in the molecular formula and in values derived from it. CCDC 951538–951543 and 1009553 contain the supplementary crystallographic data for this paper.
In Situ Infrared Spectroscopy
IR spectroscopic measurements were carried out on the B22 MIRIAM beamline at the Diamond Light Source using a Hyperion 3000 infrared microscope with a 15× objective and a liquid N2 cooled MCT detector. The sample was mounted in a Linkam FTIR600 variable-temperature gas-tight stage fitted with ZnSe windows, and ZnSe-based linear IR polarizers were used to obtain polarized IR spectra. Spectra were collected (256 scans) in the range 650–4000 cm–1 with 4 cm–1 resolution and an infrared spot size at the sample of approximately 25 by 25 μm. The sample was activated in the stage prior to measurements by heating at 150 °C under a slow flow of N2 for 10 h before being transferred to the microscope and purged under He flow at room temperature. CO2 and He were delivered to the Linkam stage using separate mass-flow controllers, the partial pressure of CO2 being controlled by varying the volumetric flow of the two gases. Both gases were predried through zeolite filters placed between the cylinders and the mass-flow controllers before being flowed through the Linkam stage.
Gas Sorption
Sorption isotherms for CO2, CH4, N2 and H2 were recorded at 77 K (liquid N2), 87 K (liquid Ar), or 273–303 K (temperature-programmed water bath from Hiden Company) on an IGA-003 system under ultrahigh vacuum from a diaphragm and turbo pumping system. All gases used were ultrapure research grade (99.999%) purchased from BOC or AIRLIQUIDE. The density of the desolvated samples used in buoyancy corrections (1.80 g cm–3) was estimated from the crystallographic density of the desolvated sample by PLATON/SOLV.30 In a typical gas adsorption experiment, ca. 50 mg of as-synthesized material was loaded into the IGA and degassed at 100 °C and high vacuum (10–10 bar) for 1 day to give fully desolvated samples.
Catalytic Reactions
All reagents and starting materials were obtained commercially from Aldrich and used without any further purification. The percentage conversion, purity, and relative yields of the final products were determined by using a Hewlett-Packard 5890 series II gas chromatograph with an FID detector and high-purity helium as the carrier gas. The products were identified by GC–MS by using a Hewlett-Packard 6890 series spectrometer. The regioselectivity of the ring-opening was determined by mass spectrometry, which shows different fragmentation patterns depending on the relative position of the nucleophilic group. In the case of 2-methoxy-2-phenylethanol, the structure was confirmed by comparison with an authentic sample.
In a typical experiment, the activated MOF catalyst (20 mg) was suspended in MeOH (5 mL), followed by the addition of 1a or benzaldehyde. The reaction was performed at the required temperature, the evolution of products was monitored by GC, and the products were analyzed by GC–MS. After the required time, the catalyst was removed by filtration, washed with MeOH, and dried at 80 °C. This dried catalyst was activated again and used for the second run with fresh 1a and methanol.
Results and Discussion
Synthesis and Structural Description
Biphenyl-3,3′5,5′-tetracarboxylic acid (Scheme 1), H4L, was reacted with Ga(NO3)3 in a 2:5:1 mixture of DMF, THF, and water (8 mL) slightly acidified with HCl in a pressure tube at 348 K for 3 days to give MFM-300(Ga2)-solv. MFM-300(Ga1.87Fe0.13)-solv was prepared in a similar manner but using an intimate mixture of Fe(NO3)3·9H2O and Ga(NO3)3 as the metal source. The structures of MFM-300(Ga2)-solv and MFM-300(Ga1.87Fe0.13)-solv were solved by single-crystal and high-resolution synchrotron powder diffraction data, respectively. Indexation of powder diffraction patterns (PXRD) of these complexes confirms that they are isomorphous to MFM-300(Al2)-solv.24
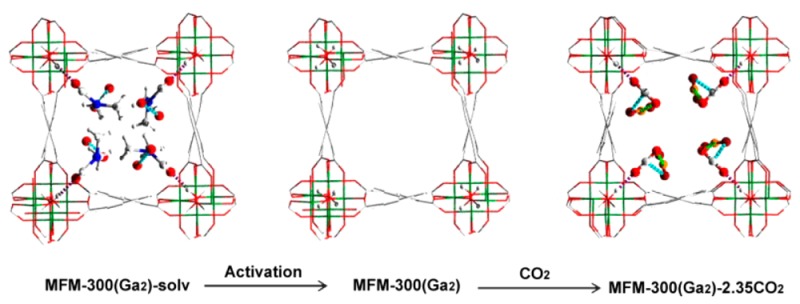
MFM-300(Ga2)-solv crystallizes in the chiral tetragonal space group I4122 and shows an open three-dimensional framework structure constructed from one-dimensional helical [Ga(OH)2O4]∞ chains bridged by tetracarboxylate ligands (Figure 2). The Ga(III) ion is octahedrally coordinated by six O-donors: four from carboxylate groups [Ga–O = 2.004(3) and 1.999(4) Å] and two from mutually cis bridging hydroxyl groups μ2-OH [Ga–O = 1.928(3) Å]. This coordination also affords a square-shaped, one-dimensional channel running through the framework parallel to the c-axis. In the analogous complex MFM-300(Al2)-solv, the four bonds from the carboxylates are slightly shorter [Al–O = 1.935(1) and 1.929(2) Å] than those in MFM-300(Ga2)-solv, but the two bonds from the μ2-OH groups are very similar [Al–O = 1.930(1) Å].24 The approximate diameter of the channel in MFM-300(Ga2)-solv, taking into account the van der Waals radii of surface atoms, is 6.7 × 6.7 Å. The channel contains uncoordinated water (2.0 per Ga) and DMF (0.8 per Ga) molecules, giving a formula of [Ga2(OH)2(C16H6O8)]·(DMF)1.6·(H2O)4 for the as-synthesized material. The DMF molecule interacts with the hydroxyl group to form a moderate hydrogen bond [O···O = 2.694(6) Å; ∠O–H···O = 180°], and two disordered water molecules form weak intermolecular hydrogen bonds [O···O = 2.94(3) Å]. The hydrogen atoms on the water molecules were not located. The total pore voids occupied by the free solvents were estimated by PLATON/SOLV30 to be 50%.
Figure 2.

Views of the crystal structure of MFM-300(Ga2). (a) View of the [Ga(OH)2O4]∞ chain; (b) projection of the structure along the a-axis; (c) view of the channel running through the framework parallel to the c-axis. (Ga: green; O: red; H: white; ligand L4– is represented by gray lines).
The solvated framework complex MFM-300(Ga1.87Fe0.13)-solv was prepared via solvothermal reaction of H4L and Ga(NO3)3/Fe(NO3)3·9H2O in DMF/THF/water containing HCl. Whereas these conditions yielded cube-shaped single crystals of MFM-300(Ga)-solv with an average size of 20–50 μm, the solvothermal synthesis of MFM-300(Ga1.87Fe0.13)-solv yielded microcrystalline solids with an approximate particle size of 5 μm rather than crystals large enough for single-crystal structure determination. Rietveld refinement confirms the phase purity of the bulk material and confirms that the Fe-doped material MFM-300(Ga1.87Fe0.13) has the same framework structure as MFM-300(Ga2), with very small differences in unit cell parameters (Table S1). This is consistent with the similar ionic radii of Ga(III) and Fe(III) and the low level of doping. A similar modulation in unit cell parameters has been observed in the Fe2+-exchanged material Mn3[(Mn4Cl)3(BTT)8]2 (BTT3– = 1,3,5-benzenetristetrazolate) following postsynthetic cation methathesis.31 Subsequently, single crystals of MFM-300(Ga1.87Fe0.13)-solv were successfully grown on SiO2 substrates as described above, and their single-crystal structure determination is entirely consistent with the structure derived from PXRD.
Attempts to form the isostructural MFM-300(Fe2)-solv have thus far failed. A nonisostructural complex was synthesized under different hydrothermal conditions using only an Fe(III) metal source to give the trinuclear Fe3-cluster-based material [Fe3O1.5(OH)(HL)(L)0.5(H2O)3.5] [denoted as MFM-310(Fe)]. MFM-310(Fe) crystallizes in the orthorhombic space group Pmn21 with a = 10.560(4) Å, b = 19.451(8) Å, and c = 11.773(5) Å and shows an entirely different framework structure to the MFM-300 series. In MFM-310(Fe), μ3-oxo-centered trinuclear iron clusters connected by L4– ligands generate a three-dimensional nonporous framework (Figure 3). This trinuclear iron cluster is connected through an oxygen positioned at the center of an isosceles triangle of 3.57(3) and 3.27(2) Å (Figure 3a). Interestingly, this trinuclear iron cluster incorporates different carboxylate connectivity compared to the more usual Fe3O(CO2)6-type cluster reported previously in the literature.32,33 PXRD data for MFM-300(Ga1.87Fe0.13)-solv confirm the absence of the MFM-310(Fe) phase, suggesting that a solid solution has been formed, rather than a mechanical binary mixture of MFM-300(Ga2)-solv, MFM-300(Fe2)-solv and MFM-310(Fe). Furthermore, no splitting or broadening of PXRD peaks attributable to the presence of a binary mixture of phases was observed, even when using a synchrotron radiation source (Δ2θ < 0.005°), supporting the formation of a fully mixed doped material (Figure S1).
Figure 3.

Views of the crystal structure of MFM-310(Fe): (a) Comparison of the common trinuclear Fe3O(O2CR)6 cluster (left)33 and the cluster present in MFM-310(Fe) (right); (b) view of the structure along the a-axis; (c) view of the structure along the c-axis. (FeO6 octahedron: green; Fe: yellow; O: red; C: gray/black).
The synthesis of MFM-300(Ga1.87Fe0.13)-solv emanated from a preparation using metal salt starting materials in an Fe:Ga molar ratio of 5:95. Characterization of the Fe content by ICP for successive isolated samples from the above synthetic procedure consistently gave metal content ratios of MFM-300(Ga1.87Fe0.13)-solv (x = 0.13) (equivalent to 6.5% doping). SEM/EDX confirmed the even spread of Fe throughout the sample.
We also targeted materials MFM-300(Ga2–xFex)-solv with higher Fe doping levels of 10, 15 and 20%. With increasing Fe content, the PXRD patterns of the isolated powders indicated the presence of more amorphous materials, and more critically, we found that the reproducibility of these higher Fe-doped materials was very poor, with significant variations in Fe content from batch to batch. Thus, only MFM-300(Ga2) doped with Fe at the 5% level in the synthesis (giving 6.5% in the product) is presented here. The partial replacement of one metal cation by another is most likely to be successful in the case of a series of isostructural compounds.23 In this case, however, the isostructural MFM-300(Fe2) cannot be isolated using the synthetic conditions reported here.
Gas Adsorption Properties
MFM-300(Ga2–xFex)-solv (x = 0, 0.13) are thermally stable up to 573 K before decomposition, as confirmed by TGA (Figure S7). The desolvated materials were prepared by soaking the as-synthesized sample in acetone for 5 days, followed by degassing under dynamic vacuum (10–10 bar) at 100 °C for 20 h, to obtain the fully desolvated samples. The CO2 isotherms at 273 K show type-I behavior with reversible uptake (Figure 4). The pore size distributions (PSDs) for both MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13) show one broad peak at approximately at 5.5 Å, close to the approximate diameter of the channel window (6.7 Å). The surface areas from the CO2 isotherms were calculated to be 392 and 491 m2 g–1 for MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13), respectively. The values for BET surface area and pore size obtained from the N2 isotherms are in good agreement with those calculated from CO2 (Table 1). These surface areas are considerably lower than that of the isostructural MFM-300(Al2) (1370 m2 g–1), taking into account the increased formula weight by ∼21% on going from Al to Ga. A similar difference in BET surface area between Al- and Ga-containing isostructural materials has been also observed in [Al(BTB)] and [Ga(BTB)] (BTB3– = benzenetribenzoate),34 where the surface areas are reported as 1045 and 62 m2 g–1, respectively. The BET surface areas for MFM-300(Ga2) are reproducible over many batches of material. This discrepancy could be due to a number of factors: not all the void space within a porous material is necessarily accessible by the gaseous substrate in the BET and gas adsorption experiments, while the bulk powder sample may not correspond precisely to the idealized structure determined by single-crystal diffraction due to partial collapse of the framework and/or blocking of pores by phase changes and different domains within the material. Notwithstanding the full structural analysis of these materials, it remains possible that residual free solvent molecules or reactant molecules remain in the pores, leading to blocking effects limiting gas entry and uptake. The phase purity of the as-synthesized bulk samples has been confirmed by high-resolution synchrotron X-ray powder diffraction data, which do not show the presence of bulk impurities (Figure S4). The total micropore volume for MFM-300(Ga2) was estimated from CO2 isotherms to be 0.201 cc g–1 increasing to 0.299 cc g–1 upon Fe-doping (Table 1). Thus, such doping has a positive effect on the micropore volume, increasing it by 49%; we ascribe this effect to increased structural integrity of the Fe-doped material giving a more ordered, and thus more porous, material with more accessible pores.
Figure 4.

CO2 adsorption isotherms for MFM-300(Ga2–xFex) (x = 0, 0.13) (a) at 195 K and (b) at 293 K up to 20 bar; (c) variation of isosteric heat of CO2 adsorption.
Table 1. Summary of Gas Adsorption Data for MFM-300(Ga2–xFex) (x = 0, 0.13).
| MFM-300(M) | pore volume (cc/g)a | pore voids (%)b | CO2 uptake at 195 K and 1 bar (mmol/g) | CO2 uptake at 273/293 K and 1 bar (mmol/g) | heat of CO2 adsorption Qst (kJ/mol)c | surface area (m2/g)d | BET surface area (m2/g)e | H2 adsorption 77 K at 20 bar (wt %) | heat of H2 adsorption Qst (kJ/mol) |
|---|---|---|---|---|---|---|---|---|---|
| Al2 | 0.375 | 42 | 3.10 | 7.00/5.40 | 30.0 | 1370 | n.a | <0.1 | 6.5 |
| Ga2 | 0.201 | 50 | 4.72 | 2.87/2.15 | 34.7 | 392 | 400 | 1.45 | 10.3 |
| Ga1.87Fe0.13 | 0.299 | 50 | 7.02 | 3.51/2.36 | 30.9 | 491 | 489 | 1.61 | 9.7 |
Calculated from CO2 data at 195 K.
Calculated from PLATON.
Heat of adsorption at low surface coverage.
Surface area calculated from CO2 isotherm at 273 K data.
BET calculated from N2 isotherm at 77 K.
Gravimetric CO2 sorption isotherms were also measured at 195 K (1 bar) and 303 K (up to 20 bar) for desolvated MFM-300(Ga2–xFex) (x = 0, 0.13) to evaluate the effect of mixing metal cations on CO2 uptake capacity. At 195 K and 1 bar, MFM-300(Ga2) shows a maximum uptake of 4.72 mmol g–1, whereas MFM-300(Ga1.87Fe0.13) shows a maximum uptake of 7.02 mmol g–1, corresponding to a 49% increase in CO2 adsorption (Figure 3). This result further suggests that MFM-300(Ga1.87Fe0.13) is a homogeneous, single-phase material: if it were simply a mechanical binary mixture of two pure M(III) (M = Ga, Fe), in order to achieve 7.02 mmol g–1 CO2 uptake for the mixture, MFM-300(Fe2) would need to have a CO2 uptake of 35.17 mol g–1, which is 3 times higher than the maximum uptake calculated based on the single-crystal structure.
At 273 K and up to 1 bar, MFM-300(Ga2–xFex) (x = 0, 0.13) show a maximum CO2 uptake of 2.87 and 3.51 mmol g–1 for x = 0 and x = 0.13, respectively. Compared with MFM-300(Ga2), the CO2 uptake in the Fe-doped MOF thus shows an increase of 22%. This value is consistent with those observed at 195 K and 1 bar, confirming the enhancement of CO2 adsorption in the doped material. Interestingly, although Fe-doping has a positive effect on CO2 uptake capacities, the heat of CO2 adsorption counterintuitively decreases upon Fe-doping [34.7 and 30.9 kJ mol–1 for MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13), respectively], indicating that the Fe-doped framework actually has a reduced interaction with regard to binding of CO2 molecules, consistent with an increased pore volume. This same effect was also observed for hydrogen adsorption, as described below, and may reflect the presence of more open and available pores within MFM-300(Ga1.87Fe0.13) rather than any specific enhanced chemical effects of doping with Fe.
MFM-300(Ga1.87Fe0.13) adsorbs more H2 at 77 K and 20 bar (1.61 wt %) than MFM-300(Ga2) (1.45 wt %). In addition, both materials display substantially higher H2 uptake capacities compared to MFM-300(Al2) (<0.2 wt %), even though the latter has the higher internal surface area.35 A slight decrease in heat of adsorption (Qst) is observed for MFM-300(Ga1.87Fe0.13) (9.7 kJ mol–1) in comparison to MFM-300(Ga2) (10.3 kJ mol–1), indicating slightly weaker H2-framework interactions as a result of Fe-doping (Figure S10). The high heat of adsorption for H2 uptake in MFM-300(Ga2–xFex) (x = 0, 0.13) is due to strong overlapping potential between the adsorbed H2 molecules and the narrow pore size (5.5 Å) of these materials.
In Situ Single-Crystal Synchrotron Infrared Spectroscopy
To investigate the nature of the CO2 interactions with the framework, we have studied the CO2 sorption in MFM-300(Ga2) by in situ IR spectroscopy on a single crystal using a high brightness synchrotron IR source coupled to an IR microscope. In the related complex MFM-300(Al2)·3.2CO2 studied by in situ PXRD at 273 K, the adsorbed CO2 molecules interact via hydrogen bonding with the framework Al–OH groups.24 We therefore monitored in MFM-300(Ga2) both the ν(OH) stretch of the MOF host and the combination bands of CO2 in the region 3500–3800 cm–1.36 The fundamental antisymmetric CO2 stretch at 2348 cm–1 has too great an absorbance at pressures above 0.2 bar in our experiment to be used as an effective probe, but the higher energy combination bands, centered at 3714 and 3612 cm–1 for gaseous CO2, are much less intense and hence more readily studied at higher uptake capacities of CO2. The activated MFM-300(Ga2) crystals show in the absence of CO2 the ν(OH) stretch at 3660 cm–1, which shifts ∼7 cm–1 to lower energy to 3653 cm–1 on increasing the partial pressure of CO2 from 0 to 1 bar (Figure 5). The difference spectra (Figure S17) clearly show a loss of the original band and the formation of a new, lower energy band, consistent with the formation of a hydrogen bond between −OH and adsorbed CO2 molecules in MFM-300(Ga2). Unsurprisingly, the combination bands of gaseous CO2 increase in intensity as the CO2 pressure increases and, on subtracting these bands from the spectra, two new lower-energy bands are revealed at 3696 and 3592 cm–1. These new bands grow concurrently with the change in the −OH absorption and are consistent with the formation of an adsorbed CO2 species in MFM-300(Ga2). We have also examined the adsorbed CO2 concentration in MFM-300(Ga2) and the changes in the −OH vibration by fitting the peaks in this region to Lorentzian line shapes. The plot of peak area vs partial pressure of CO2 (Figure S18), normalized such that the final absorption of each peak is set to 1, is entirely consistent with the isotherms obtained by gas sorption experiments and confirms that the formation of the lower energy ν(OH) band is concurrent with the rise in the adsorbed CO2 combination bands. A new absorption band was also observed at 2275 cm–1 (data not shown here), which we attribute as the fundamental vibration of adsorbed CO2, but the rest of that region (2280–2400 cm–1) is obscured by the absorbance for free CO2 gas.
Figure 5.

In situ FTIR spectra of a single crystal of MFM-300(Ga2) in the ν(OH) and CO2 combination band region on increasing the partial pressure of CO2 gas from 0 to 1 bar. The contribution from gaseous CO2 has been subtracted with reference to the blank cell as a function of CO2 loading.
To probe further the Ga–OH···O=C=O interaction within the framework, we studied a single crystal of MFM-300(Ga2) with well-defined faces using polarized IR spectroscopy. IR spectra measured using a synchrotron radiation source, which gives approximately 100 times higher photon flux density over typical laboratory sources, have much improved signal-to-noise ratios. This allows the accurate measurement of IR spectra with a polarized beam to study the orientation of the vibration modes with respect to the functional groups. The microscope was used to locate a single crystal that appeared to be lying flat on the stage window, and spectra were recorded of the sample after activation (under He) and under 1 bar of CO2. Two orthogonal directions of polarized light were employed, both parallel to a crystal edge (Figure 6c), and the spectra are shown in Figure 6a. In the absence of CO2, the IR spectrum recorded with polarization along the ab plane of the crystal (P1 as shown in Figure 6c) shows a single intense ν(OH) stretch at 3660 cm–1, as observed in the unpolarized spectra. The spectrum recorded with polarization P2 (oriented 90° to P1, along c-axis) is markedly different, with an almost complete absence of the ν(OH) absorption. In the crystal structure, all the −OH groups in the framework are found pointing into the channels (in the ab plane), with no component along the c-axis (along the channels). Therefore, the polarized IR results are consistent with the polarization in P1 being aligned with the ab plane containing the −OH groups and P2 being aligned along the c-axis of the structure, orthogonal to the −OH groups and hence hardly interacting with the −OH dipole. Having identified the directionality of the crystal, the same orientations were investigated on loading the sample with 1 bar of CO2 (Figure 6b). With polarization P1, the −OH vibration is still intense and, as with the unpolarized spectra, is shifted to lower energy (accompanied by a degree of peak broadening), indicative of a hydrogen bonding interaction with adsorbed CO2. The CO2 combination bands are again both present at 3696 and 3591 cm–1; the higher energy CO2 band is a combination of the ν1 and ν3 symmetric and antisymmetric vibrations,36 both of which are oriented along the O=C=O bond axis, leading to the potential assignment of the 3696 cm–1 band in the P1 polarized IR spectrum to the presence of a CO2 molecule hydrogen-bonded in an “end-on” fashion to and aligned along the same axis as the −OH groups. This combination band has a lower relative absorbance as a fraction of the ν(OH) band when compared with the unpolarized spectra peaks [A3696:A(OH)3660 = 0.14:1 with polarization P1 vs A3696:A(OH)3660 = 0.28:1 with unpolarized IR light], suggesting that there must be additional adsorbed CO2 present in other positions in the pore, not aligned with this polarization. Indeed, on rotating the polarization to orientation P2, the CO2-loaded spectrum changes significantly (Figure 6b). As in the activated sample, the ν(OH) stretch at 3660 cm–1 is almost completely removed, although this time a very weak, but well-defined, band at 3657 cm–1 remains (with approximately 10% of the absorbance of the corresponding peak observed for P1). The presence of this residual band indicates a degree of bending of the −OH group out of the ab plane, as a direct result of the interactions with adsorbed CO2 adjacent to the −OH group. Most notably, the CO2 combination bands increase in absorbance by a factor of ∼1.6 compared with the same bands observed at P1, commensurate with the presence of CO2 molecules in an orientation much better aligned with the P2 polarization than P1. This suggests the presence of a second (or more) binding site within the framework and that, at 1 bar, the CO2 molecules at the second site have higher occupancy than the end-on CO2 molecules hydrogen-bonded to the −OH groups. We thus sought to investigate this further by gaining more insight into the adsorption sites within the porous host by crystallographic methods.
Figure 6.

In situ polarized FTIR spectra of a single crystal of MFM-300(Ga2) in the ν(OH) and CO2 combination band region, (a) under 1 bar He and (b) under 1 bar of CO2 (the contribution from gaseous CO2 has been subtracted), recorded at two orthogonal polarizations P1 (red) and P2 (blue), each aligned with a crystal edge. Arrows indicate the direction of changes on rotating polarization from P1 to P2. The face index of the single crystal which was used for this study is shown in (c). P2 is along the c-axis, and P1 is along the ab plane.
In Situ Single-Crystal Diffraction Studies
To determine the preferred binding sites for adsorbed CO2 molecules in MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13), single crystals of as-synthesized MFM-300(Ga2)-solv and MFM-300(Ga1.87Fe0.13)-solv were studied using an in situ gas cell system on a synchrotron radiation source. Upon degassing at 393 K under high vacuum (10–4 mbar), the desolvated materials MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13) show cell volume contractions ΔV/V of 2.1% and 0.65%, respectively. The relatively small contraction for MFM-300(Ga1.87Fe0.13) is due to the synthesized crystals prepared on a SiO2 surface substrate being partially dried during the sample transfer before the experiment. No electron density was found in the pore for these desolvated samples, confirming the effectiveness of the activation procedure and the retention of the framework structure which incorporates uncoordinated −OH groups within the pore (Figures 7 and 8). The activated crystals were then exposed to 1 bar of CO2 at 195 K, and the diffraction data were collected for the resulting CO2-loaded crystal. Significant residual electron density was now found in the pore by difference Fourier map analysis and was sequentially assigned as two independent CO2 molecules populating at sites I (O1s=C1s=O2s) and II (O3s=C2s=O4s).
Figure 7.

Views of single-crystal structures of (a, d) MFM-300(Ga2)-solv; (b, e) MFM-300(Ga2) activated at 393 K and vacuum 10–4 mbar for 4 h; (c, f) MFM-300(Ga2)·2.35CO2, loaded with CO2 at 1 bar at 195 K for 2 h. The guest molecules (DMF, water, CO2) in the channels are highlighted using a ball-and-stick style (Ga: green; C: gray; O: red; H: white; N: blue). The carbon atom of CO2II is highlighted in orange. The hydrogen bonding interaction between the guest molecule and free −OH group is highlighted in purple. The electrostatic dipole interactions between CO2 molecules are highlighted in cyan (O3s···C1s) and green (O2s···C2s).
Figure 8.

Views of single-crystal structures of (a, d) MFM-300(Ga1.87Fe0.13)-solv; (b, e) MFM-300(Ga1.87Fe0.13), activated at 393 K and vacuum 10–4 mbar for 4 h; (c, f) MFM-300(Ga1.87Fe0.13)·2.0CO2, loaded with CO2 at 1 bar at 195 K for 2 h. The guest molecules (DMF, water, CO2) in the channels are highlighted as ball-and-stick models (Ga: green; C: gray; O: red; H: white; N: blue). The carbon atom of CO2II is highlighted in orange. The hydrogen bonding interaction between the guest molecule and free −OH group is highlighted in purple. The electrostatic dipole interactions between CO2 molecules are highlighted in cyan (O3s···C1s) and green (O2s···C2s).
In MFM-300(Ga2), the occupancies for these CO2 molecules at sites I and II refined to values of 0.43(3) and 0.74(3), respectively, yielding the overall formula [Ga2(OH)2(C16H6O8)]·2.35CO2 for the CO2-loaded material. This result is in excellent agreement with the experimental uptake from the CO2 isotherm at 195 K (1.18 CO2 per Ga). The CO2 at site I is ordered and binds to the −OH group in an end-on fashion via a moderate hydrogen bonding interaction [H···O1s = 1.883(10) Å; ∠O–H···O1s = 180°], entirely consistent with the interaction observed by the IR spectroscopy. Interestingly, this hydrogen bond distance is much shorter than that observed in the MFM-300(Al2)·3.2CO2 system [H···O = 2.376(13) Å; ∠O–H···O = 180°] studied by in situ PXRD at 273 K,28 indicating the formation of a stronger hydrogen bonding interaction in MFM-300(Ga)·2.35CO2. Given that MFM-300(Ga2) and MFM-300(Al2) have the same framework structure and pore surface chemistry, this difference in hydrogen bond length is most likely due to the different metal center (Al or Ga) affecting the relative acidity of the M–OH (M = Al, Ga) group. In addition to this hydrogen bond, O1s also forms weak supramolecular contacts with aromatic hydrogen atoms from the phenyl rings [O1s···H = 2.98(4), 3.14(3) Å, each occurring twice]. The CO2 at site II is disordered over two equally occupied positions. In contrast to CO2I, CO2II does not interact directly with framework atoms, instead forming two weak electrostatic dipole interactions with CO2I, between the electropositive C and electronegative O centers [C1s···O3s = 3.85(4) Å; C2s···O2s = 4.39(4) Å]. Significantly, this pattern of intermolecular interactions is distinct from the traditional “T-shaped” dipole interaction observed in solid CO2, in [Zn2(atz)2(ox)]·1.3CO2 (atz– = 3-amino-1,2,4-triazolate; ox2– = oxalate)6 and in MFM-300(Al2)·3.2CO2,24 and is, for the first time, determined here. Furthermore, both the position and the relative occupancy of these sites are in excellent agreement with the experimentally observed polarized IR absorbances for adsorbed CO2 aligned with and perpendicular to the −OH axis at room temperature. CO2 molecules at site I are aligned with the direction of P1, and CO2 molecules at site II, while not perfectly orthogonal to site I, have a significantly better alignment with P2 than P1. The increase in IR absorbance of the higher energy CO2 combination band (factor of 1.6) in P2 compared with P1 is remarkably close to the crystallographically observed ratio of site occupancies (II:I = 1.7:1), both experiments having been carried out at 1 bar CO2 pressure.
In contrast, the occupancies of the CO2 molecules at sites I and II in MFM-300(Ga1.87Fe0.13) both refined to values of 0.50 (1), yielding a formula of [Ga1.87Fe0.13(OH)2(C16H6O8)]·2.0CO2 for the CO2-loaded material. However, this result is lower than the experimental uptake from the CO2 isotherm at 195 K, which implies the formation of MFM-300(Ga1.87Fe0.13)·2.9CO2. The discrepancy could be due to two factors: (i) the kinetic effect for the population of CO2 molecules within the whole single crystal; (ii) some of the faces of the single crystals could have been blocked or damaged when it was removed from the surface substrate, thereby reducing the diffusion rate for CO2 molecules. Nevertheless, this discrepancy does not prevent us from determining the binding mechanisms. The CO2 at site I is ordered and binds to the −OH group in an end-on fashion via a weak hydrogen bonding interaction [H···O1s = 2.259(12) Å, ∠O–H···O1s = 180°]. Interestingly, this hydrogen bond distance is longer than that observed in the MFM-300(Ga2)·2.35CO2 system [H···O1s = 1.883(10) Å], indicating the formation of a weaker hydrogen bonding interaction in MFM-300(Ga1.87Fe0.13)·2.0CO2. This is consistent with the observed heat of adsorption which also decreases, indicating a weaker interaction between the mixed-metal material and the CO2 molecule. Given the identical framework structures and pore surface chemistry of MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13), this difference in hydrogen bonding is a direct result of Fe-doping into the framework structure. In addition to this hydrogen bond, O1s also forms weak supramolecular contacts with aromatic hydrogen atoms from the phenyl rings [O1s···H = 3.055(4), 3.163(3) Å, each occurring twice]. The CO2 at site II is disordered over two equally-occupied positions. However, CO2II adopts a different orientation to that observed in MFM-300(Ga2)·2.35CO2, forming a typical “T-shaped” dipole interaction with CO2I between the electropositive C center and electronegative O center [C1s···O3s = 3.207(6) Å, occurring twice]. Thus, the small percentage of Fe-doping into this solid solution has a significant effect on the CO2 binding details, including the formation of both adsorbate–adsorbent hydrogen bonds and adsorbate–adsorbate intermolecular dipole interactions. We could not of course distinguish between Ga and Fe centers in the above experiments, reflecting also the random distribution of Fe within the Ga-based host.
We have thus determined the detailed binding of CO2 in two isostructural MOFs and have confirmed the formation of hydrogen bonding, C–H and O–H supramolecular contacts, and the complementary dipole interactions and cooperative binding of CO2 molecules within the pores to form one-dimensional chains of CO2 molecules. In both CO2-loaded samples, the C=O distances lie within the range 1.176(8)–1.207(5) Å, consistent with typical C=O distances [1.155(1) Å] observed in solid CO2. The OCO angles in CO2I/CO2II are 180°/160(4)° and 180°/165(1)° for MFM-300(Ga2)·2.35CO2 and MFM-300(Ga1.87Fe0.13)·2.0CO2, respectively. The slight bending of the CO2II molecules is probably an artifact of the disorder rather than any true distortion in bonding and is comparable to that observed in [Zn2(Atz)2(ox)]·1.3CO2.6
Catalytic Reactions
Two general reactions requiring acid catalysis, namely epoxide ring-opening and the acetylation of benzaldehyde, were selected to determine the influence of the Fe-doping on the catalytic activity of MFM-300(Ga2). In the first example, the catalytic activity of as-synthesized MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13) was studied in the ring-opening of styrene oxide (1a) with methanol to yield 2-methoxy-2-phenylethanol. In the absence of catalyst, we observed 4% conversion of 1a to 2-methoxy-2-phenylethanol after 30 h at 40 °C. In contrast, the presence of heterogeneous MOF catalyst resulted in significant conversions: MFM-300(Ga2) showed 40% conversion of 1a to 2-methoxy-2-phenylethanol in 30 h while MFM-300(Ga1.87Fe0.13) resulted almost in complete conversion of 1a in the same time (Figure 9). Both materials showed no leaching of active sites, as evidenced from the hot filtration test, in which the solid catalyst was removed from the reaction mixture at 40 °C at conversions of about 20% and the clear solution in the absence of solid was then allowed to react further; no further conversion was then observed after 30 h. The stability of MFM-300(Ga1.87Fe0.13) as a solid catalyst was studied by cycling it under identical conditions at 40 °C. The percentage conversion of 1a in the first, second, and third cycles was 98, 93 and 83, respectively, in 30 h (Table 2) reflecting some gradual deactivation of the catalyst with time.
Figure 9.
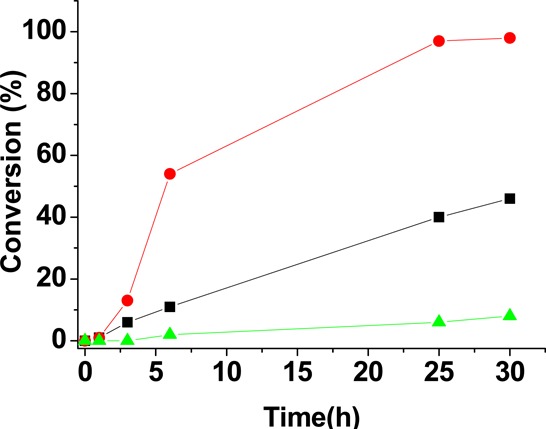
Time conversion plots for the ring-opening of 1a with MeOH using MFM-300(Ga2) (black squares) and MFM-300(Ga1.87Fe0.13) (red circles) as catalysts, and ring-opening of 1a with EtOH using MFM-300(Ga1.87Fe0.13) (green triangles) as catalyst.
Table 2. Ring-Opening of 1a to 2-Methoxy-2-phenylethanol Catalyzed by MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13) Catalystsa.
| run | catalyst | time (h) | conversion (%)b | selectivity (%)b |
|---|---|---|---|---|
| 1 | no catalyst | 30 | 4 | 98 |
| 2 | Ga2 | 30 | 46 | 97 |
| 3 | Ga2c | 6–30 | 10–12 | 97 |
| 4 | (Ga1.87Fe0.13) | 30 | 98 | 98 |
| 5 | (Ga1.87Fe0.13)d | 3–30 | 11–15 | 97 |
| 6 | (Ga1.87Fe0.13)e | 30 | 93 | 97 |
| 7 | (Ga1.87Fe0.13)f | 30 | 83 | 97 |
Reaction conditions: 1a (2.08 mmol), catalyst (20 mg) activated at 100 °C for 2 h under vacuum, methanol (5 mL), 40 °C.
Determined by GC using nitrobenzene as internal standard.
Catalyst filtered at 6 h and the reaction continued to 30 h.
Catalyst filtered at 3 h and the reaction continued to 30 h.
Second reuse.
Third reuse.
We have also studied the reaction of styrene oxide with other alcohols such as ethanol and tert-butanol. As shown in Table 3, MeOH reacts more quickly than EtOH, whereas tert-BuOH showed no reaction even after 30 h. This lack of reactivity with tert-BuOH may be due to the impeded diffusion at low temperature of this larger molecule through the pore system of the MOF, and similar results have been reported for other MOFs and with graphene oxide as heterogeneous catalysts.37 This result suggests that the catalytic sites reside in the pore rather than on the surface. The porous nature of the MOFs used in this study also plays a role in determining the reactivity of other substrates and nucleophiles, as shown in Table 3, where the scope of the catalytic activity of MFM-300(Ga1.87Fe0.13) was screened. In most of the cases using MFM-300(Ga1.87Fe0.13) as catalyst, the reaction exhibits high regioselectivity toward a single regioisomer. The alcoholysis of 2,3-epoxypropylbenzene (1b) with MFM-300(Ga1.87Fe0.13) resulted in 7% conversion with 88% selectivity for 3b, the observed regioselectivity probably reflecting the steric hindrance caused by the phenyl ring to the direction of methanol attack. Similar reactivity was observed for 1,2-epoxy-3-phenoxypropane (1c), giving 3c as the major isomer with respect to 2c. In the case of 1-hexene oxide (1d), ring-opening of epoxide using methanol as solvent favored isomer 2d over 3d. Epichlorohydrin (1e) showed 13% conversion with 3e as the major isomer, suggesting a directing inductive effect of the chloride group on the position of the attack by MeOH. The methanolysis of cyclohexene oxide (1f) resulted in 12% conversion within 30 h to 2-methoxycyclohexanol, 2f with 97% selectivity. Finally, norbornene oxide (1h) exhibited only trace conversion even after a prolonged reaction time. Overall, the unoptimized catalytic data (Table 3) show that MFM-300(Ga1.87Fe0.13) exhibits general activity as a solid Lewis acid catalyst.
Table 3. Ring-Opening of Epoxides with Various Substrates Catalyzed by MFM-300(Ga1.87Fe0.13) Using Different Nucleophilesa.


Reaction conditions: epoxide (0.250 mL), MFM-300(Ga1.87Fe0.13) activated at 100 °C for 2 h under vacuum (20 mg), methanol (5 mL), 30 h, 40 °C.
Determined by GC.
Selectivity refers to 2-methoxycyclohexanol.
To differentiate between SN1 and SN2 reaction mechanisms, 1,2-epoxy-2-methylpropane (1g) was selected as a substrate and the nucleophilic ring-opening of this epoxide was carried out with MeOH. Formation of the less-substituted alcohol 2g (90%) as the major product over the more-substituted regioisomer 3g (10%) clearly indicates that the reaction proceeds predominantly via an SN1 mechanism, with a transition state in which a considerable positive charge density has developed on the epoxide carbon atom as a consequence of the interaction of the epoxide oxygen atom with the catalyst acid sites.
Having demonstrated the catalytic activity of MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13) in the ring-opening of epoxides, we further tested them in the acetylation of benzaldehyde by MeOH. In the absence of solid catalyst, acetylation leads to 24% conversion to 1,1-dimethoxytoluene after 30 h at room temperature (Figure 10). The presence of MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13) significantly increased the conversion to 1,1-dimethoxytoluene. The two catalysts show almost identical activity, as shown by almost identical conversions of 71% and 72%, respectively, at 30 h. MFM-300(Ga1.87Fe0.13) was also tested in a leaching experiment. The reaction was started in the presence of MFM-300(Ga1.87Fe0.13), and the solid catalyst was filtered after 4 h with 11% conversion of benzaldehyde. The reaction mixture was then allowed to react further in the absence of catalyst. After 30 h, 39% (instead of 72%) of benzaldehyde was converted, consistent with the expected total conversion for the uncatalyzed reaction based on the data from blank controls in the absence of catalyst. The similar reactivity exhibited by these two MOFs could be followed by in situ IR spectroscopic analysis using CO as probe molecule (Figure 11), which showed that both MOFs contained a similar number of active sites. The IR band at 2165 cm–1 could be due to Lewis acid sites, whereas the IR band at 2145 cm–1 is attributed to hydroxyl groups. As confirmed by Figure 11, the majority of the centers interacting with CO are Brönsted acid sites, and since MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13) have similar populations of acid sites and the demand of acid strength on the benzaldehyde acetalization is low, both solids should exhibit very similar catalytic activity, as is experimentally observed. Differences in catalytic activity between MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13) are expected for other reactions such as SN1 epoxide ring-opening, which require higher acid strength. In this way, the combination of spectroscopic titration of acid sites using CO as a probe with the catalytic results for acetylation and epoxide ring-opening indicates that MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13) have the same population of acid sites, but the presence of Fe-doping increases the acid strength, enabling the use of MFM-300(Ga1.87Fe0.13) as catalyst for those reactions requiring sites with higher acid strength. This effect of Fe-doping increasing the strength of the acid sites contrasts with the weaker hydrogen bonding interaction between the framework and CO2 in single crystals of MFM-300(Ga1.87Fe0.13)·2.0CO2. It is, therefore, likely that the most active sites in catalysis for MFM-300(Ga1.87Fe0.13) are those present in defects, and these will be absent in highly crystalline samples of MFM-300(Ga2).
Figure 10.
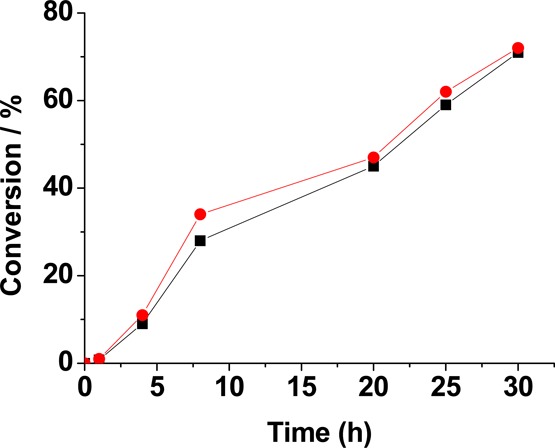
Time conversion plots for the acetylation of benzaldehyde with MeOH using MFM-300(Ga2) (black squares) and MFM-300(Ga1.87Fe0.13) (red circles) as catalysts. Reaction conditions: benzaldehyde (0.94 mmol), catalyst activated at 100 °C, 2 h (20 mg), MeOH (3 mL), room temperature.
Figure 11.
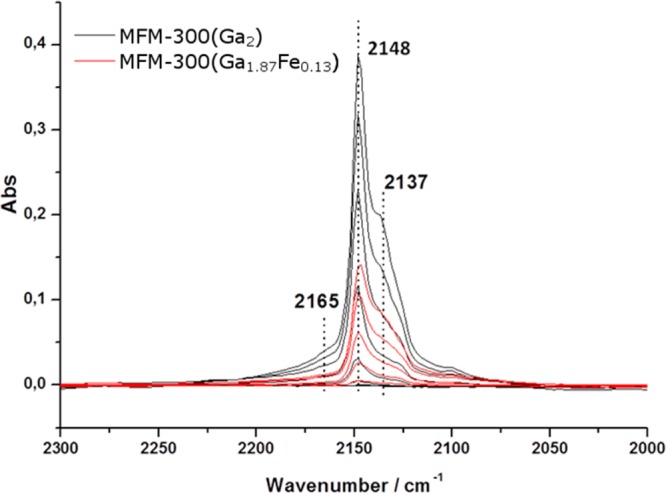
IR spectra of MFM-300(Ga2) and MFM-300(Ga1.87Fe0.13) after CO adsorption.
Conclusions
MFM-300(Ga2) and the isostructural mixed-metal solid solution MFM-300(Ga0.87Fe0.13) were prepared from homogeneous solvothermal reactions. Significant enhancement of CO2 adsorption capacity by up to 49% was observed by doping with Fe(III), reflecting the increased structural integrity of the Fe-doped material giving a more ordered material with more accessible pores. Thus, Fe-doping can be used to improve the materials properties of the host while also monitoring the effects of the heteroatom center within a parent framework without the requirement of synthesizing the isostructural MFM-300(Fe2). In situ single-crystal X-ray diffraction studies of CO2-loaded materials revealed, on a molecular level, key details into the preferred binding sites within the pores of these materials that were in excellent agreement with the results of the in situ polarized IR spectroscopic study of CO2-loaded MFM-300(Ga2). The adsorbed CO2 molecules in MFM-300(Ga2) hydrogen bond with the free −OH groups on the surface of the pores, and a previously unobserved pattern of intermolecular dipole interactions was found to stabilize the two CO2 molecules within the pores. The CO2 molecule at site I binds to the −OH group in an end-on fashion [H···O1s = 1.883(10) Å] and also interacts with the CO2 molecule at site II. In contrast, the adsorbed CO2 molecules in MFM-300(Ga1.83Fe0.13) form only very weak hydrogen bonds with free −OH groups [H···O1s = 2.259(12) Å], and a traditional “T-shaped” CO2 intermolecular dipole interaction was responsible for the stability of the CO2 molecular chain. The reduction on the strength of the hydrogen bond is also reflected in the isosteric heat of adsorption for CO2 uptake. In addition to the enhancement of the gas adsorption properties, Fe-doping has also shown a positive effect on specific catalytic reactions such as the ring-opening reactions of styrene oxide. We anticipate that the strategy developed here, based upon mixing transition and main group metal nodes in MOFs, can lead to the design and discovery of new materials with improved properties and functions.
Acknowledgments
We thank the Universities of Nottingham and Manchester for support. M.S. acknowledges receipt of an ERC Advanced Grant and EPSRC Programme Grant. We also thank EPSRC for funding of X-ray equipment. We acknowledge the use of instrumentation within the Nottingham Nanotechnology and Nanoscience Centre, and thank Dr. Christopher Parmenter for assistance. We thank Conacyt, Mexico, for funding to C.P.K. We are especially grateful to Diamond Light Source for access to Beamlines I11 (EE8943), I19 (MT7548, MT8448, MT8937), and B22 (SM11279).
Glossary
List of Materials
- MFM-300(Ga2)-solv
[Ga2(OH)2(C16H6O8)]·1.6DMF·4H2O
- MFM-300(Ga2)
[Ga2(OH)2(C16H6O8)]
- MFM-300(Ga2)·2.35CO2
[Ga2(OH)2(C16H6O8)]·2.35CO2
- MFM-300(Ga1.87Fe0.13)-solv
[Ga1.87Fe0.13(OH)2(C16H6O8)]·2.7H2O
- MFM-300(Ga1.87Fe0.13)
[Ga1.87Fe0.13(OH)2(C16H6O8)]
- MFM-300(Ga1.87Fe0.13)·2.0CO2
[Ga1.87Fe0.13(OH)2(C16H6O8)]·2.0CO2
- MFM-310(Fe)
[Fe3O1.5(OH)(HL)(L)0.5(H2O)3.5]
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.5b02108.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhou H.-C.; Long J. R.; Yaghi O. M. Chem. Rev. 2012, 112, 673–674. 10.1021/cr300014x. [DOI] [PubMed] [Google Scholar]
- Long J. R.; Yaghi O. M. Chem. Soc. Rev. 2009, 38, 1213–1214. 10.1039/b903811f. [DOI] [PubMed] [Google Scholar]
- Lin X.; Champness N. R.; Schröder M. Top. Curr. Chem. 2009, 293, 35–76. 10.1007/128_2009_21. [DOI] [PubMed] [Google Scholar]
- Furukawa H.; Kim J.; Ockwig N. W.; O’Keeffe M.; Yaghi O. M. J. Am. Chem. Soc. 2008, 130, 11650–11661. 10.1021/ja803783c. [DOI] [PubMed] [Google Scholar]
- Ramsahye N. A.; Trung T. K.; Bourrelly S.; Yang Q.; Devic T.; Maurin G.; Horcajada P.; Llewellyn P. L.; Yot P.; Serre C.; Filinchuk Y.; Fajula F.; Férey G.; Trens P. J. Phys. Chem. C 2011, 115, 18683–18695. 10.1021/jp205369t. [DOI] [Google Scholar]
- Vaidhyanathan R.; Iremonger S. S.; Shimizu G. K. H.; Boyd P. G.; Alavi S.; Woo T. K. Science 2010, 330, 650–653. 10.1126/science.1194237. [DOI] [PubMed] [Google Scholar]
- a McDonald T. M.; Lee W. R.; Mason J. A.; Wiers B. M.; Hong C. S.; Long J. R. J. Am. Chem. Soc. 2012, 134, 7056–7065. 10.1021/ja300034j. [DOI] [PubMed] [Google Scholar]; b Hu Y.; Verdegaal W. M.; Yu S. H.; Jiang H. L. ChemSusChem 2014, 7, 734–737. 10.1002/cssc.201301163. [DOI] [PubMed] [Google Scholar]
- Demessence A.; D’Alessandro D. M.; Foo M. L.; Long J. R. J. Am. Chem. Soc. 2009, 131, 8784–8785. 10.1021/ja903411w. [DOI] [PubMed] [Google Scholar]
- a Vougo-Zanda M.; Huang J.; Anokhina E.; Wang X.; Jacobson A. J. Inorg. Chem. 2008, 47, 11535–11542. 10.1021/ic800008f. [DOI] [PubMed] [Google Scholar]; b Ramsahye N. A.; Maurin G.; Bourrelly S.; Llewellyn P. L.; Serre C.; Loiseau T.; Devic T.; Férey G. J. Phys. Chem. C 2008, 112, 514–520. 10.1021/jp075782y. [DOI] [Google Scholar]; c Yan Y.; Yang S.; Blake A. J.; Schröder M. Acc. Chem. Res. 2014, 47, 296–307. 10.1021/ar400049h. [DOI] [PubMed] [Google Scholar]; d Du L.; Yang S.; Xu L.; Mina H.; Zheng B. CrystEngComm 2014, 16, 5520–5523. 10.1039/c4ce00530a. [DOI] [Google Scholar]; e Zou Y.; Yu C.; Li Y.; Lah M. S. CrystEngComm 2012, 14, 7174–7177. 10.1039/c2ce26109j. [DOI] [Google Scholar]; f Lee Y. G.; Moon H.-R.; Cheon Y. E.; Suh M. P. Angew. Chem., Int. Ed. 2008, 47, 7741–7745. 10.1002/anie.200801488. [DOI] [PubMed] [Google Scholar]; g Zheng B.; Bai J.; Duan J.; Wojtas L.; Zaworotko M. J. J. Am. Chem. Soc. 2011, 133, 748–751. 10.1021/ja110042b. [DOI] [PubMed] [Google Scholar]
- a Ibarra I. A.; Lin X.; Yang S.; Blake A. J.; Walker G. S.; Barnett S. A.; Allan D. R.; Champness N. R.; Hubberstey P.; Schröder M. Chem.—Eur. J. 2010, 16, 13671–13679. 10.1002/chem.201000926. [DOI] [PubMed] [Google Scholar]; b Yang S.; Liu L.; Sun J.; Thomas K. M.; Davies A. J.; George M. W.; Blake A. J.; Hill A. H.; Fitch A. N.; Tang C. C.; Schröder M. J. Am. Chem. Soc. 2013, 135, 4954–4957. 10.1021/ja401061m. [DOI] [PubMed] [Google Scholar]; c Hong D.-Y.; Hwang Y. K.; Serre C.; Férey G.; Chang J.-S. Adv. Funct. Mater. 2009, 19, 1537–1552. 10.1002/adfm.200801130. [DOI] [Google Scholar]; d Dhakshinamoorthy A.; Alvaro M.; Chevreau H.; Horcajada P.; Devic T.; Serre C.; Garcia H. Catal. Sci. Technol. 2012, 2, 324–330. 10.1039/C2CY00376G. [DOI] [Google Scholar]; e Volkringer C.; Loiseau T.; Guillou N.; Férey G.; Elkaïm E. Solid State Sci. 2009, 11, 1507–1512. 10.1016/j.solidstatesciences.2009.05.017. [DOI] [Google Scholar]; f Savage M.; Yang S.; Suyetin M.; Bichoutskaia E.; Lewis W.; Blake A. J.; Barnett S. A.; Schröder M. Chem.—Eur. J. 2014, 20, 8024–8029. 10.1002/chem.201304799. [DOI] [PubMed] [Google Scholar]
- a Chaplais G.; Simon-Masseron A.; Porcher F.; Lecomte C.; Bazer-Bachi D.; Bats N.; Patarin J. Phys. Chem. Chem. Phys. 2009, 11, 5241–5245. 10.1039/b822163d. [DOI] [PubMed] [Google Scholar]; b Loiseau T.; Muguerra H.; Haouas M.; Taulelle F.; Férey G. Solid State Sci. 2005, 7, 603–609. 10.1016/j.solidstatesciences.2005.01.014. [DOI] [Google Scholar]; c Volkringer C.; Loiseau T.; Guillou N.; Férey G.; Popov D.; Burghammer M.; Riekel C. Solid State Sci. 2013, 26, 38–44. 10.1016/j.solidstatesciences.2013.09.010. [DOI] [Google Scholar]; d Volkringer C.; Loiseau T.; Férey G.; Morais C. M.; Taulelle F.; Montouillout V.; Massiot D. Microporous Mesoporous Mater. 2007, 105, 111–117. 10.1016/j.micromeso.2007.05.018. [DOI] [Google Scholar]
- Furukawa H.; Ko N.; Go Y. B.; Aratani N.; Choi S. B.; Choi E.; Yazaydin A. Ö.; Snurr R. Q.; O'Keeffe M.; Kim J.; Yaghi O. M. Science 2010, 329, 424–428. 10.1126/science.1192160. [DOI] [PubMed] [Google Scholar]
- Kettner F.; Worch C.; Moellmer J.; Gläser R.; Staudt R.; Krautscheid H. Inorg. Chem. 2013, 52, 8738–8742. 10.1021/ic400857s. [DOI] [PubMed] [Google Scholar]
- Deng H.; Doonan C. J.; Furukawa H.; Ferreira R. B.; Towne J.; Knobler C. B.; Wang B.; Yaghi O. M. Science 2010, 327, 846–850. 10.1126/science.1181761. [DOI] [PubMed] [Google Scholar]
- Wang L. J.; Deng H.; Furukawa H.; Gándara F.; Cordova K. E.; Peri D.; Yaghi O. M. Inorg. Chem. 2014, 53, 5881–5883. 10.1021/ic500434a. [DOI] [PubMed] [Google Scholar]
- Breeze M. I.; Clet G.; Campo B. C.; Vimont A.; Daturi M.; Greneche J.-M.; Dent A. J.; Millange F.; Walton R. I. Inorg. Chem. 2013, 52, 8171–8182. 10.1021/ic400923d. [DOI] [PubMed] [Google Scholar]
- Fukushima T.; Horike S.; Inubushi Y.; Nakagawa K.; Kubota Y.; Takata M.; Kitagawa S. Angew. Chem., Int. Ed. 2010, 49, 4820–4824. 10.1002/anie.201000989. [DOI] [PubMed] [Google Scholar]
- Yeung H. H. M.; Li W.; Saines P. J.; Köster T. K. J.; Grey C. P.; Cheetham A. K. Angew. Chem., Int. Ed. 2013, 52, 5544–5547. 10.1002/anie.201300440. [DOI] [PubMed] [Google Scholar]
- Nouar F.; Devic T.; Chevreau H.; Guillou N.; Gibson E.; Clet G.; Daturi M.; Vimont A.; Greneche J. M.; Breeze M. I.; Walton R. I.; Llewellyn P. L.; Serre C. Chem. Commun. 2012, 48, 10237–10239. 10.1039/c2cc35348b. [DOI] [PubMed] [Google Scholar]
- Lau C. H.; Babarao R.; Hill M. R. Chem. Commun. 2013, 49, 3634–3636. 10.1039/c3cc40470f. [DOI] [PubMed] [Google Scholar]
- Takei T.; Kawashima J.; Ii T.; Maeda A.; Hasegawa M.; Kitagawa T.; Ohmura T.; Ichikawa M.; Hosoe M.; Kanoya I.; Mori W. Bull. Chem. Soc. Jpn. 2008, 81, 847–856. 10.1246/bcsj.81.847. [DOI] [Google Scholar]
- Kaye S. S.; Long J. R. J. Am. Chem. Soc. 2005, 127, 6506–6507. 10.1021/ja051168t. [DOI] [PubMed] [Google Scholar]
- Xiao D. J.; Bloch E. D.; Mason J. A.; Queen W. L.; Hudson M. R.; Planas N.; Borycz J.; Dzubak A. L.; Verma P.; Lee K.; Bonino F.; Crocellà V.; Yano J.; Bordiga S.; Truhlar D. G.; Gagliardi L.; Brown C. M.; Long J. R. Nat. Chem. 2014, 6, 590–595. 10.1038/nchem.1956. [DOI] [PubMed] [Google Scholar]
- Yang S.; Sun J.; Ramirez-Cuesta A. J.; Callear S. K.; David W. I. F.; Anderson D. P.; Newby R.; Blake A. J.; Parker J. E.; Tang C. C.; Schröder M. Nat. Chem. 2012, 4, 887–894. 10.1038/nchem.1457. [DOI] [PubMed] [Google Scholar]
- Lin X.; Telepeni I.; Blake A. J.; Dailly A.; Brown C. M.; Simmons J. M.; Zoppi M.; Walker G. S.; Thomas K. M.; Mays T. J.; Hubberstey P.; Champness N. R.; Schröder M. J. Am. Chem. Soc. 2009, 131, 2159–2171. 10.1021/ja806624j. [DOI] [PubMed] [Google Scholar]
- Arnold M.; Kortunov P.; Jones D. J.; Nedellec Y.; Kärger J.; Caro J. Eur. J. Inorg. Chem. 2007, 2007, 60–64. 10.1002/ejic.200600698. [DOI] [Google Scholar]
- Thompson S. P.; Parker J. E.; Potter J.; Hill T. P.; Birt A.; Cobb T. M.; Yuan F.; Tang C. C. Rev. Sci. Instrum. 2009, 80, 075107. 10.1063/1.3167217. [DOI] [PubMed] [Google Scholar]
- Rietveld H. M. J. Appl. Crystallogr. 1969, 2, 65–71. 10.1107/S0021889869006558. [DOI] [Google Scholar]
- Sheldrick G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Spek A. L. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 148–155. 10.1107/S090744490804362X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozek C. K.; Cozzolino A. F.; Teat S. J.; Chen Y.; Dincǎ M. Chem. Mater. 2013, 25, 2998–3002. 10.1021/cm400858d. [DOI] [Google Scholar]
- Yuan D.; Getman G. B.; Wei Z.; Snurr R. Q.; Zhou H. Chem. Commun. 2012, 48, 3297–3299. 10.1039/c2cc17168f. [DOI] [PubMed] [Google Scholar]
- Sudik A. C.; Côté A. P.; Yaghi O. M. Inorg. Chem. 2005, 44, 2998–3000. 10.1021/ic050064g. [DOI] [PubMed] [Google Scholar]
- Saha D.; Zacharia R.; Lafi L.; Cossement D.; Chahine R. Chem. Eng. J. 2011, 171, 517–525. 10.1016/j.cej.2011.04.019. [DOI] [Google Scholar]
- Yang S.; Ramirez-Cuesta A. J.; Schröder M. Chem. Phys. 2014, 428, 111–116. 10.1016/j.chemphys.2013.11.004. [DOI] [Google Scholar]
- a Yang W.; Davies A. J.; Lin X.; Suyetin M.; Matsuda R.; Blake A. J.; Wilson C.; Lewis W.; Parker J. E.; Tang C. C.; George M. G.; Hubberstey P.; Kitagawa S.; Sakamoto H.; Bichoutskaia E.; Champness N. R.; Yang S.; Schröder M. Chem. Sci. 2012, 3, 2993–2999. 10.1039/c2sc20443f. [DOI] [Google Scholar]; Burch D. E.; Gryvnak D. A.; Williams D. Appl. Opt. 1962, 1, 759–765. 10.1364/AO.1.000759. [DOI] [Google Scholar]; b Grone A.; Kapphan S. J. Phys.: Condens. Matter 1995, 7, 3051–3061. 10.1088/0953-8984/7/15/012. [DOI] [Google Scholar]
- Dhakshinamoorthy A.; Alvaro M.; Concepcion P.; Fornes V.; Garcia H. Chem. Commun. 2012, 48, 5443–5445. 10.1039/c2cc31385e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.