

Abstract
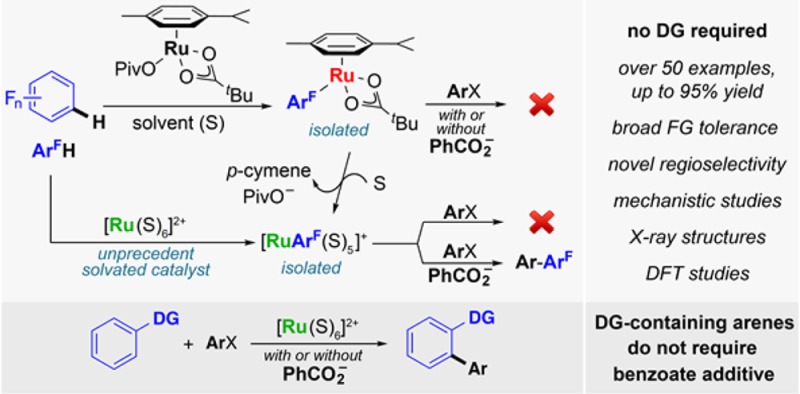
Although the ruthenium-catalyzed C–H arylation of arenes bearing directing groups with haloarenes is well-known, this process has never been achieved in the absence of directing groups. We report the first example of such a process and show that unexpectedly the reaction only takes place in the presence of catalytic amounts of a benzoic acid. Furthermore, contrary to other transition metals, the arylation site selectivity is governed by both electronic and steric factors. Stoichiometric and NMR mechanistic studies support a catalytic cycle that involves a well-defined η6-arene-ligand-free Ru(II) catalyst. Indeed, upon initial pivalate-assisted C–H activation, the aryl-Ru(II) intermediate generated is able to react with an aryl bromide coupling partner only in the presence of a benzoate additive. In contrast, directing-group-containing substrates (such as 2-phenylpyridine) do not require a benzoate additive. Deuterium labeling and kinetic isotope effect experiments indicate that C–H activation is both reversible and kinetically significant. Computational studies support a concerted metalation–deprotonation (CMD)-type ruthenation mode and shed light on the unusual arylation regioselectivity.
1. Introduction
The development of new synthesis tools for the production of biologically and industrially relevant compounds fuels the efforts of the chemistry community. In this context, the biaryl motif is ubiquitous among a wide range of compounds, many of them of industrial importance.1 Transition-metal-catalyzed cross-coupling reactions are the most powerful methods for the construction of the biaryl skeleton.2 Although highly effective, this approach requires prefunctionalization of both coupling partners. In recent years, transition-metal-catalyzed C–H activation has emerged as a promising route toward the direct functionalization of organic compounds.3,4 In particular, direct C–H arylation, the coupling of an arene (CAr–H) with an aryl halide (CAr–X), is developing as an atom- and step-economical and environmentally friendly approach to biaryls. Although using a C–H bond as a functional group surrogate is highly attractive, controlling which C–H site is activated represents a considerable challenge.4,5 The installation of a directing group (DG) on the arene, capable of coordinating the metal catalyst and facilitating C–H activation via cyclometalation, is often the most reliable option to enhance reactivity of a particular C–H bond.4h,4i However, the DG is seldom a necessity after the C–H functionalization event and may have to be removed, decreasing the overall atom economy of the method.
Among all the transition metals, palladium has been the most studied to date. Nevertheless, the chemistry of ruthenium, which has lagged behind that of palladium by almost a quarter of a century, is growing exponentially and is revealing itself to be a viable alternative.4h,6 The use of ruthenium can represent a significant economic advantage because it is more than 10 times cheaper ($258/mol) than palladium ($2798/mol).7 Developing the coordination chemistry of ruthenium allows the foremost primary and basic mechanistic factors behind the discovery of new chemical transformations to be better understood, potentially accessing unique reactivities and selectivities.
In the context of C–H activation, DGs have consistently been required for the functionalization of arenes by ruthenium ever since Lewis and Smith reported the first Ru-catalyzed C–C bond formation in 1986.8 Murai, Chatani, Kakiuchi et al., initiated the use of ruthenium(0) catalyst precursors, showing the utility of ortho-ruthenated species as catalytic intermediates for hydroalkylation and hydroalkenylation,9 silylation,10 and arylation with arylboronic esters.11 More recently, significant progress has been made considering the direct arylation of arenes possessing a chelating group, employing ruthenium(II) catalysts, by the groups of Oi,12 Ackermann,13 Maseras and Dixneuf.14
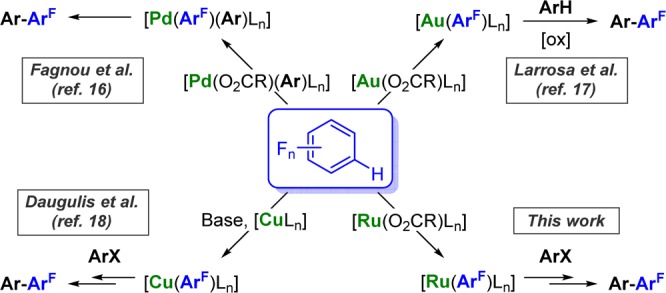
An alternative method for site-selective C–H activation is utilizing the intrinsic reactivity of a given arene.5c,15 In particular, studies with Pd,16 Au,17 and Cu18 have revealed that C–H activation on fluoroarenes occurs preferentially at the most acidic C–H bond (Scheme 1). Fundamental studies on Pd-catalyzed C–H arylation led to the hypothesis that the C–H cleavage is assisted by basic ligands such as carboxylates or carbonates in a concerted metalation-deprotonation.16,19 The groups of Maseras and Dixneuf14 and that of Ackermann20,21 also reported the benefits of such additives in aiding the chelation-assisted ruthenation of arenes. Several Ru-catalyzed C–H functionalization reactions have been reported on simple arenes, including hydroarylation of alkenes and alkynes, allylation, and hydroxylation, among others.22 However, CAr–H arylation is currently restricted to substrates bearing DGs. Therefore, using fluoroarenes as model substrates, we set out to investigate the possibility of developing a DG-free Ru-catalyzed arylation. Here we report that benzoic acids can be used as surrogates of DGs in order to enable the C–H arylation of fluoroarenes with aryl halides. Furthermore, we report mechanistic insights concerning the C–H activation and the formal oxidative addition/reductive elimination steps as well as studies toward the understanding of an unprecedented arylation site selectivity.
Scheme 1. C–H Activation/Arylation of Perfluoroarenes by Pd, Au, Cu, and Ru.
2. Results and Discussion
2.1. Optimization of the C–H Activation of Fluoroarenes with η6-Arene Ru Complexes
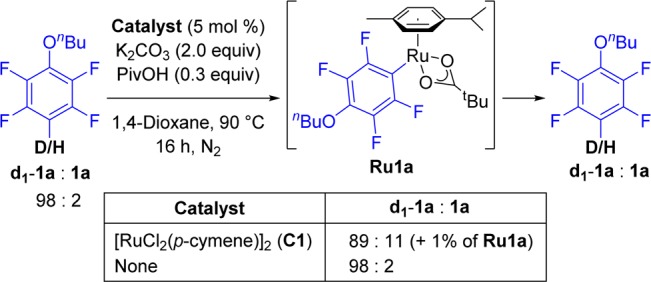
The H/D exchange processes can be used to explore the potential of a transition-metal catalyst toward the cleavage/formation of C–H bonds.23 Given the reversible nature of the C–H metalation step often observed in ruthenium catalysis,20,21,24 we decided to attempt the ruthenium-catalyzed D/H scrambling on the nonvolatile perfluorinated arene 1a. When deuterated fluoroarene d1-1a (98% deuteration) was subjected to the reaction conditions described in Scheme 2 using 5 mol % of [RuCl2(p-cymene)]2 (C1), a net 9% of H incorporation was detected. Conversely, a control experiment in the absence of catalyst C1 showed no scrambling. Furthermore, 1H and 19F NMR analysis of the reaction mixture (Figure S1) revealed the formation of an unprecedented [Ru(fluoroaryl)(OPiv)(p-cymene)] intermediate (Ru1a). This result suggests that Ru(II) complexes are able to activate C–H bonds in perfluorinated arenes, most likely involving the participation of pivalate in the metalation step.16,19
Scheme 2. D/H Scrambling of d1-1a/1a via Ru1a by Reversible C–D/C–H Activation.
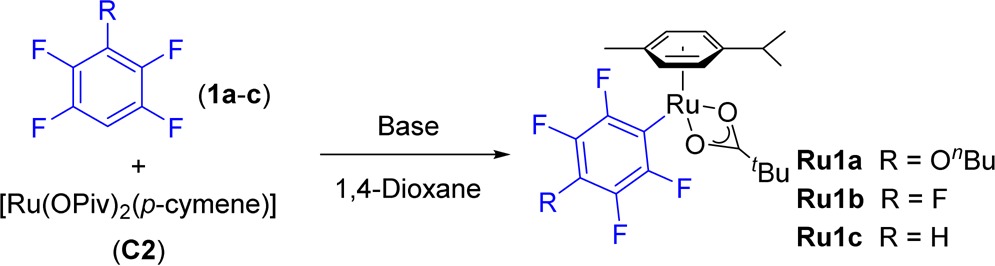
We then shifted our attention to optimizing the formation of aryl-Ru complex Ru1a by C–H activation of 1a with the well-defined complex [Ru(OPiv)2(p-cymene)] (C2, Table 1).25 We screened bases aiming to suppress protodemetalation. In the absence of base or with KOPiv (entries 1 and 2), only 6% of the desired complex was detected. Examination of alkali carbonate bases (entries 3–6) showed that although the more soluble bases led to low mass recovery26 the moderately soluble Na2CO3 (entry 4) afforded 25% of aryl-Ru Ru1a with a good balance of recovered starting pivalate complex C2. Raising the temperature to 120 °C and the concentration to 0.5 M (entry 7) led to the formation of Ru1a in 55% yield. Finally, increasing the loading of fluoroarene 1a from 5 to 20 equiv, allowed us to isolate Ru1a after column chromatography in 60% yield (entry 8). This methodology was also applied to pentafluorobenzene (1b) and 1,2,4,5-tetrafluorobenzene (1c), which afforded the corresponding aryl-Ru complexes Ru1b and Ru1c in good yields (entries 9–10). The structure of Ru1c was further confirmed by single crystal X-ray analysis (Figure 1). A kinetic isotope effect of 2.4 was observed for the formation of Ru1a, in agreement with a CMD-type C–H bond cleavage (Table S3 and Graph S1).16,19
Table 1. Optimization of the C–H Activation of Fluoroarenes with Complex C2a.
| entry | base | 1a–c (equiv) | T (°C) | C2 (%) | Ru1a–1c (%) |
|---|---|---|---|---|---|
| 1 | 1a (5) | 90 | 83 | 6 | |
| 2 | KOPiv | 1a (5) | 90 | 87 | 6 |
| 3 | Li2CO3 | 1a (5) | 90 | 78 | 12 |
| 4 | Na2CO3 | 1a (5) | 90 | 62 | 25 |
| 5 | K2CO3 | 1a (5) | 90 | 20 | 33 |
| 6 | Cs2CO3 | 1a (5) | 90 | 4 | 3 |
| 7b | Na2CO3 | 1a (5) | 120 | 33 | 55 |
| 8b,c | Na2CO3 | 1a (20) | 120 | 14 | 68 (60)d |
| 9b,c | Na2CO3 | 1b (20) | 120 | 5 | 79 (75)d |
| 10b,c | Na2CO3 | 1c (20) | 120 | 2 | 81 (77)d |
Reaction conditions: C2 (0.05 mmol), 1a (5.0 equiv), and base (2.0 equiv) in 1,4-dioxane (0.2 M) under Ar at the specified temperature for 16 h; yield is determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard.
1,4-Dioxane (0.5 M).
C2 (0.2 mmol, 1.0 equiv) and 1 (20 equiv) were used.
Yields in parentheses refer to isolated material.
Figure 1.
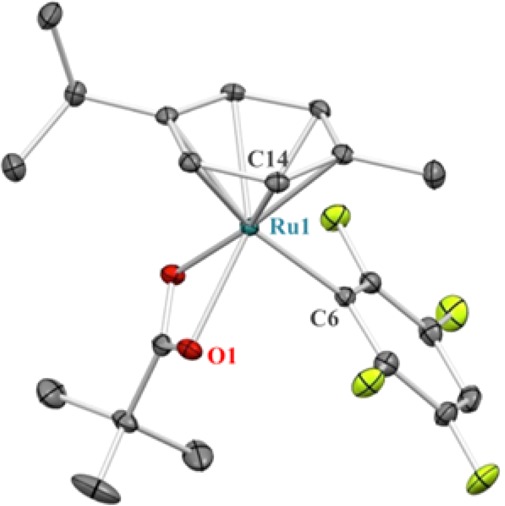
ORTEP diagram of Ru1c at 50% probability ellipsoids. Hydrogen atoms and cocrystallized CHCl3 have been omitted for clarity.
2.2. Direct Arylation of Fluoroarenes Catalyzed by η6-Arene-Ru Complexes
Having demonstrated that Ru catalysts are capable of carrying out C–H activation on electron-deficient arenes, we turned our attention to the development of a catalytic system for the C–H arylation of these substrates. Initially we investigated the direct arylation of 1a with 4-bromoanisole (2a, Table 2). When Na2CO3 was employed with 10 mol % of [Ru(OPiv)2(p-cymene)] C2, no biaryl 3aa was detected (entry 1). Encouragingly, when the novel complex [Ru(OBz)2(p-cymene)] (C3) was tested,273aa was formed in trace amounts (entry 2).
Table 2. Optimization of the Ru-Catalyzed C–H Arylation of 1a with Bromoanisole 2aa.
| entry | [Ru] | base, additives (equiv) | solvent | L (%) | 3aa (%) |
|---|---|---|---|---|---|
| 1 | C2 | Na2CO3 (2) | 1,4-dioxane | 26 | 0 |
| 2 | C3 | Na2CO3 (2) | 1,4-dioxane | 41 | <1 |
| 3 | C3 | (NMe4)OC(CF3)3 (2) | 1,4-dioxane | 69 | 19 |
| 4 | C3 | (NMe4)OC(CF3)3 (2) | MeCN | 99 | 34 |
| 5 | C3 | (NMe4)OC(CF3)3 (2) | t-BuCN | 99 | 37 |
| 6b | C3 | (NMe4)OC(CF3)3 (2) | t-BuCN | 99 | 46 |
| 7b | C3 | (NMe4)OC(CF3)3 (2), KOPiv (0.2) | t-BuCN | 99 | 54 |
| 8b | C2 | (NMe4)OC(CF3)3 (2), KOBz (0.2) | t-BuCN | 99 | 57 |
| 9b | C2 | (NMe4)OC(CF3)3 (2.2), 4-FC6H4CO2H (0.2) | t-BuCN | 99 | 67 |
| 10b | C4 | (NMe4)OC(CF3)3 (2.2), 4-FC6H4CO2H (0.2) | t-BuCN | 14 | 32 |
Reaction conditions: 2a (0.1 mmol), 1a (5.0 equiv), C2-C4 (10 mol %), base, and additive(s) stirred under N2 in a closed vessel in the specified solvent (0.5 M) at 120 °C for 16 h; yield determined by 1H NMR using 1,3-dinitrobenzene as internal standard.
t-BuCN (8.0 equiv).
We hypothesized that a weakly coordinating terminal base may prevent the formation of low-reactivity intermediates. This led us to test tetramethylammonium perfluoro-tert-butoxide, which provided 3aa in 19% yield (entry 3). The corresponding alcohol, perfluoro-tert-butyl alcohol, has a pKa of 5.4,28 so perfluoro-tert-butoxide is basic enough to deprotonate pivalic acid (pKa 5.0).29 Additionally, the low boiling point of this perfluorinated alcohol (45 °C)28 results in a high concentration of the base in solution and a low concentration of its conjugated acid at the reaction temperature of 120 °C. Furthermore, our group recently reported the ability of tetramethylammonium salts as iodide scavengers in palladium-catalyzed direct arylation.30 Similarly, (NMe)4OC(CF3)3 might be involved in catalyst regeneration via halide abstraction from a Ru–Br species. We found that among the solvents screened nitriles were the most effective with t-BuCN being slightly more effective than MeCN (37% yield, entry 5, compared to 34%, entry 4). Decreasing the loading of t-BuCN to 8 equiv increased formation of 3aa to 46% (entry 6), presumably because of strong coordination of the nitriles to ruthenium. The addition of KOPiv (entry 7) further improved the biaryl yield to 54%. When 10 mol % of the pivalate catalyst C2 was used in combination with a substoichiometric amount of KOBz (entry 8), 3aa was formed in 57% yield. After an extensive screening of carboxylic acid cocatalysts (Table S5), 4-fluorobenzoic acid was found to be the most suitable, leading to 3aa in 67% yield (entry 9). Among all the benzoic acids tested, a reactivity trend could not be found, other than a negative effect of ortho substitution in the phenyl ring.31
2.3. Role of the η6-Arene Ligand and Development of a New Catalyst
During the optimization of the reaction, we observed that nitrile solvents promoted quantitative dissociation of the p-cymene ligand of the ruthenium catalyst at the end of the reaction (L, Table 2).32 Dixneuf, Jutand, and co-workers had previously observed that a p-cymene ligand was necessary for the cross-coupling of cycloruthenated phenylpyridine complexes with aryl halides. Thus, they established that although [Ru(o-C6H4-Py)(p-cymene)MeCN]+ was able to react with aryl halides to form the cross-coupled biaryl the p-cymene-free complex [Ru(o-C6H4-Py)(MeCN)4]+ was inert under the same conditions.24 In view of this precedent, we attempted to improve the efficiency of our catalytic system by using a less labile η6-arene-ligand such as hexamethylbenzene. We found that [Ru(OPiv)2(η6-C6Me6)] (C4) was significantly more stable under the reaction conditions as only 14% of the total C6Me6 ligand was detected as the free arene at the end of the reaction. Despite this, only 32% yield of 3aa was detected after 16 h (Table 2, entry 10).
This led us to hypothesize that under our reaction conditions dissociation of the arene ligand could be necessary for the reaction to take place. To test this, the kinetic profile of the reaction was examined (Figure 2 and Table S7).33 As shown in Figure 2, when 10 mol % of the p-cymene-containing catalyst C2 was employed, an induction period was observed in the formation of biaryl 3aa (purple squares). This induction period correlated with the dissociation of the p-cymene ligand (black circles). 1H NMR analysis of the reaction mixture (Figure 3) showed rapid disappearance of the pivalate complex C2 (5 min) with formation of several unknown p-cymene-containing complexes along with aryl-Ru complex Ru1a. More importantly, quantitative dissociation of the p-cymene ligand had taken place within the first 30 min of the reaction. 19F NMR revealed that along with Ru1a there formed a new aryl-Ru species which became the only detectable fluoroaryl-Ru complex after 30 min. Independent synthesis by thermal replacement of the p-cymene ligand of Ru1a with t-BuCN confirmed the newly formed species to be p-cymene-free aryl-Ru complex Ru2a (Scheme 3). Furthermore, the structure was confirmed by X-ray analysis of the analogous complex Ru2c (Figure 4, left).
Figure 2.
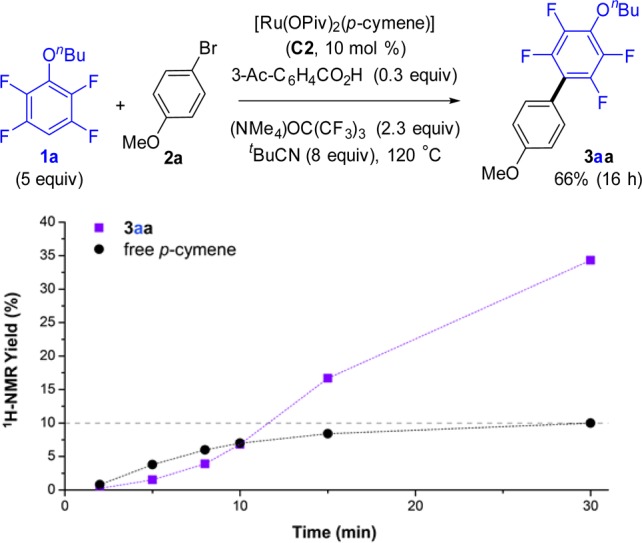
Cross-coupling of 1a and 2a with p-cymene-containing catalyst C2 (top). Profile for the formation over time of 3aa and free p-cymene, determined by 1H NMR analysis (bottom).
Figure 3.
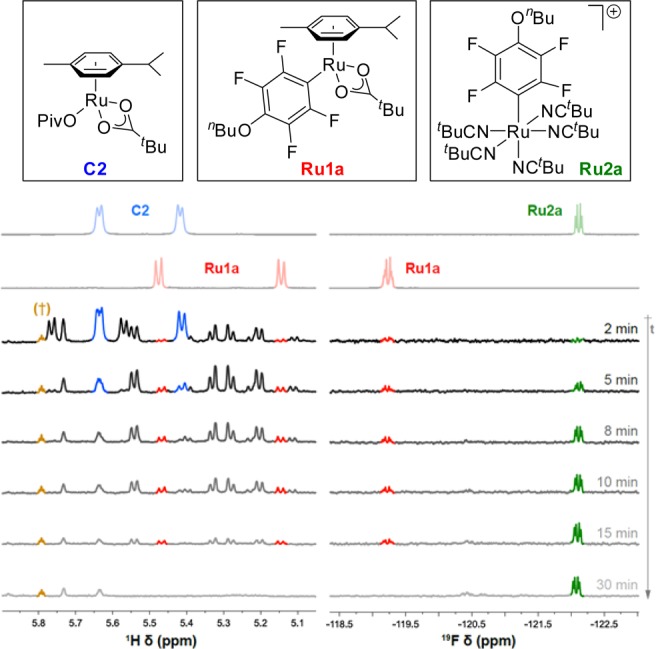
Time-dependent NMR experiments (reaction conditions as Figure 2). Left-hand side. 1H NMR expansion of the AB system region of Ru-coordinated p-cymene: C2 (blue), Ru1a (red). Right-hand side. 19F NMR expansion of fluoroaryl-ruthenium complexes region: Ru1a (red), Ru2a (green). Signal marked with † corresponds to a satellite peak of 1,3,5-trimethoxybenzene, used as an internal standard in these experiments.
Scheme 3. Synthesis of Ru2a and Ru2c by Thermal Ligand Substitution.

Figure 4.
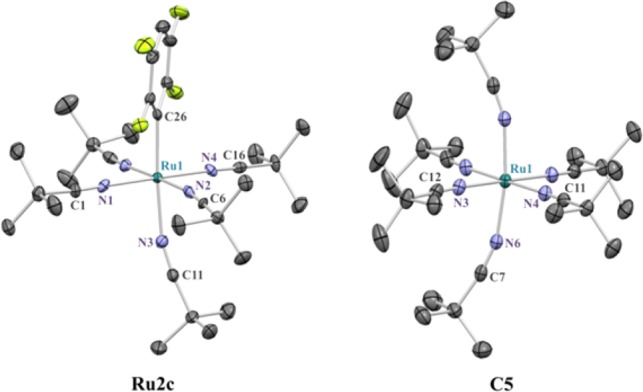
ORTEP diagrams of Ru2c and C5 at 50% probability ellipsoids. Hydrogen atoms and BF4– counterion(s) have been omitted for clarity.
These data indicate that p-cymene complex C2 is able to carry out C–H activation on 1a generating Ru1a in t-BuCN. Ru1a, in turn, must lose its p-cymene ligand forming cationic intermediate Ru2a, which then reacts with bromoarene 2a to yield product 3aa (Figures 2 and 3). Formation of 3aa continues after complete loss of p-cymene, suggesting that a nitrile-coordinated Ru(II) species lacking the p-cymene unit can also carry out C–H activation.
This experimental observation led us to design a η6-arene-free Ru(II) catalyst to simplify and potentially improve our catalytic system.34 Hence, we synthesized [Ru(t-BuCN)6][BF4]2 (C5) from RuCl3·xH2O (Scheme 4). The structure of C5 was confirmed by single-crystal X-ray analysis (Figure 4, right).
Scheme 4. Synthesis of Hexakis(pivalonitrile-κ-N)ruthenium(II) Bis(tetrafluoroborate).

Ru complex C5 was a suitable catalyst for the cross-coupling between 1a and 2a (Figure 5 and Table S8) under analogous conditions to those previously used for the p-cymene complex C2.33 Furthermore, NMR monitoring of the reaction showed no appreciable induction period in the formation of 3aa. 19F NMR spectra in Figure 5 show p-cymene-free Ru2a (Scheme 3) as the only detectable aryl-Ru species throughout the course of the reaction. Inspection of the 19F NMR spectrum of the reaction mixture after 30 min, using an internal standard (Figure S3), revealed that 50% of the ruthenium loaded at the beginning of the reaction was in the form of Ru2a, suggesting that this complex may be a resting state of the catalytic cycle.
Figure 5.
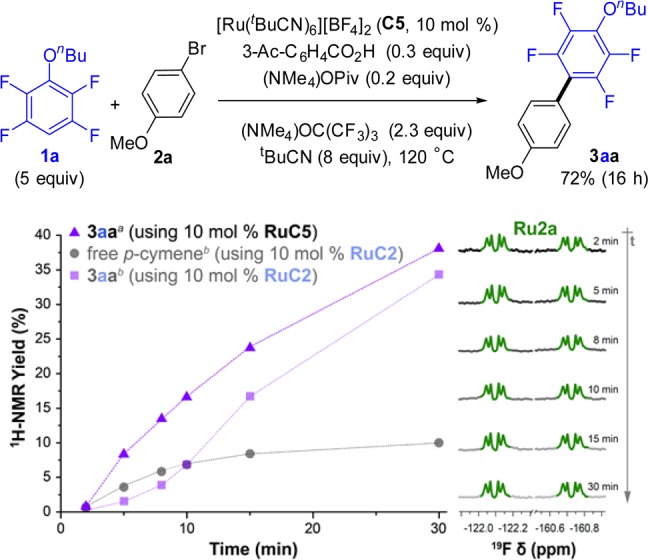
Cross-coupling of 1a and 2a with p-cymene-free catalyst C5 (top). Time profile for the formation of 3aa, determined by 1H NMR (bottom left). Profile of the reaction with C2 (Figure 2) showed in faded color for comparison. 19F NMR expansions of the aryl-Ru region of the reaction mixtures at different times (bottom right).
2.4. Optimization of the Direct Arylation Catalyzed by η6-Arene-Free Ru Catalyst C5
With the more active η6-arene-free complex C5 in hand, we returned to the optimization of the catalytic process (Table 3). Under the conditions previously used for p-cymene complex C2, the new catalyst provided 59% of 3ab from the cross-coupling of 1a and bromobenzene (2b, entry 1). Addition of a substoichiometric amount of (NMe4)OPiv (entry 2) resulted in 72% yield of 3ab. Fine-tuning of the stoichiometry for each individual component of the process (entries 3–4) allowed further optimization of the arylation while lowering the catalyst loading to 4 mol %. With the optimal loadings of 3.0 equiv of 1a, 4 mol % of C5, 3.0 equiv of t-BuCN, 0.4 equiv of (NMe4)OPiv, and 0.35 equiv of (NMe4)(4-fluorobenzoate), 3ab was isolated in 76% yield (entry 4). The arylation is equally efficient under rigorously anhydrous conditions (entry 5), but it is not compatible with the presence of oxygen or with TEMPO (entries 6–7). Importantly, the arylation does not take place without the benzoate additive (entry 8; see below for further discussion).
Table 3. Optimization of the Ru-Catalyzed C–H Arylation of 1a with Bromobenzene 2b.
| entry | C5 (mol %) | (NMe4)OC(CF3)3 (equiv) | (NMe4)(4-FC6H4CO2) (equiv) | (NMe4)OPiv (equiv) | t-BuCN (equiv) | 3ab (%)a |
|---|---|---|---|---|---|---|
| 1b | 10 | 2 | 0.2 | − | 8 | 59 |
| 2b | 10 | 2 | 0.2 | 0.2 | 8 | 72 |
| 3 | 4 | 2.2 | 0.6 | 0.45 | 3 | 80 |
| 4c | 4 | 2.5 | 0.35 | 0.4 | 3 | 82 (76)g |
| 5c,d | 4 | 2.5 | 0.35 | 0.4 | 3 | 81 |
| 6c,e | 4 | 2.5 | 0.35 | 0.4 | 3 | 0 |
| 7c,f | 4 | 2.5 | 0.35 | 0.4 | 3 | <1 |
| 8c | 4 | 2.5 | − | 0.4 | 3 | 0 |
Yield determined by 1H NMR using 1,3-dinitrobenzene as internal standard.
1a (5 equiv).
Temperature: 115 °C.
Reaction carried out in a glovebox.
Reaction under air.
TEMPO (1.0 equiv) was added.
Yield in parentheses refers to isolated material.
2.5. Insight on the Role of the Benzoate Additive
To probe further the surprising role of the benzoate additive, we attempted a stoichiometric reaction between the preformed tetrafluorophenyl-Ru complex Ru2c and 5-bromo-m-xylene (2c) (Scheme 5a; see also Figure S4). Interestingly, Ru2c does not react with the aryl bromide unless the benzoate additive is present in the mixture. This implies that the benzoate additive is fundamental for the formal oxidative addition step but not for the metalation of the substrate. Furthermore, in stark contrast with the observations by Dixneuf and Jutand’s group,24 a p-cymene-free aryl-Ru(II) complex can react with aryl halides.35 On view of these results, it is tempting to speculate that the benzoate ligand may be playing an important role in facilitating an oxidative addition or a SET process. However, further mechanistic studies will be necessary to understand its role fully.
Scheme 5. Reactivity of Ru2c with Bromoarene 2c under Stoichiometric and Catalytic Conditions.
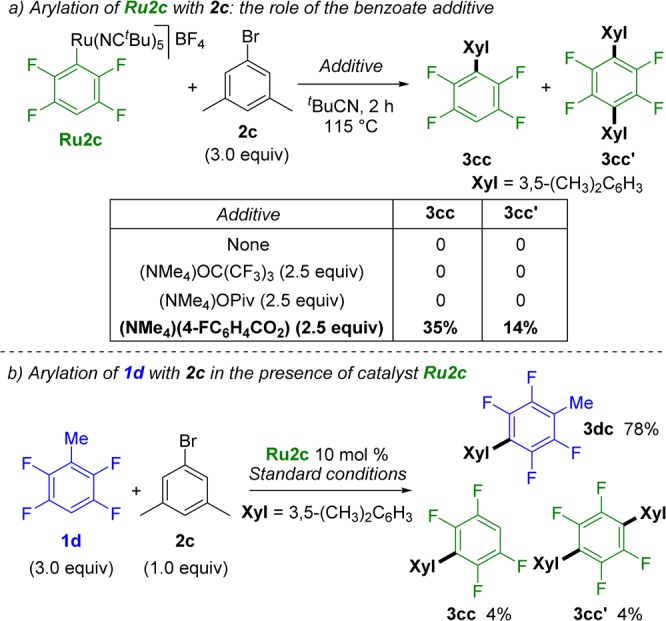
Standard conditions: 2c (0.1 mmol), 1d (3 equiv), (NMe4)(4-FC6H4CO2) (0.35 equiv), (NMe4)OPiv (0.4 equiv), (NMe4)(OC(CF3)3) (2.5 equiv), Ru2c (10 mol %), and t-BuCN (3 equiv) stirred at 115 °C under N2 in a closed vessel for 16 h.
To confirm the intermediacy of the fluoroaryl-Ru complexes (Ru2) in the reaction, the direct arylation of tetrafluorotoluene 1d with 2c was studied using as the catalyst complex Ru2c, which contains a tetrafluorophenyl unit. Even in the presence of a large excess of 1d (30 equiv with respect to the catalyst), nearly quantitative formation of the products from arylation of tetrafluorobenzene (3cc and 3cc′) was observed (Scheme 5b; see also Figure S5). This result strongly supports the idea that the fluoroaryl-Ru Ru2 complex is an active intermediate in the reaction.36
2.6. Scope of the Ru-Catalyzed Direct Arylation
With the optimal reaction conditions in hand (Table 3, entry 4), we explored the compatibility of our protocol with a variety of functionalities on the aryl bromide coupling partner for the arylation of pentafluorobenzene (1b) (Scheme 6). The reaction works in the presence of a wide range of substituents at the ortho, meta, and para positions, affording the corresponding biaryl products 3ba–3bv in moderate to excellent yields. Generally, ortho-substituted (3be, 3bg, and 3bi) and electron-poor aryl bromides (3bj, 3bk, 3bn, and 3bo) are less reactive. Notably, sensitive functional groups such as COOMe (3bo), COMe (3bn), SMe (3bl), and NMe2 (3bm) as well as terminal (3br) and internal alkenes (3bs) are tolerated. Some reactivity is also observed with cyano- (3bv) or nitro- (3bw) substituents; however, −CHO and −OH groups and terminal alkynes (3bx–3bz) are not compatible with the system. (See Supporting Information for details.) In addition to aryl bromides, iodides and chlorides can also be used as coupling partners (3bb, 3ba, and 3bk), although with reduced efficiency. Conversely, pseudohalides PhOTs and PhOMs did not afford biaryl 3bb.38o,o-Disubstituted aryl halides are unreactive in our system, allowing the iodine-containing product 3bq to be formed in good yield.39 The latter could be further functionalized by other coupling reactions. Finally, bromoarenes 2a′–c′, which are prone to cyclize under palladium40 or base/phenanthroline41 catalysis to form the five- or six-membered adducts via intramolecular C–H arylation, exhibited instead complete selectivity toward the intermolecular process (3ba′–3bc′) displaying a unique characteristic of our system.
Scheme 6. Scope of the Ru-Catalyzed Arylation of 1b with Bromoarenes 2a–z, a′–c′.
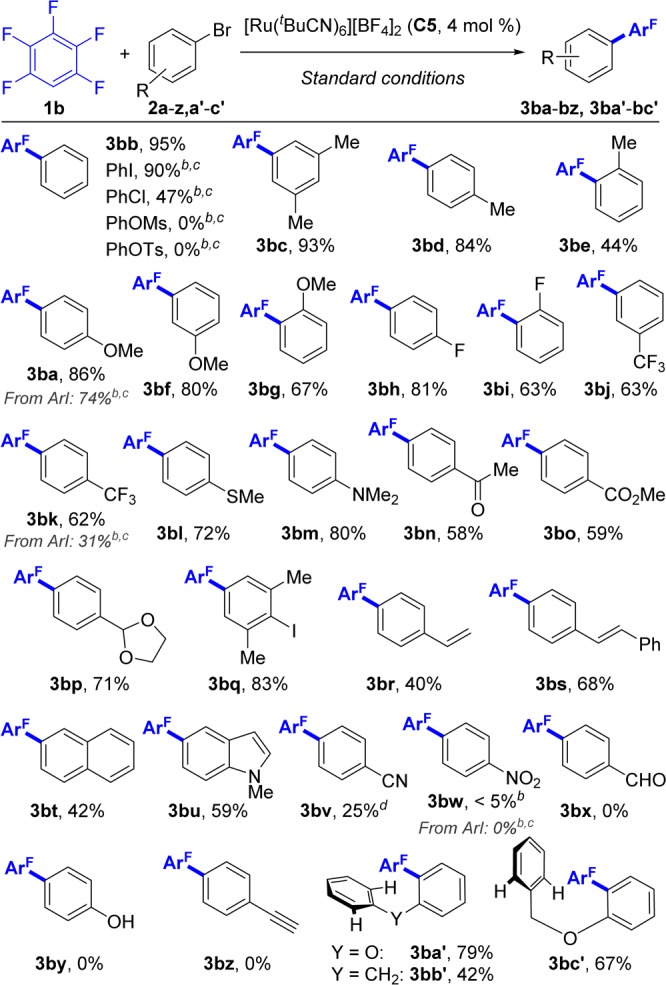
Standard conditions: bromoarene 2 (0.5 mmol), 1b (3 equiv), (NMe4)(4-FC6H4CO2) (0.35 equiv), (NMe4)OPiv (0.4 equiv), (NMe4)(OC(CF3)3) (2.5 equiv), C5 (4 mol %), and t-BuCN (3 equiv) stirred at 115 °C under N2 in a closed vessel for 16 h. Yields are of pure, isolated products.
Yield evaluated by 1H NMR with dibromomethane as internal standard.
Carried out with 1.0 equiv of the stated ArX instead of 2b.
C5 (8 mol %), reaction time 1 h.
We then turned our attention to the generality of this methodology with respect to the (fluoro)arene partner (Scheme 7). The functional group tolerance is similar to that of the aryl bromides (3dc–3ic). Tetrafluorobenzene derivatives (3cc–3kc) displayed excellent reactivity. 1,2,4,5-Tetrafluorobenzene provided the arylated product in good yield (3cc, 74%) along with a small amount of bisarylation (10%), whereas both 1,2,3,5- and 1,2,3,4-tetrafluorobenzene provided exclusively monoarylation products (3jc and 3kc). Interestingly, both 1,2,3,5- and 1,2,3,4-tetrafluorobenzene have been reported to undergo bisarylation under Pd catalysis.16 This is also the case for 1,2,3,5-tetrafluorobenzene under Cu catalysis, whereas the 1,2,3,4- analogue was poorly reactive.18 Further differences between the Ru-based system and those of Pd or Cu can be observed for the tri- and difluorobenzene series (3lc–3oc, 3uc, and 3vc). Symmetric 1,3,5-trifluorobenzene and 1,4-difluorobenzene did not give bis-/tris-arylation (3lc and 3uc) as otherwise seen with Pd.16 1,2,4-Trifluorobenzene provided three isomers in a 1:0.95:0.13 ratio (3mc, 3mc′, and 3mc″) with 5% of bisarylated adduct (3mc‴), whereas both with Pd and Cu only arylation between the two fluorine atoms has been reported.42 1,2,3-Trifluorobenzene afforded two isomers in a 1:0.3 ratio (3nc and 3nc′), and 1,3-difluorobenzene generated three isomers in a 1:0.63:0.25 ratio (3vc, 3vc′, and 3vc″, respectively). A less electron-deficient arene such as fluorobenzene provided only traces of its arylation product 3wc, whereas benzene (3xc) was completely inert. Regioselectivity with Pd and Cu catalysts for polyfluoroarenes has been shown to correlate with the acidity of the different C–H positions.19d,19e,18b We hypothesized that the observed differences could be explained by a more pronounced influence of steric factors when using the octahedral Ru catalyst, thus making the preference for the most acidic proton less relevant in some cases. A similar behavior has been described for octahedral rhodium and iridium complexes, which display sterically controlled selectivity.4e,43
Scheme 7. Scope of the Ru-Catalyzed Arylation of (Hetero)Aromatics 1c–z, 4–7 with Bromoarenes 2a–b.
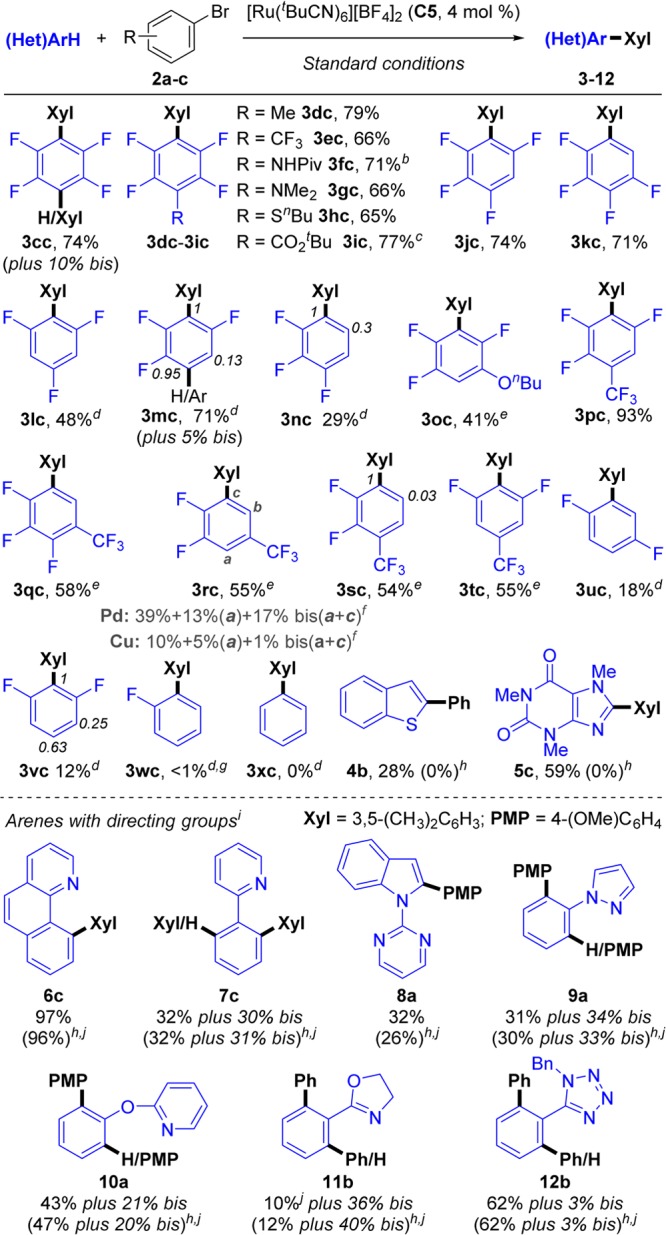
Standard conditions: bromoarene 2 (0.5 mmol), (fluoro)arene 1 (3 equiv), (NMe4)(4-FC6H4CO2) (0.35 equiv), (NMe4)OPiv (0.4 equiv), (NMe4)(OC(CF3)3) (2.5 equiv), C5 (4 mol %), and t-BuCN (3 equiv) stirred at 115 °C under N2 in a closed vessel for 16 h. Yields are of pure, isolated products.
t-BuCN (6 equiv).
Reaction time was 2 h.
1 (10 equiv).
1 (5 equiv).
See Figures 7 and S8 and Table S12.
Detected in traces by 19F NMR.
Yields in brackets are from reactions carried out without (NMe4)(4-FC6H4CO2).
(Hetero)arene (0.5 mmol) and bromoarene 2 (1 equiv).
Yield determined by 1H NMR analysis.
To test this hypothesis, we decided to study substrates containing a CF3 substituent, a strongly electron-withdrawing group significantly more bulky than fluorine. Substrates containing both C–H bonds placed between two fluorine substituents and C–H bonds placed between a fluorine and a CF3 showed complete selectivity for reaction at the least hindered position (3pc and 3tc). More remarkably, C–H bonds having only one ortho-fluorine were also preferred, with complete selectivity, over positions with both fluorine and CF3 (3rc). Finally, high selectivity was obtained for reaction ortho to a fluorine in the presence of positions with only one ortho-CF3 (3qc and 3sc). In comparison, arylation of 1r under Pd catalysis provided a mixture of 3rc and the product from arylation between the fluorine and the CF3 (3rc′) and bisarylation (3rc″) in 39, 13, and 17%, respectively (Figure 7). Under Cu catalysis, the same substrate provided low yields of mixed 3rc, 3rc′, and 3rc″ (10, 5, and 1%, respectively; Table S12).
Figure 7.
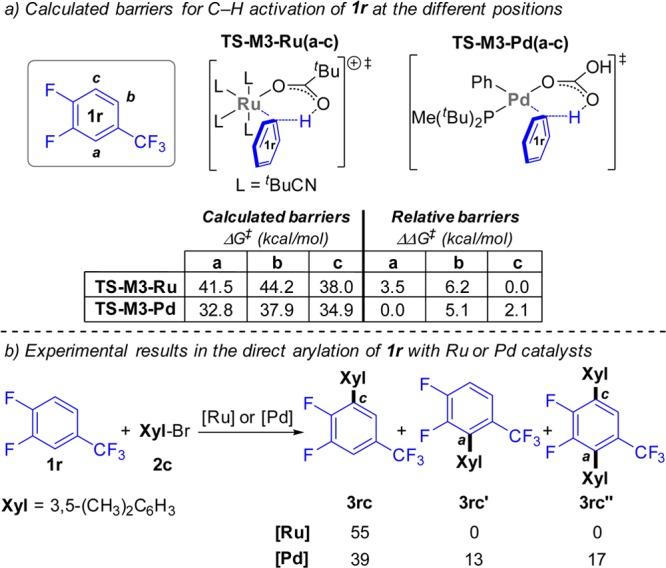
Calculated and experimental regioselectivities for the C–H activation of 1r with Ru or Pd catalysts.
Heteroaromatic compounds bearing an acidic proton, such as benzo[b]thiophene and caffeine, underwent arylation under identical reaction conditions (4b and 5c), proving to be unreactive when (NMe4)(4-fluorobenzoate) was omitted.
Substrates bearing nitrogenated directing groups were also arylated (6–12). Remarkably, these substrates did not require the benzoate additive, providing similar yields and product distributions with or without it (results in brackets in Scheme 7).44
2.7. Kinetic Isotope Effect
To assess the reversibility of the C–H activation without altering the arylation conditions, equimolar amounts of d1-1a and 2,3,5,6-tetrafluoroanisole (1y) were subjected to reaction with bromobenzene (2b) (Scheme 8; see also Figure S7). Biaryls 3ab and 3yb as well as d1-tetrafluoroanisole (d1-1y) and nondeuterated 1a were detected, confirming the reversible nature of the ruthenation step.45 In addition to the H/D scrambling, comparison of the arylation products 3ab and 3yb allowed calculation of a KIE of 2.2. This shows that the C–H activation, albeit reversible, is kinetically relevant. This was confirmed by additional experiments with 1a and d1-1a, affording a KIE of 2.4 (measured in parallel experiments, Table S11 and Graph S2).
Scheme 8. H/D Exchange and Competition KIE Experiment.
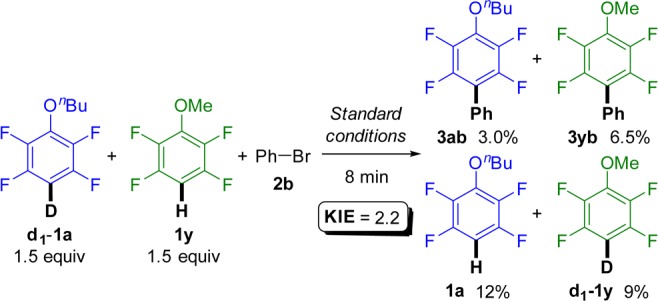
Standard conditions: bromoarene 2b (0.5 mmol), d1-1a (1.5 equiv), 1y (1.5 equiv), (NMe4)(4-FC6H4CO2) (0.35 equiv), (NMe4)OPiv (0.4 equiv), (NMe4)(OC(CF3)3) (2.5 equiv), [Ru(t-BuCN)6][BF4]2 (C5, 4 mol %), and t-BuCN (3 equiv) stirred at 115 °C under N2 in a closed vessel.
2.8. Preliminary DFT Studies
To obtain more information on the factors influencing the Ru-promoted C–H activation, DFT was used to study the CMD-type C–H activation mechanism on several potentially relevant model Ru(II)complexes.46,47 Ru(OAc)2(η6-benzene) (M1) was used as a model of C2, whereas monocationic [Ru(OAc)(MeCN)4]+ (M2) was used to model the η6-arene-free complex likely formed from reaction of C5 with pivalate.48,49 For both models, CMD-type transition states with pentafluorobenzene 1b (TS-M1 and TS-M2, respectively, Figure 6) were found with very similar energies (29.5 and 28.8 kcal/mol, respectively), consistent with the previous observation that both complexes are active in the C–H activation reaction (see above). Also, both aryl-Ru(II) products (P-M1 and P-M2) were less stable than the starting materials, which explains the experimental requirement for a stoichiometric base to drive the process forward.
Figure 6.
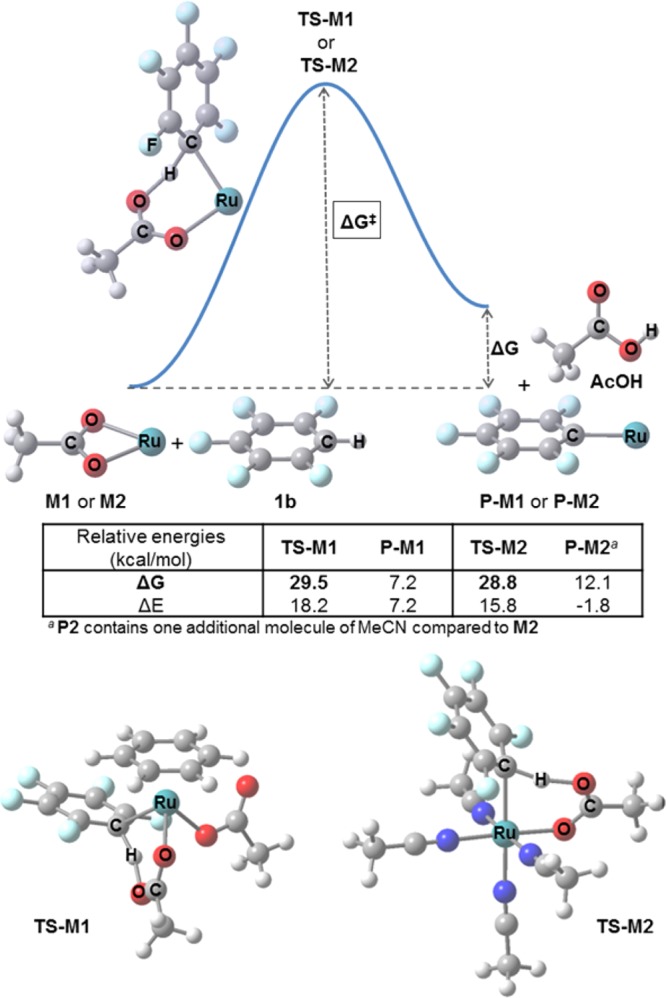
Top: Schematic profile for the C–H activation step (ligands on Ru omitted for clarity) and calculated energies. Bottom: Optimized structures for the transition states with the p-cymene-containing catalyst (TS-M1) and the cationic η6-arene-free catalyst (TS-M2).
Regioselectivity was studied using substrate 1r (Figure 7; see also Figure S13 and Table S14), which has three C–H bonds with different electronic and steric properties (labeled a–c). To assess steric factors, the real system was used for these calculations. With [Ru(t-BuCN)4(OPiv)]+ (M3-Ru), the computed barriers for each position matched the experimental results, with the least sterically hindered position c being favored, followed by the most electron-deficient one (a, 3.5 kcal/mol higher; see data in Figure 7a). Interestingly, analogous calculations with a Pd-based system16 [Pd(OCOH)(Ph)(P(t-Bu)2Me)] (M3-Pd) provided a different selectivity, favoring the most electron-deficient position a although with a lower selectivity over c (2.1 kcal/mol).50 Experimentally, both Ru and Pd16 catalysts provided 3rc as the major product, resulting from mono arylation at position c. Although this was the only product obtained using Ru, a significant amount of arylation at position a (Figure 7b, products 3rc′ and 3rc″) was obtained with Pd.51 Thus, although with some degree of inaccuracy, the calculations acceptably reproduce the observed higher trend of Pd to react at position a.
In an attempt to rationalize the observed and calculated differences in selectivity between Ru and Pd systems, we studied a deconvolution of the factors contributing to these calculated barriers. The energy barrier can be decomposed in two main contributions: (i) the energy cost of distorting each of the individual reactants to the geometry they adopt in the TS (ΔEdist(ArH) and ΔEdist(M) for the arene and the metal complex, respectively) and (ii) the energy gain of the interaction between the two individual distorted fragments (ΔEint).52 Application of this analysis provided the deconvoluted energy data displayed in Table 4. For easier comparison, the relative barriers (ΔΔG⧧) and distortion energies of the arene (ΔΔEdist(ArH)) and interaction enegies (ΔΔEint) are displayed in Figure 8, together with the relative deprotonation energies of the three positions of the arene (ΔΔEdeprot).
Table 4. Distortion and Interaction Energies for the C–H Activation of 1r with Ru and Pd Catalysts, And Elongation of the C–H Bond at the TS Geometrya.
| TS-M3 | ΔG⧧ | ΔE⧧ | ΔEdist(M) | ΔEdist(ArFH) | ΔEint | ΔdC–H |
|---|---|---|---|---|---|---|
| Ru(a) | 41.5 | 24.8 | 28.6 | 26.9 | –30.7 | 0.235 |
| Ru(b) | 44.2 | 28.2 | 29.0 | 29.5 | –30.4 | 0.245 |
| Ru(c) | 38.0 | 22.3 | 28.0 | 25.7 | –31.5 | 0.221 |
| Pd(a) | 32.8 | 21.0 | 21.1 | 34.3 | –34.4 | 0.291 |
| Pd(b) | 37.9 | 26.3 | 21.6 | 39.8 | –35.0 | 0.318 |
| Pd(c) | 34.9 | 24.7 | 21.7 | 37.9 | –34.9 | 0.313 |
Calculated energies in kcal/mol, and C–H elongations (referred to as the C–H distance in the arene ground state) in Å.
Figure 8.
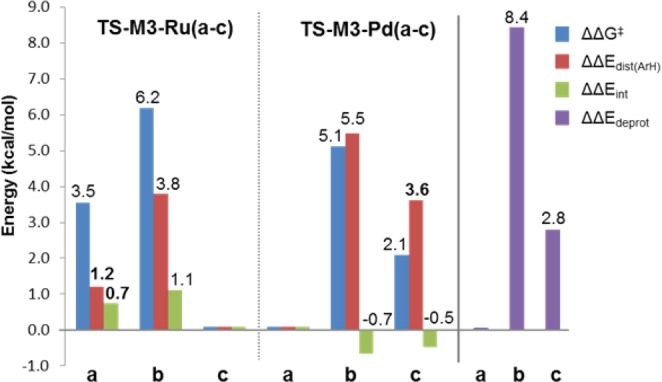
Relative barriers (ΔΔG⧧) and arene distortion (ΔΔEdist(ArH)) and interaction (ΔΔEint) energies for C–H activation of 1r with Ru and Pd, compared to the deprotonation energies of 1r (ΔΔEdeprot).
With the Pd catalyst, the energy cost of distorting the arene to the TS geometry (ΔEdist(ArH)) showed a good correlation with the calculated barriers. Thus, palladation at position b had a ΔEdist(ArH) 5.5 kcal/mol higher than palladation at position a, and ΔΔG⧧ was 5.1 kcal/mol. For position c ΔΔEdist(ArH) was 3.6 kcal/mol and ΔΔG⧧ was 2.1 kcal/mol. In both these cases, the differences in interaction energy ΔΔEint were comparatively small and actually favoring isomers b (−0.7 kcal/mol) and c (−0.5 kcal/mol) over a (compared to ΔΔEdist(ArH) = 5.5 and 3.6 kcal/mol for b and c, respectively). In summary, it can be said that the regioselectivity for this substrate with Pd catalysis was controlled by ΔEdist(ArH).19d,19e
In contrast, the behavior of the Ru-based model could not so easily be rationalized. Both ΔEdist(ArH) and ΔEint favored the experimentally observed metalation at position c. For position b, a relatively large contribution of ΔEdist(ArH) was observed (ΔΔEdist(ArH) = 3.8 kcal/mol and ΔΔEint = 1.1 kcal/mol), whereas for position a, both distortion and interaction had similar contributions (ΔΔEdist(ArH) = 1.2 kcal/mol and ΔΔEint = 0.8 kcal/mol).
The trend observed for the calculated barriers leading to different regioisomers with Pd (ΔG⧧: a < c < b) fits that for ΔEdist(ArH) and for the deprotonation energy of the arene (ΔEdeprot, Figure 8). This is not surprising because a large contribution to the arene distortion energy stems from the elongation of the C–H bond; thus, it is related to the heterolytic bond cleavage energy. This relationship has been observed before for Pd-catalyzed C–H activation of polyfluoroarenes.19d,19e
With Ru, the correlation between ΔG⧧ and ΔEdist(ArH) (c < a < b, Figure 8) was observed, however, this trend did not match that of ΔEdeprot (a < c < b), which is inherent to the arene. To explain this difference, we analyzed the extent of the geometrical distortion of the arene in the different TS structures. The elongations of the C–H bonds respect of the ground geometry were consistently larger with Pd (0.291–0.318 Å) than with Ru (0.221–0.245 Å), which fits with the higher values of ΔEdist(ArH) calculated with Pd. For both metals, the regioisomer providing the lowest barrier was also the one with the smallest elongation. The less distorted arene in the TS with Ru is consistent with a stronger influence on the selectivity of the interaction between the arene and the cationic complex.
A higher influence of interaction could be explained by the higher steric hindrance around the Ru center compared to that around Pd, the cationic character of the Ru catalyst, or a combination of both. Clarifying the relative weights of these factors and establishing general trends requires a more extensive study.
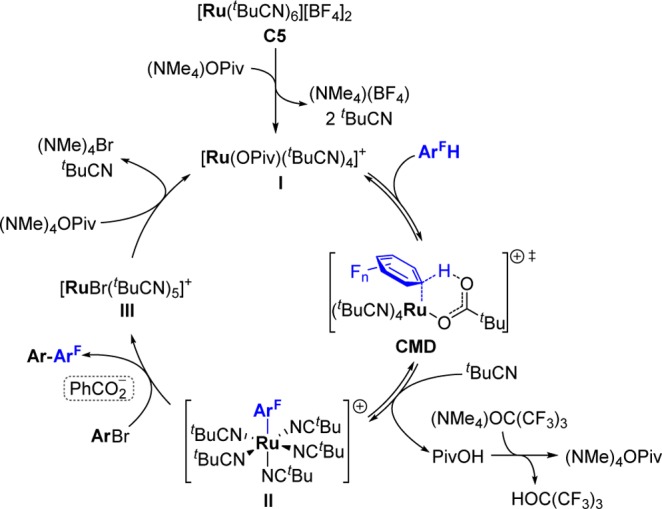
On the basis of our experimental and computational observations, a plausible catalytic cycle is described in Scheme 9. The cationic ruthenium intermediate I, generated from precatalyst C5 after precipitation of (NMe4)BF4 and ligand replacement by pivalate, carries out reversible C–H activation on the perfluoroarene (ArFH) via concerted metalation–deprotonation (CMD). The resulting aryl-Ru species II becomes redox-active in the presence of the benzoate additive, undergoing a formal oxidative addition/reductive elimination step with the aryl halide. This forms the biaryl product (Ar–ArF) and complex III, which after halide abstraction regenerates I, closing the cycle.
Scheme 9. Proposed Catalytic Cycle.
3. Conclusions
We have described the first ruthenium-catalyzed system capable of C–H arylation of arenes without the need for a directing group. This methodology is applicable to a wide array of (hetero)arenes bearing relatively acidic protons and presents a broad functional group tolerance. Contrary to previous reports on Pd, Cu, and Au catalysis, the metalation/arylation site selectivity on the fluoroarene seems to be governed by both electronic and steric factors.
19F NMR and 1H NMR studies have demonstrated that Ru(II)biscarboxylate(η6-arene) complexes are able to carry out C–H activation on arenes without a chelating group, but the resulting aryl ruthenium(II) species do not react with the electrophile unless the (η6-arene)-ligand is displaced by the solvent. The synthesis and characterization of unprecedented aryl-Ru(II) intermediates confirmed the spectroscopic data and led to a more effective η6-arene-free Ru(II) catalyst. The aryl-Ru(II) species, generated from metalation of the perfluorinated arene, reacts with aryl halides to form biaryls in both single-turnover and catalytic manners but only if a benzoate additive is present in the reaction mixture. In contrast, the benzoate additive did not have any significant effect on the results obtained with substrates having a directing group. Although further investigation is required, this suggests that the benzoic acid triggers the redox susceptibility of the ruthenated species in a fashion similar to that of ortho-metalated Ru(II) complexes.
D/H labeling experiments demonstrated the reversibility of the C–H activation under the reaction conditions. Nevertheless, KIE experiments revealed the ruthenation of the substrate to be kinetically significant.
The present catalytic system shows relevant differences in terms of regioselectivity with previously described Pd catalysts. DFT calculations suggest that the difference between both systems arise mainly from a larger relevance of steric factors in the Ru system.
Further investigations on the mechanism aimed to understand the role of action of the benzoate additive as well as kinetic and in-depth computational studies are ongoing in our laboratories.
Acknowledgments
We gratefully acknowledge the Engineering and Physical Sciences Research Council (EPSRC, EP/I038578/1 and EP/K039547/1) for funding and the European Research Council for a Starting Grant (to I.L.). We are grateful to EPSRC National Mass Spectrometry Service (Swansea), and Majid Motevalli (QMUL), and Iñigo J. Vitorica-Yrezabal (UoM) for X-ray crystallography.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b01615.
The authors declare no competing financial interest.
Supplementary Material
References
- a Bringmann G.; Günther C.; Ochse M.; Schupp O.; Tasler S.. Biaryls in Nature: A Multi-Facetted Class of Stereochemically, Biosynthetically, and Pharmacologically Intriguing Secondary Metabolites. In Progress in the Chemistry of Organic Natural Products; Herz W., Falk H., Kirby G. W., Moore R. E., Eds.; Springer: Vienna, Austria, 2001; Vol. 82, pp 1–249. [DOI] [PubMed] [Google Scholar]; b Hassan J.; Sévignon M.; Gozzi C.; Schulz E.; Lemaire M. Chem. Rev. 2002, 102, 1359–1470. 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]; c Horton D. A.; Bourne G. T.; Smythe M. L. Chem. Rev. 2003, 103, 893–930. 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]; d Bringmann G.; Price Mortimer A. J.; Keller P. A.; Gresser M. J.; Garner J.; Breuning M. Angew. Chem., Int. Ed. 2005, 44, 5384–5427. 10.1002/anie.200462661. [DOI] [PubMed] [Google Scholar]
- de Meijere A.; Diederich F.. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- a Trost B. M. Angew. Chem., Int. Ed. Engl. 1995, 34, 259–281. 10.1002/anie.199502591. [DOI] [Google Scholar]; b Labinger J. A.; Bercaw J. E. Nature 2002, 417, 507–514. 10.1038/417507a. [DOI] [PubMed] [Google Scholar]; c Wencel-Delord J.; Glorius F. Nat. Chem. 2013, 5, 369–375. 10.1038/nchem.1607. [DOI] [PubMed] [Google Scholar]
- Representative reviews on C–H activation:; a Alberico D.; Scott M. E.; Lautens M. Chem. Rev. 2007, 107, 174–238. 10.1021/cr0509760. [DOI] [PubMed] [Google Scholar]; b Ackermann L.; Vicente R.; Kapdi A. R. Angew. Chem., Int. Ed. 2009, 48, 9792–9826. 10.1002/anie.200902996. [DOI] [PubMed] [Google Scholar]; c Gutekunst W. R.; Baran P. S. Chem. Soc. Rev. 2011, 40, 1976–1991. 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]; d Lyons T. W.; Sanford M. S. Chem. Rev. 2010, 110, 1147–1169. 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Mkhalid I. A. I.; Barnard J. H.; Marder T. B.; Murphy J. M.; Hartwig J. F. Chem. Rev. 2010, 110, 890–931. 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]; f Boorman T. C.; Larrosa I. Chem. Soc. Rev. 2011, 40, 1910–1925. 10.1039/C0CS00098A. [DOI] [PubMed] [Google Scholar]; g Wencel-Delord J.; Droge T.; Liu F.; Glorius F. Chem. Soc. Rev. 2011, 40, 4740–4761. 10.1039/c1cs15083a. [DOI] [PubMed] [Google Scholar]; h Arockiam P. B.; Bruneau C.; Dixneuf P. H. Chem. Rev. 2012, 112, 5879–5918. 10.1021/cr300153j. [DOI] [PubMed] [Google Scholar]; i Engle K. M.; Mei T.-S.; Wasa M.; Yu J.-Q. Acc. Chem. Res. 2012, 45, 788–802. 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Girard S. A.; Knauber T.; Li C. J. Angew. Chem., Int. Ed. 2014, 53, 74–100. 10.1002/anie.201304268. [DOI] [PubMed] [Google Scholar]; k Kakiuchi F.; Kochi T.; Murai S. Synlett 2014, 25, 2390–2414. 10.1055/s-0034-1379210. [DOI] [Google Scholar]; l Tani S.; Uehara T. N.; Yamaguchi J.; Itami K. Chem. Sci. 2014, 5, 123–135. 10.1039/C3SC52199K. [DOI] [Google Scholar]; m Zhang X.-S.; Chen K.; Shi Z.-J. Chem. Sci. 2014, 5, 2146–2159. 10.1039/c3sc53115e. [DOI] [Google Scholar]
- a Juliá-Hernández F.; Simonetti M.; Larrosa I. Angew. Chem., Int. Ed. 2013, 52, 11458–11460. 10.1002/anie.201306425. [DOI] [PubMed] [Google Scholar]; b Schranck J.; Tlili A.; Beller M. Angew. Chem., Int. Ed. 2014, 53, 9426–9428. 10.1002/anie.201405714. [DOI] [PubMed] [Google Scholar]; c Yu D.-G.; de Azambuja F.; Glorius F. Angew. Chem., Int. Ed. 2014, 53, 7710–7712. 10.1002/anie.201403894. [DOI] [PubMed] [Google Scholar]
- a Trost B. M.; Toste F. D.; Pinkerton A. B. Chem. Rev. 2001, 101, 2067–2096. 10.1021/cr000666b. [DOI] [PubMed] [Google Scholar]; b De Sarkar S.; Liu W.; Kozhushkov S. I.; Ackermann L. Adv. Synth. Catal. 2014, 356, 1461–1479. 10.1002/adsc.201400110. [DOI] [Google Scholar]
- Palladium and Ruthenium, Monthly Average Prices between 21st July 2012 and 21st Jul 2015. Johnson Matthey Precious Metals Management Home Page. http://www.platinum.matthey.com/prices/price-charts (accessed Jul 21, 2015).
- Lewis L. N.; Smith J. F. J. Am. Chem. Soc. 1986, 108, 2728–2735. 10.1021/ja00270a036. [DOI] [Google Scholar]
- a Murai S.; Kakiuchi F.; Sekine S.; Tanaka Y.; Kamatani A.; Sonoda M.; Chatani N. Nature 1993, 366, 529–531. 10.1038/366529a0. [DOI] [Google Scholar]; b Kakiuchi F.; Yamamoto Y.; Chatani N.; Murai S. Chem. Lett. 1995, 681–682. 10.1246/cl.1995.681. [DOI] [Google Scholar]; c Kakiuchi F.; Murai S. Acc. Chem. Res. 2002, 35, 826–834. 10.1021/ar960318p. [DOI] [PubMed] [Google Scholar]
- a Kakiuchi F.; Matsumoto M.; Sonoda M.; Fukuyama T.; Chatani N.; Murai S.; Furukawa N.; Seki Y. Chem. Lett. 2000, 750–751. 10.1246/cl.2000.750. [DOI] [Google Scholar]; b Kakiuchi F.; Igi K.; Matsumoto M.; Chatani N.; Murai S. Chem. Lett. 2001, 422–423. 10.1246/cl.2001.422. [DOI] [Google Scholar]
- a Kakiuchi F.; Kan S.; Igi K.; Chatani N.; Murai S. J. Am. Chem. Soc. 2003, 125, 1698–1699. 10.1021/ja029273f. [DOI] [PubMed] [Google Scholar]; b Kakiuchi F.; Matsuura Y.; Kan S.; Chatani N. J. Am. Chem. Soc. 2005, 127, 5936–5945. 10.1021/ja043334n. [DOI] [PubMed] [Google Scholar]
- a Oi S.; Fukita S.; Hirata N.; Watanuki N.; Miyano S.; Inoue Y. Org. Lett. 2001, 3, 2579–2581. 10.1021/ol016257z. [DOI] [PubMed] [Google Scholar]; b Oi S.; Ogino Y.; Fukita S.; Inoue Y. Org. Lett. 2002, 4, 1783–1785. 10.1021/ol025851l. [DOI] [PubMed] [Google Scholar]
- a Ackermann L. Org. Lett. 2005, 7, 3123–3125. 10.1021/ol051216e. [DOI] [PubMed] [Google Scholar]; b Ackermann L.; Althammer A.; Born R. Angew. Chem., Int. Ed. 2006, 45, 2619–2622. 10.1002/anie.200504450. [DOI] [PubMed] [Google Scholar]; For a related alkylation, see; c Ackermann L.; Novák P.; Vicente R.; Hofmann N. Angew. Chem., Int. Ed. 2009, 48, 6045–6048. 10.1002/anie.200902458. [DOI] [PubMed] [Google Scholar]
- Özdemir I.; Demir S.; Çetinkaya B.; Gourlaouen C.; Maseras F.; Bruneau C.; Dixneuf P. H. J. Am. Chem. Soc. 2008, 130, 1156–1157. 10.1021/ja710276x. [DOI] [PubMed] [Google Scholar]
- a Gorelsky S. I.; Lapointe D.; Fagnou K. J. Am. Chem. Soc. 2008, 130, 10848–10849. 10.1021/ja802533u. [DOI] [PubMed] [Google Scholar]; b Lapointe D.; Fagnou K. Chem. Lett. 2010, 39, 1118–1126. 10.1246/cl.2010.1118. [DOI] [Google Scholar]; c Sandtorv A. H. Adv. Synth. Catal. 2015, 357, 2403–2435. 10.1002/adsc.201500374. [DOI] [Google Scholar]
- Lafrance M.; Rowley C. N.; Woo T. K.; Fagnou K. J. Am. Chem. Soc. 2006, 128, 8754–8756. 10.1021/ja062509l. [DOI] [PubMed] [Google Scholar]
- a Lu P.; Boorman T. C.; Slawin A. M. Z.; Larrosa I. J. Am. Chem. Soc. 2010, 132, 5580–5581. 10.1021/ja101525w. [DOI] [PubMed] [Google Scholar]; b Ahlsten N.; Perry G. J. P.; Cambeiro X. C.; Boorman T. C.; Larrosa I. Catal. Sci. Technol. 2013, 3, 2892–2897. 10.1039/c3cy00240c. [DOI] [Google Scholar]; c Cambeiro X. C.; Boorman T. C.; Lu P.; Larrosa I. Angew. Chem., Int. Ed. 2013, 52, 1781–1784. 10.1002/anie.201209007. [DOI] [PubMed] [Google Scholar]; d Cambeiro X. C.; Ahlsten N.; Larrosa I. J. Am. Chem. Soc. 2015, 137, 15636–15639. 10.1021/jacs.5b10593. [DOI] [PubMed] [Google Scholar]
- a Do H.-Q.; Daugulis O. J. Am. Chem. Soc. 2008, 130, 1128–1129. 10.1021/ja077862l. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Do H.-Q.; Khan R. M. K.; Daugulis O. J. Am. Chem. Soc. 2008, 130, 15185–15192. 10.1021/ja805688p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Davies D. L.; Donald S. M. A.; Macgregor S. A. J. Am. Chem. Soc. 2005, 127, 13754–13755. 10.1021/ja052047w. [DOI] [PubMed] [Google Scholar]; b García-Cuadrado D.; Braga A. A. C.; Maseras F.; Echavarren A. M. J. Am. Chem. Soc. 2006, 128, 1066–1067. 10.1021/ja056165v. [DOI] [PubMed] [Google Scholar]; c García-Cuadrado D.; de Mendoza P.; Braga A. A. C.; Maseras F.; Echavarren A. M. J. Am. Chem. Soc. 2007, 129, 6880–6886. 10.1021/ja071034a. [DOI] [PubMed] [Google Scholar]; d Gorelsky S. I. Coord. Chem. Rev. 2013, 257, 153–164. 10.1016/j.ccr.2012.06.016. [DOI] [Google Scholar]; e Gorelsky S. I.; Lapointe D.; Fagnou K. J. Org. Chem. 2012, 77, 658–668. 10.1021/jo202342q. [DOI] [PubMed] [Google Scholar]
- Ackermann L. Acc. Chem. Res. 2014, 47, 281–295. 10.1021/ar3002798. [DOI] [PubMed] [Google Scholar]
- a Ackermann L.; Vicente R.; Althammer A. Org. Lett. 2008, 10, 2299–2302. 10.1021/ol800773x. [DOI] [PubMed] [Google Scholar]; b Ackermann L.; Vicente R.; Potukuchi H. K.; Pirovano V. Org. Lett. 2010, 12, 5032–5035. 10.1021/ol102187e. [DOI] [PubMed] [Google Scholar]; c Ackermann L. Chem. Rev. 2011, 111, 1315–1345. 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]
- For other Ru-catalyzed CAr–H functionalization reactions without directing groups, see the following examples. (1) Intra- and intermolecular hydroarylation of alkenes and alkynes with aromatic compounds:; a Lail M.; Arrowood B. N.; Gunnoe T. B. J. Am. Chem. Soc. 2003, 125, 7506–7507. 10.1021/ja035076k. [DOI] [PubMed] [Google Scholar]; b Youn S. W.; Pastine S. J.; Sames D. Org. Lett. 2004, 6, 581–584. 10.1021/ol036385i. [DOI] [PubMed] [Google Scholar]; c Pittard K. A.; Lee J. P.; Cundari T. R.; Gunnoe T. B.; Petersen J. L. Organometallics 2004, 23, 5514–5523. 10.1021/om049508r. [DOI] [Google Scholar]; d Cadierno V.; Francos J.; Gimeno J. Chem. Commun. 2010, 46, 4175–4177. 10.1039/c002804e. [DOI] [PubMed] [Google Scholar]; e Tan S. T.; Teo Y. C.; Fan W. Y. J. Organomet. Chem. 2012, 708–709, 58–64. 10.1016/j.jorganchem.2012.02.017. [DOI] [Google Scholar]; (2) Allylation of arenes with Ru(IV)-based catalysts:; f Nishibayashi Y.; Yamanashi M.; Takagi Y.; Hidai M. Chem. Commun. 1997, 859–860. 10.1039/a701115f. [DOI] [Google Scholar]; g Fernandez I.; Hermatschweiler R.; Breher F.; Pregosin P. S.; Veiros L. F.; Calhorda M. J. Angew. Chem., Int. Ed. 2006, 45, 6386–6391. 10.1002/anie.200600447. [DOI] [PubMed] [Google Scholar]; h Bruneau C.; Achard M. Coord. Chem. Rev. 2012, 256, 525–536. 10.1016/j.ccr.2011.10.018. [DOI] [Google Scholar]; (3) Propargylation of aromatic compounds with propargyl alcohols:; i Bustelo E.; Dixneuf P. H. Adv. Synth. Catal. 2005, 347, 393–397. 10.1002/adsc.200404219. [DOI] [Google Scholar]; j Fischmeister C.; Toupet L.; Dixneuf P. H. New J. Chem. 2005, 29, 765–768. 10.1039/b501305d. [DOI] [Google Scholar]; k Thies N.; Hrib C. G.; Haak E. Chem. - Eur. J. 2012, 18, 6302–6308. 10.1002/chem.201200188. [DOI] [PubMed] [Google Scholar]; (4) Oxidative cross-coupling of benzene derivatives with cycloalkanes:; l Guo X.; Li C.-J. Org. Lett. 2011, 13, 4977–4979. 10.1021/ol202081c. [DOI] [PubMed] [Google Scholar]; (5) Oxidative coupling of arenes with alkenes using bimolecular oxygen as terminal oxidant:; m Weissman H.; Song X.; Milstein D. J. Am. Chem. Soc. 2001, 123, 337–338. 10.1021/ja003361n. [DOI] [PubMed] [Google Scholar]; (6) C3 selective C–H silylation of indoles:; n Klare H. F.; Oestreich M.; Ito J.; Nishiyama H.; Ohki Y.; Tatsumi K. J. Am. Chem. Soc. 2011, 133, 3312–3315. 10.1021/ja111483r. [DOI] [PubMed] [Google Scholar]; (7) Dehydrogenative C–N cross- and homocoupling of carbazole-type derivatives:; o Louillat M.-L.; Patureau F. W. Org. Lett. 2013, 15, 164–167. 10.1021/ol303216u. [DOI] [PubMed] [Google Scholar]; p Louillat M. L.; Biafora A.; Legros F.; Patureau F. W. Angew. Chem., Int. Ed. 2014, 53, 3505–3509. 10.1002/anie.201308601. [DOI] [PubMed] [Google Scholar]; and (8) para-Hydroxylation of anisole-type substrates with iodine(III)species:; q Liu W.; Ackermann L. Org. Lett. 2013, 15, 3484–3486. 10.1021/ol401535k. [DOI] [PubMed] [Google Scholar]
- a Jessop P. G.; Morris R. H. Coord. Chem. Rev. 1992, 121, 155–284. 10.1016/0010-8545(92)80067-2. [DOI] [Google Scholar]; b Prechtl M. H.; Holscher M.; Ben-David Y.; Theyssen N.; Loschen R.; Milstein D.; Leitner W. Angew. Chem., Int. Ed. 2007, 46, 2269–2272. 10.1002/anie.200603677. [DOI] [PubMed] [Google Scholar]
- Ferrer Flegeau E.; Bruneau C.; Dixneuf P. H.; Jutand A. J. Am. Chem. Soc. 2011, 133, 10161–10170. 10.1021/ja201462n. [DOI] [PubMed] [Google Scholar]
- Arockiam P. B.; Fischmeister C.; Bruneau C.; Dixneuf P. H. Angew. Chem., Int. Ed. 2010, 49, 6629–6632. 10.1002/anie.201002870. [DOI] [PubMed] [Google Scholar]
- If the concentration of carbonates in solution is too high, then they presumably replace the pivalate ligand(s) generating Ru(II) carbonate/bicarbonate p-cymene complexes, which are supposedly less capable of C–H activation (Figure S2). 19F NMR did not show any aryl-Ru(II) complex other than Ru1a in the mixture.
- The related [Ru(p-cymene)(O2CMes)2] (Mes = mesityl) has been used previously in the direct arylation of several directing-group-containing arenes:Ackermann L.; Pospech J.; Potukuchi H. K. Org. Lett. 2012, 14, 2146–2149. 10.1021/ol300671y. [DOI] [PubMed] [Google Scholar]
- Dyatkin B. L.; Mochalina E. P.; Knunyants I. L. Tetrahedron 1965, 21, 2991–2995. 10.1016/S0040-4020(01)96918-2. [DOI] [Google Scholar]
- Lomas J. S. J. Phys. Org. Chem. 2012, 25, 620–627. 10.1002/poc.1953. [DOI] [Google Scholar]
- Arroniz C.; Denis J. G.; Ironmonger A.; Rassias G.; Larrosa I. Chem. Sci. 2014, 5, 3509–3514. 10.1039/C4SC01215A. [DOI] [Google Scholar]
- A recent report has shown a beneficial effect of electron-poor, fluorinated benzoates in alkyne aminoarylation reactions:Yang F.; Ackermann L. J. Org. Chem. 2014, 79, 12070–12082. 10.1021/jo501884v. [DOI] [PubMed] [Google Scholar]
- For previous reports on the thermal and photo-substitution of (η6-arenes) in Ru(II) complexes by σ- or π-donor ligands, see:; a Zelonka R. A.; Baird M. C. J. Organomet. Chem. 1972, 44, 383–389. 10.1016/S0022-328X(00)82927-7. [DOI] [Google Scholar]; b Bennett M. A.; Smith A. K. J. Chem. Soc., Dalton Trans. 1974, 233–241. 10.1039/dt9740000233. [DOI] [Google Scholar]; c Weber W.; Ford P. C. Inorg. Chem. 1986, 25, 1088–1092. 10.1021/ic00228a007. [DOI] [Google Scholar]; For substitution of (η6-arene) in ortho-ruthenated 2-phenylpyridine complexes by nitriles see:; d Fernandez S.; Pfeffer M.; Ritleng V.; Sirlin C. Organometallics 1999, 18, 2390–2394. Moreover, in the same report, substitution did not take place on ortho-ruthenated N,N-dimethylbenzylamine complexes, leading the authors to suggest a relevant role of the strongly donating pyridine ligand 10.1021/om9901067. [DOI] [Google Scholar]
- 3-Acetylbenzoic acid, used in these experiments, showed activity identical to that of 4-fluorobenzoic acid (Tables S4 and S5).
- Precedents on the use of η6-arene-free Ru precatalysts in direct arylation with nitrogen-containing directing groups:; a Ackermann L.; Born R.; Álvarez-Bercedo P. Angew. Chem., Int. Ed. 2007, 46, 6364–6367. 10.1002/anie.200701727. [DOI] [PubMed] [Google Scholar]; b Ackermann L.; Althammer A.; Born R. Synlett 2007, 2007, 2833–2836. 10.1055/s-2007-990838. [DOI] [Google Scholar]; c Ackermann L.; Althammer A.; Born R. Tetrahedron 2008, 64, 6115–6124. 10.1016/j.tet.2008.01.050. [DOI] [Google Scholar]; d Ackermann L.; Vicente R. Org. Lett. 2009, 11, 4922–4925. 10.1021/ol9020354. [DOI] [PubMed] [Google Scholar]; e Luo N.; Yu Z. Chem. - Eur. J. 2010, 16, 787–791. 10.1002/chem.200902612. [DOI] [PubMed] [Google Scholar]; f Seki M. ACS Catal. 2011, 1, 607–610. 10.1021/cs200044t. [DOI] [Google Scholar]
- Although the reason behind this behavior is still unclear, the difference between our observations and those of Dixneuf and Jutand (ref (24)) could be explained by the use of different solvents (NMP or t-BuCN) or the use of iodoarenes in their report and bromoarenes here. In addition, Pilarski et al. recently reported that in the [RuCl2(p-cymene)]2-catalyzed arylation of N-pyrimidine-protected indoles with boronic acids, some catalytic activity is maintained even after full dissociation of the p-cymene ligand:Sollert C.; Devaraj K.; Orthaber A.; Gates P. J.; Pilarski L. T. Chem. - Eur. J. 2015, 21, 5380–5386. 10.1002/chem.201405931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- When subjected to the reaction conditions described in Scheme 5b, Ru1c was less efficient than Ru2c, possibly because of the need for losing its p-cymene ligand before entering in the arylation catalytic cycle (Table S10 and Figure S5).
- Pd-catalysed direct arylation of fluoroarenes with aryltosylates has been described:Ackermann L.; Fenner S. Chem. Commun. 2011, 47, 430–432. 10.1039/C0CC02360D. [DOI] [PubMed] [Google Scholar]
- The sluggish reactivity of ortho-substituted haloarenes towards direct arylation has been previously exploited to favor oxidative coupling products. See Ackermann L.; Novák P.; Vicente R.; Pirovano V.; Potukuchi H. K. Synthesis 2010, 2010, 2245–2253. No oxidative coupling was observed under our conditions. 10.1055/s-0029-1220010. [DOI] [Google Scholar]
- Campeau L.-C.; Parisien M.; Jean A.; Fagnou K. J. Am. Chem. Soc. 2006, 128, 581–590. 10.1021/ja055819x. [DOI] [PubMed] [Google Scholar]
- Sun C. L.; Li H.; Yu D. G.; Yu M.; Zhou X.; Lu X. Y.; Huang K.; Zheng S. F.; Li B. J.; Shi Z. J. Nat. Chem. 2010, 2, 1044–1049. 10.1038/nchem.862. [DOI] [PubMed] [Google Scholar]
- a Chang J. W. W.; Chia E. Y.; Chai C. L. L.; Seayad J. Org. Biomol. Chem. 2012, 10, 2289–2299. 10.1039/c2ob06840k. [DOI] [PubMed] [Google Scholar]; b Lesieur M.; Lazreg F.; Cazin C. S. J. Chem. Commun. 2014, 50, 8927–8929. 10.1039/C4CC03201B. [DOI] [PubMed] [Google Scholar]
- a Cho J.-Y.; Iverson C. N.; Smith M. R. J. Am. Chem. Soc. 2000, 122, 12868–12869. 10.1021/ja0013069. [DOI] [Google Scholar]; b Cho J.-Y.; Tse M. K.; Holmes D.; Maleczka R. E.; Smith M. R. Science 2002, 295, 305–308. 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]; c Cheng C.; Hartwig J. F. Science 2014, 343, 853–857. 10.1126/science.1248042. [DOI] [PubMed] [Google Scholar]
- Previous studies have shown a beneficial effect of secondary phosphine oxides, carboxylates, phosphines, and other additives in related direct arylation reactions:; a Ackermann L.; Diers E.; Manvar A. Org. Lett. 2012, 14, 1154–1157. 10.1021/ol3000876. [DOI] [PubMed] [Google Scholar]; b Ackermann L.; Lygin A. V. Org. Lett. 2011, 13, 3332–3335. 10.1021/ol2010648. [DOI] [PubMed] [Google Scholar]; c Diers E.; Phani Kumar N. Y.; Mejuch T.; Marek I.; Ackermann L. Tetrahedron 2013, 69, 4445–4453. 10.1016/j.tet.2013.01.006. [DOI] [Google Scholar]; d Hubrich J.; Himmler T.; Rodefeld L.; Ackermann L. ACS Catal. 2015, 5, 4089–4093. 10.1021/acscatal.5b00939. [DOI] [Google Scholar]; e Ackermann L.; Born R.; Vicente R. ChemSusChem 2009, 2, 546–549. 10.1002/cssc.200900014. [DOI] [PubMed] [Google Scholar]; f Ackermann L.; Mulzer M. Org. Lett. 2008, 10, 5043–5045. 10.1021/ol802252m. [DOI] [PubMed] [Google Scholar]
- Additional experiments with d1-1a and pentafluorobenzene (1b) further confirm this. See Scheme S1 and Figure S6.
- Calculations were carried out with Gaussian 09 using B3LYP functional and 6-31G basis set with polarization functions for C, H, O, N, and F atoms, SDD base and ECPs for Ru.; Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision d.01; Gaussian, Inc.: Wallingford, CT, 2009.
- An alternative mechanism with C–H activation aided by an external base was also studied for two model complexes. The calculated barriers were significantly higher (by ca. 15 kcal/mol) than those of CMD-type mechanisms; thus, further studies were centered on the latter (Figure S12 and Table S13).
- Other models studied are fac- and mer-Ru(OAc)2(MeCN)2, and Ru[(OC(CF3)3](OAc)(MeCN)3. All of them provided similar barriers (Figure S12 and Table S13).
- Although the complex analogous to M3 could not be isolated experimentally, in situ 1H NMR spectra of a mixture of C5 and (NMe4)OPiv showed the appearance of new OPiv and t-BuCN signals, along with decoordinated t-BuCN (Figures S9 and S10).
- For details, including the effect of dispersion corrections on the model and real systems, see Figure S13 and Table S14.
- A different experimental regioselectivity for substrate 1r was also found with Cu (Table S12 and Figure S8).
- a Legault C. Y.; Garcia Y.; Merlic C. A.; Houk K. N. J. Am. Chem. Soc. 2007, 129, 12664–12665. 10.1021/ja075785o. [DOI] [PubMed] [Google Scholar]; For the application of this analysis to Pd-catalyzed C–H activation reactions, see also refs (15), (16), (19d),19e, and; b Ricci P.; Krämer K.; Cambeiro X. C.; Larrosa I. J. Am. Chem. Soc. 2013, 135, 13258–13261. 10.1021/ja405936s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.