

Abstract
Although reducing low-density lipoprotein-cholesterol (LDL-C) levels with lipid-lowering agents (statins) decreases cardiovascular disease (CVD) risk, a substantial residual risk (up to 70% of baseline) remains after treatment in most patient populations. High-density lipoprotein (HDL) is a potential contributor to residual risk, and low HDL-cholesterol (HDL-C) is an established risk factor for CVD. However, in contrast to conventional lipid-lowering therapies, recent studies show that pharmacologic increases in HDL-C levels do not bring about clinical benefits. These observations have given rise to the concept of dysfunctional HDL where increases in serum HDL-C may not be beneficial because HDL loss of function is not corrected by or even intensified by the therapy. Chronic kidney disease (CKD) increases CVD risk, and patients whose CKD progresses to end-stage renal disease (ESRD) requiring dialysis are at the highest CVD risk of any patient type studied. The ESRD population is also unique in its lack of significant benefit from standard lipid-lowering interventions. Recent studies indicate that HDL-C levels do not predict CVD in the CKD population. Moreover, CKD profoundly alters metabolism and composition of HDL particles and impairs their protective effects on functions such as cellular cholesterol efflux, endothelial protection, and control of inflammation and oxidation. Thus, CKD-induced perturbations in HDL may contribute to the excess CVD in CKD patients. Understanding the mechanisms of vascular protection in renal disease can present new therapeutic targets for intervention in this population.
Keywords: Cardiovascular disease, Chronic kidney disease, Residual cardiovascular risk, HDL, Statins, Cholesterol efflux
Introduction
Whereas low-density lipoprotein (LDL) continues to provide evidence of its value as a cardiovascular disease (CVD) risk marker and therapeutic target for CVD benefits, increasing knowledge of high-density lipoprotein (HDL) is casting doubts about its role in CVD risk prediction with reservations arising from recent natural randomization studies and absence of clinical trial support for its role as a therapeutic target. However, in view of the recognition establishing that HDL performs multiple important functions from extracting cellular cholesterol to toning-down inflammation and oxidation, it is possible that the key in utilizing it as risk marker or therapeutic target is not in determining levels of HDL cholesterol (HDL-C) but rather in assessing its functionality. Raising HDL-C levels may be appropriate when HDL is functional but not when HDL is dysfunctional. The difficulty is identifying HDL that is functional and developing interventions that increase its functionality. Renal disease has been known to affect HDL levels, but more recent evidence suggests that its strongest effect is in influencing HDL composition and affecting its functions.
CVD in Renal Patients
Chronic kidney disease (CKD) is an independent risk factor for CVD. The American College of Cardiology/American Heart Association (ACC/AHA) and the National Kidney Foundation (NKF) recommend that CKD be considered equivalent to pre-existing coronary artery disease (CAD) as risk predictor (1,2). Using a large population-based study, Go et al. first documented that a decline in the glomerular filtration rate (GFR) is the main independent risk factor for CV events including hospitalization secondary to peripheral artery disease (PAD), CAD, congestive heart failure (CHF), or stroke (3). Similar conclusions were drawn from a systematic review encompassing ~1.4 million adults showing that a gradual fall of GFR is associated with an increased risk of death, and that individuals with lowest baseline GFR are at the highest risk of all-cause mortality (4). CKD not only increases CVD prevalence but also conveys poorer prognosis after CV events. Compared to non-CKD patients whose in-hospital mortality is ~2%, individuals with modest reduction in GFR (50–75 mL/min) have 6% mortality, those with GFR 35–50 mL/min have 14% mortality, those with GFR of <35 mL/min have 21% mortality, and those requiring dialysis have a 30% mortality rate (5). Other studies confirm the dramatically decreased survival of CKD patients following acute myocardial events, estimating that the risk of sudden cardiac death is increased by 11% for every 10 mL/min decline in GFR (6). In individuals with established ESRD, only half are expected to survive an acute CV event (4,7). Compared to age-adjusted CVD mortality in the non-CKD population, this estimate is 15–30 times higher, a discrepancy that increases among younger CKD cohorts (8). Importantly, although prevention of progressive deterioration in kidney function has been the primary concern in CKD, it is now clear that acute cardiovascular events and death are more common than CKD progression to ESRD. In a study of ~30,000 CKD patients, the 5-year CVD mortality rate was 19.5%, 24.3%, and 45.7% in those with CKD stages 2, 3, or 4, respectively, compared with a much lower risk of progressing to ESRD (1.1%, 1.3%, and 19.9%, respectively) (9). This is significant because early CKD now affects some 10–16% of the population worldwide, a figure that is projected to rise (10,11).
Recent studies underscore that cardiovascular mortality in CKD is due to many causes including myocardial dysfunction, valvular disease, and arrhythmias; however, atherosclerotic CAD is significantly higher in CKD patients than in the general population (12,13). Even early kidney disease has been linked to early signs of atherosclerosis including subendothelial lipid deposition, upregulation of adhesion molecules, recruitment of monocytes, conversion of macrophages to foam cells, and elastolysis (14,15). The prevalence as well as the progression of atherosclerotic disease is increased in the CKD population. Thus, CKD patients with an initially normal angiography develop myocardial infarction (MI) more frequently than non-CKD subjects (5.2 vs. 0.7%), whereas >50% of ESRD patients on dialysis develop significant new coronary stenosis (>50%) over a period of 30 months (16). Approximately 10–20% of CAD deaths in dialysis patients are due to acute MI, with a third occurring in the first year of dialysis treatment (17,18). Overall, atherosclerotic CAD is a significant cause of morbidity and mortality over the entire spectrum of CKD, and GFR <60 mL/min is as good at predicting future cardiac events as are previous history of MI, diabetes, angiographic evidence of obstructive CAD, and a positive stress test (16). This is a challenging circumstance because the presentation of acute CVD event in advanced CKD patients is often atypical, carries a poor prognosis, and is constrained by limited therapeutic options.
Traditional and Nontraditional Risk Factors and Residual Cardiovascular Risk
Traditional CVD risk factors described in the Framingham study (19) include dyslipidemia, diabetes mellitus, hypertension, smoking, older age, male gender, physical inactivity and family history of premature CVD. All these risk factors are common in CKD. However, it is not known the extent to which each factor adds to the incidence of CVD in CKD patients. For example, elevated levels of LDL-C, the primary driver of CVD in the general population, are not consistently present in CKD. Indeed, the degree and pattern of dyslipidemia are not uniform across CKD stages and are heavily influenced by the degree of renal dysfunction, the underlying etiology, and whether nephrotic syndrome is present (Table 1). For example, nephrotic patients are characterized by dramatically increased total and LDL-C, high triglycerides and normal or decreased HDL-C. Non-nephrotic CKD patients typically have normal or increased total and LDL-C, high triglycerides and decreased HDL-C. Patients whose C KD has advanced to ESRD on hemodialysis have normal or decreased total and LDL-C, high triglyceride and decreased HDL-C, whereas ESRD patients on peritoneal dialysis have increased total and LDL-C, very high triglycerides and decreased HDL-C (20,21). The paradoxical divergence between LDL-C levels and CVD becomes particularly apparent as renal dysfunction progresses to ESRD (22–24). Other risk factors relevant in the general population such as hypertension and increased BMI also lose their prognostic value in the setting of advancing CKD (23,25). Such observations have prompted a search for nontraditional risk factors specific to CKD. The CKD-associated risks now include malnutrition, low albumin, inflammation, high oxidative stress, anemia, elevated homocysteine, and dysregulated calcium/phosphorus metabolism. Although there is experimental and clinical support for each of these possibilities, none of the factors have been definitively proven as causal in the accelerated CVD occurring in the CKD population.
Table 1.
Lipid and apolipoprotein profile across CKD
| Pre-dialysis CKD | Nephrotic syndrome | Hemodialysis | Peritoneal dialysis | |
|---|---|---|---|---|
| Total cholesterol | →↑ | ↑↑ | →↓ | ↑ |
| LDL cholesterol | →↑ | ↑↑ | →↓ | ↑ |
| Small dense LDL | ↑ | ↑ | ↑ | ↑ |
| ApoB | → | ↑↑ | ↓→↑ | ↑ |
| HDL | ↓ | →↓ | ↓ | ↓ |
| Triglyceride | ↑ | ↑ | ↑ | ↑↑ |
| Lp(a) | →↑ | ↑↑ | ↑ | ↑↑ |
| ApoA-I | →↓ | →↑ | ↓ | ↓ |
CKD, chronic kidney disease; LDL, low-density lipoprotein.
Assessments that included statistical adjustment for traditional and nontraditional risk factors have led to the very important recognition that CKD itself is a powerful independent predictor of future coronary events, CVD, and total mortality. A large-scale meta-analysis of >1 million participants encompassing the general population as well as high-risk and chronic disease populations emphasize the concept that CKD increases the relative risk of mortality in individuals with or without hypertension (11) and with or without diabetes (26). A separate study compared CAD rates in individuals with diabetes vs. those with CKD (27). The incidence of myocardial infarction was similar in diabetics and in non-diabetics with CKD stages 1–4. Individuals with more advanced CKD, especially those with more severe proteinuria, had markedly heightened cardiovascular risk compared with diabetics without CKD. Together these studies emphasize that in addition to the traditional and nontraditional risk factors, CKD itself is a powerful independent risk factor for future coronary events and mortality. The data also illustrate the importance of adding information on renal function to CV risk prediction tools. It is worth noting that the Framingham 10-year risk prediction formula heavily underestimates risk in CKD patients (28).
Observational studies, clinical trials and meta-analyses have clearly shown that lowering LDL-C by HMG-CoA reductase inhibitors (statins) reduces CVD. Nonetheless, despite the widespread use of statin therapy, alone or in combination with other agents, the potential for sizable additional reduction in residual risk exists. Even aggressive statin therapy that achieves LDL-C <70 mg/dL leaves a considerable residual risk (29–31). The major statin trials have reported relative risk reductions between 25 and 45%, which means a residual risk of 55–75% (Figure 1) (32). A meta-analysis of 38 studies that included >37,000 CKD patients not on dialysis found that statin therapy reduced major cardiovascular events (33). Compared to placebo, statins significantly decrease death and cardiovascular events by ~20%, thus leaving an ~80% residual risk. By contrast, CKD patients who progress to ESRD requiring dialysis are uniquely resistant to the risk-reduction power of statins. The 4D study (Die Deutsche Diabetes Dialyse) of diabetics on maintenance hemodialysis reported a nonsignificant 8% reduction in cardiovascular death, nonfatal MI and stroke (34). The AURORA study (A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Hemodialysis: An Assessment of Survival and Cardiovascular Events) reported a nonsignificant 4% reduction in the primary outcome of cardiovascular death, nonfatal myocardial infarction or nonfatal stroke or all-cause mortality (35). The SHARP study, the only randomized placebo-controlled analysis of lipid-lowering therapy (simvastatin and ezetimibe) in a CKD population (1/3 of subjects on dialysis) revealed a significant 17% reduction in major atherosclerotic events among treated subjects vs. controls (22). However, no benefits were observed among those with ESRD requiring dialysis, despite robust reductions in LDL-C levels. Thus, patients with ESRD on dialysis show limited responsiveness to lipid-lowering therapy, which suggests a role of additional factors in the residual risk of these patients. Contributors to residual risk include insulin resistance, pro-coagulable state, elevated triglycerides, preponderance of atherogenic LDL, accumulation of remnant particles, and low HDL-C levels.
Figure 1.
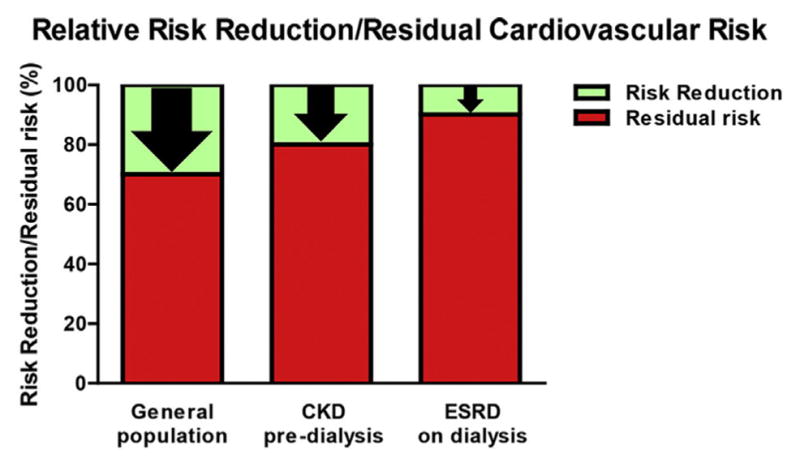
Relative risk reduction and residual cardiovascular risk in the general population and patients with chronic kidney disease (CKD) or end-stage renal disease (ESRD) on maintenance dialysis. (A color figure can be found in the online version of this article.)
Role of HDL in Residual Cardiovascular Risk
A function of HDL accepted to be of benefit to vascular health is the ability to extract cellular cholesterol and facilitate reverse cholesterol transport (RCT), a multi-step, multi-organ process that shuttles excess tissue cholesterol to the liver for excretion in bile (36,37). Modulation of the first step in RCT, namely, cholesterol efflux, can affect atherosclerotic plaque burden and composition (37–40). In animals, macrophage cholesterol efflux is inversely correlated with the extent of atherosclerosis. In humans, the capacity of HDL to promote cholesterol efflux from cultured macrophage-derived foam cells has been shown to predict subclinical atherosclerosis and CAD in a non-CKD population (39). Further support to regard cholesterol efflux capacity as a new biomarker of cardiovascular risk came from a recent study describing a large, multiethnic cohort without a history of cardiovascular disease followed for 9.4 years (40). This study showed that increasing cholesterol efflux capacity (highest vs. lowest quartile) inversely correlates with atherosclerotic cardiovascular disease rates even after adjustment of HDL-C level and HDL particle concentration. Furthermore, CVD risk prediction was significantly improved by adding cholesterol efflux capacity to traditional risk factors. These studies suggest that HDL-C levels do not predict HDL function. This is noteworthy because genome-wide association studies have failed to prove that genetic factors increasing HDL-C levels are associated with CAD rates (41). Further, genetic variations in the HDL metabolic pathway that decrease or increase the concentration of HDL-C do not influence CV risk in a way that confirms the inverse relationship seen in observational studies. For example, low HDL-C levels due to mutation in apoA-I, i.e., apoAIMilano, is not associated with increased atherosclerosis; by contrast, high HDL-C levels due to CETP mutations are not associated with decreased atherosclerosis (42–44). Finally, the disappointing clinical trials showing that significantly raised HDL-C levels do not provide vascular protection (inhibition of proatherogenic cholesteryl ester transfer protein (CETP) inhibitor torcetrapib in ILLUMINATE and dalcetrapib in dal-OUTCOMES as well as niacin treatment in AIM-HIGH) further underscore that, in isolation, levels of HDL-C may be insufficient as a marker of anti-atherogenic effects or therapeutic target (45–47). Instead, the studies suggest that various circumstances and disease states such as CKD impart important qualitative and functional differences not reflected in HDL-C.
HDL-C and Risk of CVD and Mortality in CKD
The inverse correlation between reduced HDL-C and increased risk of CVD has not been a consistent finding in cohorts of CKD patients. The multiethnic study of atherosclerosis (MESA) study did find that the association between lower HDL-C and carotid intima-media thickness (CIMT) become stronger with lower eGFR (48). HDL-C has also been reported to predict incident MI in hemodialysis patients without history of CVD (49) and was associated with enhanced mortality in short-term outcome in CKD patients with metabolic syndrome (50). By contrast, other recent studies have found no association between HDL-C and cardiovascular or all-cause mortality in maintenance hemodialysis patients (51,52). A post hoc analysis of the German Diabetes Dialysis study found no association between HDL-C and a composite outcome of cardiac death, MI, stroke, and all-cause mortality (53). This study also found that levels of ApoA-I and ApoC-III were not associated with outcomes. Similarly, a very large study involving >33,000 maintenance hemodialysis patients found that although the majority of patients had low HDL-C, a U-shaped association best characterized the relationship of HDL-C levels and cardiovascular and all-cause mortality (54). Patients with HDL-C <30 mg/dL or >60 mg/dL had a significant increase in cardiovascular and all-cause mortality, whereas those with HDL-C between 30 and 60 mg/dL had the highest survival.
Low HDL-C levels also do not predict mortality risk or severity of CAD in patients with moderate CKD. A cohort of >3000 participants, most of whom had preexisting CAD, referred for coronary angiography were followed for a median of 9.9 years. Among those with normal eGFR, an inverse relationship between HDL-C or Apo-AI level and cardiovascular mortality was found (55). However, even modest reductions in eGFR (60–89 mL/min) eliminated the association between HDL-C and CVD. Similar to the findings in dialysis populations (53), in individuals with more advanced renal impairment (<60 mL/min) higher HDL-C levels showed a tendency to predict increased, rather than reduced, risk of CVD. It is most interesting that such a paradoxical relationship between high HDL-C and increased CVD and mortality has also been observed in other high-risk populations including patients with rheumatoid arthritis, non-ST-elevation MI, or CAD (56–59). The implication of these findings is that in circumstances that adversely affect the composition or functionality of HDL particles, increasing HDL-C levels may be detrimental. Complexity of HDL particles is increasingly recognized to reflect changes in the structure, composition and biochemical characteristics of its components.
CKD Modulates HDL-C: Biogenesis, Maturation, and Catabolism
Low levels of HDL-C characteristic of CKD reflect the effects of impaired kidney function on the synthesis, assembly, maturation and catabolism of HDL components (20,60–65). Biogenesis of HDL begins with hepatic and intestinal production of ApoA-I. Paralleling HDL-C, plasma levels of ApoA-I are decreased in humans and animal models of CKD (66,67). Intestinal biogenesis contributes ~30% of plasma ApoA-I, but whether CKD affects this process is currently unknown. Also unsettled is if CKD affects hepatic production of ApoA-I. Experimental models and in vitro studies have reported impairment in hepatic synthesis and secretion of ApoA-I through mechanisms that include reduced mRNA stability (67–70). However, metabolic turnover studies in human subjects with CKD find increased ApoA-I catabolism without a change in the production rate (71–75).
Assembly and maturation of HDL particles starts with the lipidation of nascent, lipid-poor pre-β discs. ApoA-I interaction with the ATP-binding cassette transporter A1 (ABCA1) forms nascent pre-β-HDL, which then interacts with lecithin-cholesterol acyltransferase (LCAT), an essential step in esterification of free cholesterol on the surface of HDL and formation of cholesteryl ester-rich spherical HDL. This critical step is a very rapid process and explains why most HDLs in plasma are spherical and not discoidal. CKD impairs lipidation of ApoA-I and maturation of HDL (76). CKD decreases ABCA1 (77,78), which compromises lipidation of ApoA-I. Clinical and experimental studies show that CKD reduces circulating LCAT levels and activity, and downregulates hepatic LCAT gene expression (79–82). Reduced LCAT expression and activity also affects HDL maturation and increases degradation of HDL via hepatic endocytic receptor, beta chain of ATP synthase, diverting the mature HDL away form SRBI-dependent selective HDL uptake (83). Interestingly, reduced expression of SRBI and upregulation of endocytic HDL receptors was observed in livers of mice with nephrotic syndrome, predicting increased catabolism and diminished recycling of ApoA-I (67,84). Triglyceride enrichment of HDL characteristic in CKD reflects impairment in the activities of enzymes such as hepatic lipase and endothelial lipase resulting from downregulation of the enzymes, genes and presence of lipase inhibitors in plasma (84,85). Triglyceride enrichment of HDL increases its susceptibility to binding to the hepatic endocytic receptors and intracellular degradation. Maturation of HDL continues in the circulation and involves acquisition of triglycerides from apoB-containing lipoproteins (VLDL, IDL, LDL) in exchange for cholesteryl ester, processes mediated by cholesteryl ester transfer protein (CETP) and phospholipid transfer protein (PLTP). It remains unsettled whether levels of CETP are altered in CKD patients (86–88). However, a recent comparison of ESRD maintained on hemo- or peritoneal dialysis found PLTP activity to be almost double the levels observed in normal controls (89). The increased PLTP activity correlated with a reduction in serum phospholipids, apoA-I, apoA-II, increase in apoC-II and apoC-III and inversely correlated with paraoxonase activity of HDL, suggesting that PLTP has an important role in remodeling HDL composition in this setting.
The kidney is a major site of HDL homeostasis (72). In animals, between 30 and 70% of injected lipid-free ApoA-I is cleared by the kidney (90–92). ApoA-I and small HDL3 are filtered by the glomerular capillaries and taken up by the cubilin-megalin-amnionless complex in the proximal tubule (93–96). Cubilin deficiency and proximal tubular re-absorption failure due to Fanconi syndrome both increase urinary excretion of ApoA-I (73,94). Patients with Fanconi syndrome also show increased urinary loss of ApoA-IV, but not of ApoA-II, which is associated with larger cholesterol-rich HDL particles (73). These findings suggest that the glomerular filter is critical in preventing loss of all but the largest HDL particles. Thus, disorders that impede lipidation and maturation of ApoA-I or conditions that encourage its dissociation from the mature spherical HDL, permit it to get across the glomerular filter, which then exposes the particles to uptake and catabolism in the proximal tubules. Supporting this concept are experimental studies showing that inactivation of Abca1 increases plasma removal and renal degradation of lipid-free ApoA-I (97,98). Patients with Tangier’s disease due to ABCA1 gene mutations have renal hypercatabolism of HDL, which results in low plasma Apo-AI and HDL (99,100). Individuals with LCAT deficiency have impaired maturation of pre-β-HDL, but their low levels of HDL also reflect increased renal catabolism of the immature, small HDL particles (97,101). Cubilin-deficient mice have reduced proximal tubule uptake and increased urinary loss of albumin and ApoA-I, with significant decrease in plasma levels of albumin, ApoA-I and HDL3, but not of the larger, mature HDL2 (102). Together these observations support the novel concept that kidney processes including glomerular filtration and tubular reabsorption can influence ApoA-I levels and HDL subclass distribution.
Importantly, disruption in the glomerular filtration barrier by common renal disorders such as diabetic nephropathy, focal and segmental glomerulosclerosis, or tubular injury could permit filtration of greater quantity and greater variety of HDL particles. Indeed, patients with moderate glomerular impairment have increased fractional catabolic rate of ApoA-I and small HDL3 (71,72). Patients with proteinuria but without diabetes show a shift in HDL size distribution towards large particles consistent with selective renal loss of small HDL particles (103). Even in the absence of reduced GFR, proteinuria causes lipid abnormalities including reduced levels of HDL-C (104–108). For example, animal studies show that even modest proteinuria decreases ApoA-IV and increases ApoA-II and ApoC-III levels in HDL, whereas more severe proteinuria causes HDL enrichment in oxidation products such as epoxides and diols, which can profoundly affect HDL functionality (107,109). These considerations are relevant because proteinuria is a strong risk factor for CVD and mortality (110–114). The predictive power of proteinuria remains strong for microalbuminuria, primary or secondary renal disease, and presence or absence of GFR changes (10,21,110,115). The link between proteinuria and CVD is commonly attributed to endothelial injury, but proteinuria also signals changes in renal metabolism of ApoA-I affecting levels, composition, and function of HDL particles. It is likely that HDL is affected differently in different stages of CKD. This concept is illustrated in Figure 2. For example, mild CKD with proteinuria may increase renal catabolism and urinary loss of ApoA-I and small HDL particles, whereas advanced CKD may mostly trigger extra-renal effects of increased hepatic uptake and catabolism of HDL.
Figure 2.

The kidney and high-density lipoprotein (HDL) metabolism. (A) Under normal conditions, ApoA-I and small HDL3 cross the glomerular filtration barrier and are taken up by the cubilin-megalin complex to undergo catabolism, be reabsorbed and brought back into the circulation, or be lost in the urine. (B) In proteinuric conditions, greater quantity of ApoA-I, HDL3 as well as larger HDL particles such as HDL2 cross the glomerular filtration barrier. (C) In CKD, as GFR falls, fewer HDL particles can be filtered and reduced HDL-C may reflect increased catabolism in the liver. (A color figure can be found in the online version of this article.)
CKD Alters HDL Composition
There is no a specific footprint for HDL of patients with CKD. The HDL proteome of dialysis patients has reduced levels of ApoA-I, ApoA-II, ApoM, and higher levels of SAA1, ApoC-II, ApoC-III, ApoA-IV, albumin, lipoprotein-associated phospholipase A2 (Lp-PLA2), surfactant protein B (SP-B), and α-1-microglobulin/bikunin precursor (116–119). Levels of SP-B, SAA1 and pigment epithelium-derived factor progressively increased in HDL of patients whose CKD progresses from moderate to severe to ESRD (116). A study of HDL from kidney transplant patients with reduced (eGFR >40 mL/min) or poor renal function (eGFR <30 mL/min) and patients on hemodialysis revealed that HDL protein distribution of transplanted patients with preserved renal function was similar to controls, whereas that of transplanted patients with poor renal function was similar to that of hemodialysis patients (120). These results suggest that alterations in the protein cargo of HDL are linked to kidney function. Alterations in the HDL lipidome have also been documented in CKD patients and include increased triglycerides and lysophospholipids and decreased phospholipids and cholesterol (117).
Posttranslational modifications of HDL proteins and lipids by reactive oxygen/nitrogen species or the resulting reactive carbonyls can profoundly affect its function (76,121–125). CKD increases reactive oxygen/nitrogen species and increases myeloperoxidase (MPO) levels and activity, which affects HDL composition and alters functions such as ABCA1-mediated cholesterol efflux, activation of LCAT, and endothelial cell survival (76,126–128). MPO-catalyzed lipoprotein carbamylation involves formation of cyanate (a product of urea) and ε-carbamyl-lysine homocitrulline (HCit) (129,130). Levels of serum HCit and carbamylated albumin predict mortality in ESRD-HD patients (131,132). Carbamylated HDL was increased in atherosclerotic plaques of CKD patients (129). Protein carbamylation generated by peroxidase-catalyzed oxidation of thiocyanate also predicts CV risk in nonuremic individuals (129,133). Exposure of human coronary artery endothelial cells to cyanate promotes protein carbamylation to levels observed in uremic patients, reduces expression of endothelial nitric oxide synthase, and increases tissue factor and plasminogen activator inhibitor-1 expression in aortic tissue (134). Posttranslational modification by glycation is another mechanism potentially causing dysfunctional HDL in CKD, especially in individuals with CKD and diabetes, by reducing the ability of LCAT to esterify cholesterol and decreasing PON1 levels (101,135–140). It must be noted that HDL modifications in CKD may be subtle and detection of oxidized species not easy to accomplish when using the entire HDL compartment rather than specific HDL subfractions (141). Indeed, Weichhart et al. failed to detect increased oxidation of ApoA-I in CKD patients compared with healthy controls (116).
CKD Alters Multiple HDL Functions
Cholesterol efflux
CKD impairs extraction of cellular lipids by HDL. In patients with ESRD on hemodialysis, HDL had a dramatically reduced ability to accept cholesterol from lipid-loaded macrophages (142). Interestingly, HDL from ESRD-HD diabetics showed profound efflux impairment compared with HDL from diabetics without kidney disease. The fact that this effect was not modified by statin use is consistent with the concept that statins do not influence efflux and provide no benefit against CVD events in dialysis patients even in the face of robust LDL-C reduction (22,34,35). Similarly impaired capacity of HDL to promote efflux has been reported in other cohorts of dialysis patients (117,120,143).
Human monocyte THP-1 cells treated with a liver X receptor (LXR) agonist that increased expression of ABCA1, and ATP-binding cassette G1 (ABCG1) significantly increased cholesterol efflux to control HDL (142). Significantly, LXR-activation of macrophage ABCA1/G1 also increased cholesterol efflux to HDL from ESRD-HD patients. This is interesting because uremic serum added to cultured coronary artery endothelial cells dramatically reduced expression of ABCA1 (77), and uninephrectomy in mice aggravates atherosclerosis by impairing ABCA1 expression and cholesterol efflux (144).
The scenario may be different in subjects with more moderate renal disease. ApoB-depleted serum from adults with stage 3 CKD had similar ABCA1-mediated cholesterol efflux but reduced SR-BI-mediated efflux than controls (145). By contrast, preliminary studies have reported significantly reduced efflux to HDL from stages 3–4 CKD patients (146). Children with stages 2–5 CKD showed a progressive reduction in cholesterol efflux capacity of apoB-depleted serum (143). Interestingly, HDL-mediated endothelial dysfunction correlated with degree of renal impairment and with levels of circulating markers of vascular dysfunction (urate, angiopoietin-2, IL-6), endothelial dysfunction (nitric oxide production, superoxide production, vascular cell adhesion molecule-1 expression), and with clinical measures of arterial disease (aortic pulse wave velocity, carotid intima-media thickness) (143). Although renal transplantation and recovery of renal function (eGFR ~50 mL/min) improved markers of endothelial and vascular function improved, cholesterol efflux capacity remained depressed. Similarly, adults whose kidney function was restored by transplantation did not correct the impairment in the capacity of HDL for cholesterol acceptor (120). Even after stratification into transplant recipients with good graft function and poor graft function, the cholesterol acceptor capacity remained profoundly depressed in both groups and was not different than in ESRD patients on hemodialysis.
These observations raise the possibility that cholesterol efflux capacity of HDL represents a more severe or advanced disruption of normal HDL functionality, or requires more long-standing disease and/or comorbidities. It is therefore notable that children with CKD or ESRD requiring dialysis who do not have long-standing comorbidities or risk factors characteristic of adults with CKD (diabetes, obesity, pre-existing CVD) have HDL that is consistently revealed to have profound impairment in anti-inflammatory, anti-oxidative and endothelial protection functions (143,147,148). By contrast, impairment in cholesterol efflux capacity in not consistently observed. Thus, compared to children with no significant reduction in cholesterol efflux capacity, the cohort with reduced cholesterol efflux was older and already had demonstrable vasculopathy (abnormal aortic pulse wave velocity and increased carotid intima-media thickness) (143,147). Although differences in cholesterol efflux capacity in these studies could reflect different methodology for HDL isolation and efflux measurement, it is also possible that reduced cholesterol efflux in the older cohort reflects established CVD. In this connection, although reduced cholesterol efflux was associated with prevalent CAD in adults with CKD, increased rather than decreased cholesterol efflux was associated with risk of future MI, stroke or death (149).
Anti-oxidant and anti-inflammatory effects
Anti-oxidant and anti-inflammatory effects of HDL reflect the levels and activity of constituent HDL components that bind to and dispose of endotoxins and oxidized phospholipids (52,119,150–152). The capacity of HDL from CKD patients to prevent LDL oxidation is markedly diminished (52,119,150,152). Compared to normal controls, HDL from CKD patients show decreased levels of paraoxonase (30%), glutathione peroxidase (50%), LCAT (60%) and ApoA-I (41%) (78). Even though proteomic analysis of uremic HDL did not show a significant difference in PON1 (153), enzyme activity was about half of that observed in HDL of controls (120). These observations complement an earlier study reporting that individuals with low antioxidant activity of HDL had more co-morbidities and increased risk of cardiovascular and all-cause mortalities than individuals with normal HDL (52). In a recent prospective study of >400 dialysis patients, high levels of oxidized HDL were associated with increased CIMT, whereas a combination of high ox-HDL and high IL-6 predicted greater risk for CVD events and CVD-related mortality (154). PON and arylesterase activity are low even in moderate CKD and appear to associate with rates of nonfatal myocardial infarction, stroke, and death (155).
The antioxidant and anti-inflammatory activity of HDL may be linked. THP-1 macrophages treated with HDL isolated from ESRD patients on dialysis showed significantly greater inflammatory response with increased interleukin-1 β (IL-1β), interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) production, compared with the response elicited by normal HDL (142). Moreover, whereas HDL from control subjects reduced MCP-1-induced migration of THP-1 macrophages, HDL from hemodialysis patients provided no antichemotactic effect. Similarly, amplified cytokine response was noted in macrophages exposed to HDL isolated from children with dialysis-requiring CKD (147). HDL from CKD patients has defective anti-inflammatory function. Among the 49 proteins altered in HDL of ESRD patients, only SAA levels inversely correlated with its anti-inflammatory potency (116). Similarly, dramatic enrichment with SAA in HDL of dialysis patients was associated with lower anti-inflammatory capacity and linked this effect to activation of formyl-peptide receptor 2 (119). In subjects with normal kidney function, SAA can displace both ApoA-I and PON1, thus explaining reduced anti-oxidative and anti-inflammatory activity (156). Because CKD is a chronic inflammation state, SAA enrichment may become a marker of HDL dysfunction. A post-hoc analysis of the 4D study showed that HDL enrichment in SAA was associated with risk of cardiovascular events, whereas HDL enrichment in SP-B was associated with all-cause mortality (120).
Several studies have reported that SAA-enriched HDL reduces cholesterol efflux capacity (117,157,158) although there is no consensus in the literature (61,159,160). No correlation was found between macrophage cholesterol efflux capacity and markers of systemic inflammation such as high sensitivity CRP or between cholesterol efflux capacity of HDL and inflammatory macrophage response elicited by HDL of patients with ESRD-HD (142). Additionally, statin therapy abolished the difference in cytokine response between HDL of ESRD patients and normal controls, whereas impaired efflux capacity of uremic HDL persisted under statin treatment. Interestingly, ATF3, a transcriptional repressor linked to inflammation and cellular stress responses, is a novel mechanism by which HDL may exert anti-inflammatory effects that are independent of its cholesterol transport capacity (161). HDL induces changes in ATF3 causing decreased transcription of cytokines such as IL-6, IL-12β, and TNFα. Interestingly, although the anti-inflammatory properties of HDL were lost in mice and cells lacking Atf3, no differences in cholesterol transport were observed. Thus, it is possible that separate and distinct pathways affect HDL-mediated cholesterol handling and inflammation control.
Endothelial support
HDL protects the vascular endothelium. Early studies showed that HDL vasodilates precontracted aortic segments, an effect linked to activation of endothelial nitric oxide synthase and production of nitric oxide (NO). HDL is also known to stimulate endothelial cell proliferation, migration, adhesion molecule production, and cell survival and repair (14,162–164). HDL of CKD patients has shown impairment in several of these functions, as they are less effective in suppressing expression of vascular cell adhesion molecules, have impaired ability to support endothelial cell survival and repair, and promote rather than decrease monocyte adhesion to the endothelium (147,148). HDL of CKD patients are also less effective in restoring endothelial cell proliferation following TNF-α stimulus, results that are in line with observations that uremic serum impairs endothelial cell proliferation (165,166).
HDL from adults and children with stages 2–4 CKD promote endothelial dysfunction by stimulating generations of reactive oxygen species and inhibiting endothelial NO bioavailability. The underlying mechanism appears to involve symmetric dimethylarginine (SDMA), the structural isomer of asymmetric dimethylarginine (ADMA), endogenous products of protein methylation that accumulate as kidney function decreases. Increased levels of SDMA/ADMA have been linked to increased CVD risk (148). HDL of patients with CKD show increased content in SDMA, and SDMA-containing HDL interact with endothelial TLR2 leading to enhanced NADP-dependent ROS production and reduced endothelial NO bioavailability in vitro. In mice, SDMA in HDL causes hypertension and impaired re-endothelialization after carotid injury. Notably, infusion of SDMA-enriched HDL into TLR2-deficient mice does not cause hypertension, confirming a key role of TLR-2 in mediating the effects of dysfunctional HDL. In a separate study, levels of HDL-associated SDMA were inversely related to endothelial cell production of NO (143). Further, HDL-induced endothelial NO production is progressively reduced in individuals with stages 2–5 CKD, with the most profound changes in dialysis patients. Overall, these observations suggest that SDMA accumulation on HDL may be the mechanism that links endothelial dysfunction, hypertension, and atherosclerosis in CKD patients (167,168).
Conclusions
Many systemic diseases can influence HDL composition and function. CKD, particularly in advanced stages, disrupts the ability of HDL to extract cellular cholesterol, control inflammation and oxidation, and protect the endothelium, likely contributing to the exaggerated rate of CVD in renal patients. The fact that dialysis patients are not protected by statin therapy is compatible with the notion that an aberrant HDL disallows the CVD benefits of drastic LDL reductions. Identifying the structural and functional characteristics of the HDL of CKD patients may go a long way in developing therapeutic strategies based more on reinstating proper HDL function than in doggedly increasing levels of HDL cholesterol.
References
- 1.Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction–executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1999 Guidelines for the Management of Patients With Acute Myocardial Infarction) Circulation. 2004;110:588–636. doi: 10.1161/01.CIR.0000134791.68010.FA. [DOI] [PubMed] [Google Scholar]
- 2.Briasoulis A, Bakris GL. Chronic kidney disease as a coronary artery disease risk equivalent. Curr Cardiol Rep. 2013;15:340. doi: 10.1007/s11886-012-0340-4. [DOI] [PubMed] [Google Scholar]
- 3.Go AS, Chertow GM, Fan D, et al. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–1305. doi: 10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- 4.Tonelli M, Wiebe N, Culleton B, et al. Chronic kidney disease and mortality risk: a systematic review. J Am Soc Nephrol. 2006;17:2034–2047. doi: 10.1681/ASN.2005101085. [DOI] [PubMed] [Google Scholar]
- 5.Wright RS, Reeder GS, Herzog CA, et al. Acute myocardial infarction and renal dysfunction: a high-risk combination. Ann Intern Med. 2002;137:563–570. doi: 10.7326/0003-4819-137-7-200210010-00007. [DOI] [PubMed] [Google Scholar]
- 6.Gibson CM, Pinto DS, Murphy SA, et al. Association of creatinine and creatinine clearance on presentation in acute myocardial infarction with subsequent mortality. J Am Coll Cardiol. 2003;42:1535–1543. doi: 10.1016/j.jacc.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Schiffrin EL, Lipman ML, Mann JF. Chronic kidney disease: effects on the cardiovascular system. Circulation. 2007;116:85–97. doi: 10.1161/CIRCULATIONAHA.106.678342. [DOI] [PubMed] [Google Scholar]
- 8.Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis. 1998;32:S112–S119. doi: 10.1053/ajkd.1998.v32.pm9820470. [DOI] [PubMed] [Google Scholar]
- 9.Keith DS, Nichols GA, Gullion CM, et al. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med. 2004;164:659–663. doi: 10.1001/archinte.164.6.659. [DOI] [PubMed] [Google Scholar]
- 10.Levey AS, de Jong PE, Coresh J, et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int. 2011;80:17–28. doi: 10.1038/ki.2010.483. [DOI] [PubMed] [Google Scholar]
- 11.Mahmoodi BK, Matsushita K, Woodward M, et al. Associations of kidney disease measures with mortality and end-stage renal disease in individuals with and without hypertension: a meta-analysis. Lancet. 2012;380:1649–1661. doi: 10.1016/S0140-6736(12)61272-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baber U, Stone GW, Weisz G, et al. Coronary plaque composition, morphology, and outcomes in patients with and without chronic kidney disease presenting with acute coronary syndromes. JACC Cardiovasc Imaging. 2012;5:S53–S61. doi: 10.1016/j.jcmg.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 13.Weiner DE, Tighiouart H, Elsayed EF, et al. The relationship between nontraditional risk factors and outcomes in individuals with stage 3 to 4 CKD. Am J Kidney Dis. 2008;51:212–223. doi: 10.1053/j.ajkd.2007.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCullough PA, Li S, Jurkovitz CT, et al. Chronic kidney disease, prevalence of premature cardiovascular disease, and relationship to short-term mortality. Am Heart J. 2008;156:277–283. doi: 10.1016/j.ahj.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 15.Suganuma E, Zuo Y, Ayabe N, et al. Antiatherogenic effects of angiotensin receptor antagonism in mild renal dysfunction. J Am Soc Nephrol. 2006;17:433–441. doi: 10.1681/ASN.2005080883. [DOI] [PubMed] [Google Scholar]
- 16.Yiu KH, de Graaf FR, Schuijf JD, et al. Prognostic value of renal dysfunction for the prediction of outcome versus results of computed tomographic coronary angiography. Am J Cardiol. 2011;108:968–972. doi: 10.1016/j.amjcard.2011.05.031. [DOI] [PubMed] [Google Scholar]
- 17.Collins AJ, Foley RN, Herzog C, et al. US Renal Data System 2012 Annual Data Report. Am J Kidney Dis. 2013;61:e1–e476. doi: 10.1053/j.ajkd.2012.11.031. [DOI] [PubMed] [Google Scholar]
- 18.Herzog CA, Ma JZ, Collins AJ. Poor long-term survival after acute myocardial infarction among patients on long-term dialysis. N Engl J Med. 1998;339:799–805. doi: 10.1056/NEJM199809173391203. [DOI] [PubMed] [Google Scholar]
- 19.Kannel WB, Dawber TR, Kagan A, et al. Factors of risk in the development of coronary heart disease–six year follow-up experience. The Framingham Study. Ann Intern Med. 1961;55:33–50. doi: 10.7326/0003-4819-55-1-33. [DOI] [PubMed] [Google Scholar]
- 20.Kaysen GA. Lipid and lipoprotein metabolism in chronic kidney disease. J Ren Nutr. 2009;19:73–77. doi: 10.1053/j.jrn.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 21.Chronic Kidney Disease Prognosis Consortium. Matsushita K, van der Velde M, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet. 2010;375:2073–2081. doi: 10.1016/S0140-6736(10)60674-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baigent C, Landray MJ, Reith C, et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet. 2011;377:2181–2192. doi: 10.1016/S0140-6736(11)60739-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kovesdy CP, Anderson JE, Kalantar-Zadeh K. Inverse association between lipid levels and mortality in men with chronic kidney disease who are not yet on dialysis: effects of case mix and the malnutrition-inflammation-cachexia syndrome. J Am Soc Nephrol. 2007;18:304–311. doi: 10.1681/ASN.2006060674. [DOI] [PubMed] [Google Scholar]
- 24.Chawla V, Greene T, Beck GJ, et al. Hyperlipidemia and long-term outcomes in nondiabetic chronic kidney disease. Clin J Am Soc Nephrol. 2010;5:1582–1587. doi: 10.2215/CJN.01450210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shah DS, Polkinghorne KR, Pellicano R, et al. Are traditional risk factors valid for assessing cardiovascular risk in end-stage renal failure patients? Nephrology (Carlton) 2008;13:667–671. doi: 10.1111/j.1440-1797.2008.00982.x. [DOI] [PubMed] [Google Scholar]
- 26.Fox CS, Matsushita K, Woodward M, et al. Associations of kidney disease measures with mortality and end-stage renal disease in individuals with and without diabetes: a meta-analysis. Lancet. 2012;380:1662–1693. doi: 10.1016/S0140-6736(12)61350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tonelli M, Muntner P, Lloyd A, et al. Risk of coronary events in people with chronic kidney disease compared with those with diabetes: a population-level cohort study. Lancet. 2012;380:807–814. doi: 10.1016/S0140-6736(12)60572-8. [DOI] [PubMed] [Google Scholar]
- 28.van der Zee S, Baber U, Elmariah S, et al. Cardiovascular risk factors in patients with chronic kidney disease. Nat Rev Cardiol. 2009;6:580–589. doi: 10.1038/nrcardio.2009.121. [DOI] [PubMed] [Google Scholar]
- 29.Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–1504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- 30.Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 31.LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352:1425–1435. doi: 10.1056/NEJMoa050461. [DOI] [PubMed] [Google Scholar]
- 32.Kones R. Molecular sources of residual cardiovascular risk, clinical signals, and innovative solutions: relationship with subclinical disease, undertreatment, and poor adherence: implications of new evidence upon optimizing cardiovascular patient outcomes. Vasc Health Risk Manag. 2013;9:617–670. doi: 10.2147/VHRM.S37119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palmer SC, Navaneethan SD, Craig JC, et al. HMG CoA reductase inhibitors (statins) for people with chronic kidney disease not requiring dialysis. Cochrane Database Syst Rev. 2014;5:CD007784. doi: 10.1002/14651858.CD007784.pub2. [DOI] [PubMed] [Google Scholar]
- 34.Wanner C, Krane V, Marz W, et al. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med. 2005;353:238–248. doi: 10.1056/NEJMoa043545. [DOI] [PubMed] [Google Scholar]
- 35.Fellstrom BC, Jardine AG, Schmieder RE, et al. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N Engl J Med. 2009;360:1395–1407. doi: 10.1056/NEJMoa0810177. [DOI] [PubMed] [Google Scholar]
- 36.Tan HC, Tai ES, Sviridov D, et al. Relationships between cholesterol efflux and high-density lipoprotein particles in patients with type 2 diabetes mellitus. J Clin Lipidol. 2011;5:467–473. doi: 10.1016/j.jacl.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 37.Naik SU, Wang X, Da Silva JS, et al. Pharmacological activation of liver X receptors promotes reverse cholesterol transport in vivo. Circulation. 2006;113:90–97. doi: 10.1161/CIRCULATIONAHA.105.560177. [DOI] [PubMed] [Google Scholar]
- 38.Tardif JC, Gregoire J, L’Allier PL, et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA. 2007;297:1675–1682. doi: 10.1001/jama.297.15.jpc70004. [DOI] [PubMed] [Google Scholar]
- 39.Khera AV, Cuchel M, de la Llera-Moya M, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rohatgi A, Khera A, Berry JD, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–2393. doi: 10.1056/NEJMoa1409065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Voight BF, Peloso GM, Orho-Melander M, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johannsen TH, Kamstrup PR, Andersen RV, et al. Hepatic lipase, genetically elevated high-density lipoprotein, and risk of ischemic cardiovascular disease. J Clin Endocrinol Metab. 2009;94:1264–1273. doi: 10.1210/jc.2008-1342. [DOI] [PubMed] [Google Scholar]
- 43.Calabresi L, Baldassarre D, Simonelli S, et al. Plasma lecithin: cholesterol acyltransferase and carotid intima-media thickness in European individuals at high cardiovascular risk. J Lipid Res. 2011;52:1569–1574. doi: 10.1194/jlr.P014977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sirtori CR, Calabresi L, Franceschini G, et al. Cardiovascular status of carriers of the apolipoprotein A-I (Milano) mutant: the Limone sul Garda study. Circulation. 2001;103:1949–1954. doi: 10.1161/01.cir.103.15.1949. [DOI] [PubMed] [Google Scholar]
- 45.Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz GG, Olsson AG, Abt M, et al. dal-OUTCOMES Investigators. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 47.Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 48.Lamprea-Montealegre JA, Astor BC, McClelland RL, et al. CKD, plasma lipids, and common carotid intima-media thickness: results from the Multi-Ethnic Study of Atherosclerosis. Clin J Am Soc Nephrol. 2012;7:1777–1785. doi: 10.2215/CJN.02090212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shoji T, Masakane I, Watanabe Y, et al. Elevated non-high-density lipoprotein cholesterol (non-HDL-C) predicts atherosclerotic cardiovascular events in hemodialysis patients. Clin J Am Soc Nephrol. 2011;6:1112–1120. doi: 10.2215/CJN.09961110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Navaneethan SD, Schold JD, Kirwan JP, et al. Metabolic syndrome, ESRD, and death in CKD. Clin J Am Soc Nephrol. 2013;8:945–952. doi: 10.2215/CJN.09870912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kilpatrick RD, McAllister CJ, Kovesdy CP, et al. Association between serum lipids and survival in hemodialysis patients and impact of race. J Am Soc Nephrol. 2007;18:293–303. doi: 10.1681/ASN.2006070795. [DOI] [PubMed] [Google Scholar]
- 52.Kalantar-Zadeh K, Kopple JD, Kamranpour N, et al. HDL-inflammatory index correlates with poor outcome in hemodialysis patients. Kidney Int. 2007;72:1149–1156. doi: 10.1038/sj.ki.5002491. [DOI] [PubMed] [Google Scholar]
- 53.Silbernagel G, Genser B, Drechsler C, et al. HDL cholesterol, apolipoproteins, and cardiovascular risk in hemodialysis patients. J Am Soc Nephrol. 2015;26:484–492. doi: 10.1681/ASN.2013080816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moradi H, Streja E, Kashyap ML, et al. Elevated high-density lipoprotein cholesterol and cardiovascular mortality in maintenance hemodialysis patients. Nephrol Dial Transplant. 2014;29:1554–1562. doi: 10.1093/ndt/gfu022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zewinger S, Speer T, Kleber ME, et al. HDL cholesterol is not associated with lower mortality in patients with kidney dysfunction. J Am Soc Nephrol. 2014;25:1073–1082. doi: 10.1681/ASN.2013050482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duffy D, Holmes DN, Roe MT, et al. The impact of high-density lipoprotein cholesterol levels on long-term outcomes after non-ST-elevation myocardial infarction. Am Heart J. 2012;163:705–713. doi: 10.1016/j.ahj.2012.01.029. [DOI] [PubMed] [Google Scholar]
- 57.Corsetti JP, Zareba W, Moss AJ, et al. Elevated HDL is a risk factor for recurrent coronary events in a subgroup of non-diabetic postinfarction patients with hypercholesterolemia and inflammation. Atherosclerosis. 2006;187:191–197. doi: 10.1016/j.atherosclerosis.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 58.Corsetti JP, Salzman P, Ryan D, et al. Plasminogen activator inhibitor-2 polymorphism associates with recurrent coronary event risk in patients with high HDL and C-reactive protein levels. PLoS One. 2013;8:e68920. doi: 10.1371/journal.pone.0068920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Myasoedova E, Crowson CS, Kremers HM, et al. Lipid paradox in rheumatoid arthritis: the impact of serum lipid measures and systemic inflammation on the risk of cardiovascular disease. Ann Rheum Dis. 2011;70:482–487. doi: 10.1136/ard.2010.135871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Attman PO, Samuelsson O, Alaupovic P. Lipoprotein metabolism and renal failure. Am J Kidney Dis. 1993;21:573–592. doi: 10.1016/s0272-6386(12)80030-8. [DOI] [PubMed] [Google Scholar]
- 61.de Boer IH, Astor BC, Kramer H, et al. Lipoprotein abnormalities associated with mild impairment of kidney function in the multiethnic study of atherosclerosis. Clin J Am Soc Nephrol. 2008;3:125–132. doi: 10.2215/CJN.03390807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vaziri ND. Causes of dysregulation of lipid metabolism in chronic renal failure. Semin Dial. 2009;22:644–651. doi: 10.1111/j.1525-139X.2009.00661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vaziri ND, Navab M, Fogelman AM. HDL metabolism and activity in chronic kidney disease. Nat Rev Nephrol. 2010;6:287–296. doi: 10.1038/nrneph.2010.36. [DOI] [PubMed] [Google Scholar]
- 64.Miida T, Miyazaki O, Hanyu O, et al. LCAT-dependent conversion of prebeta1-HDL into alpha-migrating HDL is severely delayed in hemodialysis patients. J Am Soc Nephrol. 2003;14:732–738. doi: 10.1097/01.asn.0000046962.43220.8a. [DOI] [PubMed] [Google Scholar]
- 65.Rahman M, Yang W, Akkina S, et al. Relation of serum lipids and lipoproteins with progression of CKD: The CRIC study. Clin J Am Soc Nephrol. 2014;9:1190–1198. doi: 10.2215/CJN.09320913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Attman PO, Alaupovic P. Lipid and apolipoprotein profiles of uremic dyslipoproteinemia–relation to renal function and dialysis. Nephron. 1991;57:401–410. doi: 10.1159/000186303. [DOI] [PubMed] [Google Scholar]
- 67.Vaziri ND, Deng G, Liang K. Hepatic HDL receptor, SR-B1 and Apo A-I expression in chronic renal failure. Nephrol Dial Transplant. 1999;14:1462–1466. doi: 10.1093/ndt/14.6.1462. [DOI] [PubMed] [Google Scholar]
- 68.Moradi H, Said HM, Vaziri ND. Post-transcriptional nature of uremia-induced downregulation of hepatic apolipoprotein A-I production. Transl Res. 2013;161:477–485. doi: 10.1016/j.trsl.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kamanna VS, Kashyap ML, Pai R, et al. Uremic serum subfraction inhibits apolipoprotein A-I production by a human hepatoma cell line. J Am Soc Nephrol. 1994;5:193–200. doi: 10.1681/ASN.V52193. [DOI] [PubMed] [Google Scholar]
- 70.Shah GM, Lin ZL, Kamanna VS, et al. Effect of serum subfractions from peritoneal dialysis patients on Hep-G2 cell apolipoprotein A-I and B metabolism. Kidney Int. 1996;50:2079–2087. doi: 10.1038/ki.1996.532. [DOI] [PubMed] [Google Scholar]
- 71.Batista MC, Welty FK, Diffenderfer MR, et al. Apolipoprotein A-I, B-100, and B-48 metabolism in subjects with chronic kidney disease, obesity, and the metabolic syndrome. Metabolism. 2004;53:1255–1261. doi: 10.1016/j.metabol.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 72.Marsh JB, Welty FK, Schaefer EJ. Stable isotope turnover of apolipoproteins of high-density lipoproteins in humans. Curr Opin Lipidol. 2000;11:261–266. doi: 10.1097/00041433-200006000-00006. [DOI] [PubMed] [Google Scholar]
- 73.Graversen JH, Castro G, Kandoussi A, et al. A pivotal role of the human kidney in catabolism of HDL protein components apolipoprotein A-I and A-IV but not of A-II. Lipids. 2008;43:467–470. doi: 10.1007/s11745-008-3169-2. [DOI] [PubMed] [Google Scholar]
- 74.Ikewaki K. In vivo kinetic studies to further understand pathogenesis of abnormal lipoprotein metabolism in chronic kidney disease. Clin Exp Nephrol. 2014;18:261–264. doi: 10.1007/s10157-013-0881-x. [DOI] [PubMed] [Google Scholar]
- 75.Ooi EM, Chan DT, Watts GF, et al. Plasma apolipoprotein C-III metabolism in patients with chronic kidney disease. J Lipid Res. 2011;52:794–800. doi: 10.1194/jlr.M011163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shao B, Heinecke JW. Impact of HDL oxidation by the myeloperoxidase system on sterol efflux by the ABCA1 pathway. J Proteomics. 2011;74:2289–2299. doi: 10.1016/j.jprot.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cardinal H, Raymond MA, Hebert MJ, et al. Uraemic plasma decreases the expression of ABCA1, ABCG1 and cell-cycle genes in human coronary arterial endothelial cells. Nephrol Dial Transplant. 2007;22:409–416. doi: 10.1093/ndt/gfl619. [DOI] [PubMed] [Google Scholar]
- 78.Moradi H, Pahl MV, Elahimehr R, et al. Impaired antioxidant activity of high-density lipoprotein in chronic kidney disease. Transl Res. 2009;153:77–85. doi: 10.1016/j.trsl.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 79.Calabresi L, Pisciotta L, Costantin A, et al. The molecular basis of lecithin:cholesterol acyltransferase deficiency syndromes: a comprehensive study of molecular and biochemical findings in 13 unrelated Italian families. Arterioscler Thromb Vasc Biol. 2005;25:1972–1978. doi: 10.1161/01.ATV.0000175751.30616.13. [DOI] [PubMed] [Google Scholar]
- 80.Calabresi L, Simonelli S, Conca P, et al. Acquired lecithin:cholesterol acyltransferase deficiency as a major factor in lowering plasma HDL levels in chronic kidney disease. J Intern Med. 2015;277:552–561. doi: 10.1111/joim.12290. [DOI] [PubMed] [Google Scholar]
- 81.Vaziri ND, Liang K, Parks JS. Down-regulation of hepatic lecithin: cholesterol acyltransferase gene expression in chronic renal failure. Kidney Int. 2001;59:2192–2196. doi: 10.1046/j.1523-1755.2001.00734.x. [DOI] [PubMed] [Google Scholar]
- 82.Mekki K, Bouchenak M, Remaoun M, et al. Effect of long-term hemodialysis on plasma lecithin: cholesterol acyltransferase activity and the amounts and compositions of HDL2 and HDL3 in hemodialysis-treated patients with chronic renal failure: a 9-year longitudinal study. Med Sci Monit. 2004;10:CR439–CR446. [PubMed] [Google Scholar]
- 83.Fabre AC, Vantourout P, Champagne E, et al. Cell surface adenylate kinase activity regulates the F(1)-ATPase/P2Y (13)-mediated HDL endocytosis pathway on human hepatocytes. Cell Mol Life Sci. 2006;63:2829–2837. doi: 10.1007/s00018-006-6325-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liang K, Vaziri ND. Down-regulation of hepatic lipase expression in experimental nephrotic syndrome. Kidney Int. 1997;51:1933–1937. doi: 10.1038/ki.1997.263. [DOI] [PubMed] [Google Scholar]
- 85.Vaziri ND, Yuan J, Ni Z, et al. Lipoprotein lipase deficiency in chronic kidney disease is accompanied by down-regulation of endothelial GPIHBP1 expression. Clin Exp Nephrol. 2012;16:238–243. doi: 10.1007/s10157-011-0549-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pahl MV, Ni Z, Sepassi L, et al. Plasma phospholipid transfer protein, cholesteryl ester transfer protein and lecithin:cholesterol acyltransferase in end-stage renal disease (ESRD) Nephrol Dial Transplant. 2009;24:2541–2546. doi: 10.1093/ndt/gfp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kimura H, Miyazaki R, Imura T, et al. Hepatic lipase mutation may reduce vascular disease prevalence in hemodialysis patients with high CETP levels. Kidney Int. 2003;64:1829–1837. doi: 10.1046/j.1523-1755.2003.00285.x. [DOI] [PubMed] [Google Scholar]
- 88.de Sain-van der Velden MG, Kaysen GA, Barrett HA, et al. Increased VLDL in nephrotic patients results from a decreased catabolism while increased LDL results from increased synthesis. Kidney Int. 1998;53:994–1001. doi: 10.1111/j.1523-1755.1998.00831.x. [DOI] [PubMed] [Google Scholar]
- 89.Holzer M, Schilcher G, Curcic S, et al. Dialysis modalities and HDL composition and function. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2014030309. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Braschi S, Neville TA, Maugeais C, et al. Role of the kidney in regulating the metabolism of HDL in rabbits: evidence that iodination alters the catabolism of apolipoprotein A-I by the kidney. Biochemistry. 2000;39:5441–5449. doi: 10.1021/bi9919504. [DOI] [PubMed] [Google Scholar]
- 91.Glass CK, Pittman RC, Keller GA, et al. Tissue sites of degradation of apoprotein A-I in the rat. J Biol Chem. 1983;258:7161–7167. [PubMed] [Google Scholar]
- 92.Woollett LA, Spady DK. Kinetic parameters for high density lipoprotein apoprotein AI and cholesteryl ester transport in the hamster. J Clin Invest. 1997;99:1704–1713. doi: 10.1172/JCI119334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hammad SM, Barth JL, Knaak C, et al. Megalin acts in concert with cubilin to mediate endocytosis of high density lipoproteins. J Biol Chem. 2000;275:12003–12008. doi: 10.1074/jbc.275.16.12003. [DOI] [PubMed] [Google Scholar]
- 94.Kozyraki R, Fyfe J, Kristiansen M, et al. The intrinsic factor-vitamin B12 receptor, cubilin, is a high-affinity apolipoprotein A-I receptor facilitating endocytosis of high-density lipoprotein. Nat Med. 1999;5:656–661. doi: 10.1038/9504. [DOI] [PubMed] [Google Scholar]
- 95.Moestrup SK, Nielsen LB. The role of the kidney in lipid metabolism. Curr Opin Lipidol. 2005;16:301–306. doi: 10.1097/01.mol.0000169350.45944.d4. [DOI] [PubMed] [Google Scholar]
- 96.Dallinga-Thie GM, Van’t Hooft FM, Van Tol A. Tissue sites of degradation of high density lipoprotein apolipoprotein A-IV in rats. Arteriosclerosis. 1986;6:277–284. doi: 10.1161/01.atv.6.3.277. [DOI] [PubMed] [Google Scholar]
- 97.Lee JY, Timmins JM, Mulya A, et al. HDLs in apoA-I transgenic Abca1 knockout mice are remodeled normally in plasma but are hyper-catabolized by the kidney. J Lipid Res. 2005;46:2233–2245. doi: 10.1194/jlr.M500179-JLR200. [DOI] [PubMed] [Google Scholar]
- 98.Timmins JM, Lee JY, Boudyguina E, et al. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J Clin Invest. 2005;115:1333–1342. doi: 10.1172/JCI23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schaefer EJ, Blum CB, Levy RI, et al. Metabolism of high-density lipoprotein apolipoproteins in Tangier disease. N Engl J Med. 1978;299:905–910. doi: 10.1056/NEJM197810262991701. [DOI] [PubMed] [Google Scholar]
- 100.Oram JF. Tangier disease and ABCA1. Biochim Biophys Acta. 2000;1529:321–330. doi: 10.1016/s1388-1981(00)00157-8. [DOI] [PubMed] [Google Scholar]
- 101.Rader DJ, Ikewaki K, Duverger N, et al. Markedly accelerated catabolism of apolipoprotein A-II (ApoA-II) and high density lipoproteins containing ApoA-II in classic lecithin: cholesterol acyltransferase deficiency and fish-eye disease. J Clin Invest. 1994;93:321–330. doi: 10.1172/JCI116962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Aseem O, Smith BT, Cooley MA, et al. Cubilin maintains blood levels of HDL and albumin. J Am Soc Nephrol. 2014;25:1028–1036. doi: 10.1681/ASN.2013060671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Soto-Miranda E, Carreon-Torres E, Lorenzo K, et al. Shift of high-density lipoprotein size distribution toward large particles in patients with proteinuria. Clin Chim Acta. 2012;414:241–245. doi: 10.1016/j.cca.2012.09.028. [DOI] [PubMed] [Google Scholar]
- 104.Chaturvedi N, Fuller JH, Taskinen MR, et al. Differing associations of lipid and lipoprotein disturbances with the macrovascular and microvascular complications of type 1 diabetes. Diabetes Care. 2001;24:2071–2077. doi: 10.2337/diacare.24.12.2071. [DOI] [PubMed] [Google Scholar]
- 105.Kahri J, Groop PH, Elliott T, et al. Plasma cholesteryl ester transfer protein and its relationship to plasma lipoproteins and apolipoprotein A-I-containing lipoproteins in IDDM patients with microalbuminuria and clinical nephropathy. Diabetes Care. 1994;17:412–419. doi: 10.2337/diacare.17.5.412. [DOI] [PubMed] [Google Scholar]
- 106.Shearer GC, Couser WG, Kaysen GA. Nephrotic livers secrete normal VLDL that acquire structural and functional defects following interaction with HDL. Kidney Int. 2004;65:228–237. doi: 10.1111/j.1523-1755.2004.00373.x. [DOI] [PubMed] [Google Scholar]
- 107.Shearer GC, Newman JW, Hammock BD, et al. Graded effects of proteinuria on HDL structure in nephrotic rats. J Am Soc Nephrol. 2005;16:1309–1319. doi: 10.1681/ASN.2004080644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shearer GC, Stevenson FT, Atkinson DN, et al. Hypoalbuminemia and proteinuria contribute separately to reduced lipoprotein catabolism in the nephrotic syndrome. Kidney Int. 2001;59:179–189. doi: 10.1046/j.1523-1755.2001.00478.x. [DOI] [PubMed] [Google Scholar]
- 109.Newman JW, Kaysen GA, Hammock BD, et al. Proteinuria increases oxylipid concentrations in VLDL and HDL but not LDL particles in the rat. J Lipid Res. 2007;48:1792–1800. doi: 10.1194/jlr.M700146-JLR200. [DOI] [PubMed] [Google Scholar]
- 110.Sarnak MJ, Astor BC. Implications of proteinuria: CKD progression and cardiovascular outcomes. Adv Chronic Kidney Dis. 2011;18:258–266. doi: 10.1053/j.ackd.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 111.Gerstein HC, Mann JF, Yi Q, et al. Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic individuals. JAMA. 2001;286:421–426. doi: 10.1001/jama.286.4.421. [DOI] [PubMed] [Google Scholar]
- 112.Diercks GF, van Boven AJ, Hillege HL, et al. Microalbuminuria is independently associated with ischaemic electrocardiographic abnormalities in a large non-diabetic population. The PREVEND (Prevention of REnal and Vascular ENdstage Disease) study. Eur Heart J. 2000;21:1922–1927. doi: 10.1053/euhj.2000.2248. [DOI] [PubMed] [Google Scholar]
- 113.Hillege HL, Fidler V, Diercks GF, et al. Urinary albumin excretion predicts cardiovascular and noncardiovascular mortality in general population. Circulation. 2002;106:1777–1782. doi: 10.1161/01.cir.0000031732.78052.81. [DOI] [PubMed] [Google Scholar]
- 114.Arnlov J, Evans JC, Meigs JB, et al. Low-grade albuminuria and incidence of cardiovascular disease events in nonhypertensive and nondiabetic individuals: the Framingham Heart Study. Circulation. 2005;112:969–975. doi: 10.1161/CIRCULATIONAHA.105.538132. [DOI] [PubMed] [Google Scholar]
- 115.Hemmelgarn BR, Manns BJ, Lloyd A, et al. Relation between kidney function, proteinuria, and adverse outcomes. JAMA. 2010;303:423–429. doi: 10.1001/jama.2010.39. [DOI] [PubMed] [Google Scholar]
- 116.Weichhart T, Kopecky C, Kubicek M, et al. Serum amyloid A in uremic HDL promotes inflammation. J Am Soc Nephrol. 2012;23:934–947. doi: 10.1681/ASN.2011070668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Holzer M, Birner-Gruenberger R, Stojakovic T, et al. Uremia alters HDL composition and function. J Am Soc Nephrol. 2011;22:1631–1641. doi: 10.1681/ASN.2010111144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mange A, Goux A, Badiou S, et al. HDL proteome in hemodialysis patients: a quantitative nanoflow liquid chromatography-tandem mass spectrometry approach. PLoS One. 2012;7:e34107. doi: 10.1371/journal.pone.0034107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tolle M, Huang T, Schuchardt M, et al. High-density lipoprotein loses its anti-inflammatory capacity by accumulation of pro-inflammatory-serum amyloid A. Cardiovasc Res. 2012;94:154–162. doi: 10.1093/cvr/cvs089. [DOI] [PubMed] [Google Scholar]
- 120.Kopecky C, Haidinger M, Birner-Grunberger R, et al. Restoration of renal function does not correct impairment of uremic HDL properties. J Am Soc Nephrol. 2014;26:565–575. doi: 10.1681/ASN.2013111219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shao B, Pennathur S, Pagani I, et al. Modifying apolipoprotein A-I by malondialdehyde, but not by an array of other reactive carbonyls, blocks cholesterol efflux by the ABCA1 pathway. J Biol Chem. 2010;285:18473–18484. doi: 10.1074/jbc.M110.118182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shao B, Tang C, Heinecke JW, et al. Oxidation of apolipoprotein A-I by myeloperoxidase impairs the initial interactions with ABCA1 required for signaling and cholesterol export. J Lipid Res. 2010;51:1849–1858. doi: 10.1194/jlr.M004085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shao B, O’Brien KD, McDonald TO, et al. Acrolein modifies apolipoprotein A-I in the human artery wall. Ann NY Acad Sci. 2005;1043:396–403. doi: 10.1196/annals.1333.046. [DOI] [PubMed] [Google Scholar]
- 124.Shao B, Oda MN, Bergt C, et al. Myeloperoxidase impairs ABCA1-dependent cholesterol efflux through methionine oxidation and site-specific tyrosine chlorination of apolipoprotein A-I. J Biol Chem. 2006;281:9001–9004. doi: 10.1074/jbc.C600011200. [DOI] [PubMed] [Google Scholar]
- 125.Shao B. Site-specific oxidation of apolipoprotein A-I impairs cholesterol export by ABCA1, a key cardioprotective function of HDL. Biochim Biophys Acta. 2012;1821:490–501. doi: 10.1016/j.bbalip.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zheng L, Nukuna B, Brennan ML, et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest. 2004;114:529–541. doi: 10.1172/JCI21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wu Z, Wagner MA, Zheng L, et al. The refined structure of nascent HDL reveals a key functional domain for particle maturation and dysfunction. Nat Struct Mol Biol. 2007;14:861–868. doi: 10.1038/nsmb1284. [DOI] [PubMed] [Google Scholar]
- 128.Undurti A, Huang Y, Lupica JA, et al. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J Biol Chem. 2009;284:30825–30835. doi: 10.1074/jbc.M109.047605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang Z, Nicholls SJ, Rodriguez ER, et al. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med. 2007;13:1176–1184. doi: 10.1038/nm1637. [DOI] [PubMed] [Google Scholar]
- 130.Kraus LM, Kraus AP., Jr Carbamoylation of amino acids and proteins in uremia. Kidney Int Suppl. 2001;78:S102–S107. doi: 10.1046/j.1523-1755.2001.59780102.x. [DOI] [PubMed] [Google Scholar]
- 131.Koeth RA, Kalantar-Zadeh K, Wang Z, et al. Protein carbamylation predicts mortality in ESRD. J Am Soc Nephrol. 2013;24:853–861. doi: 10.1681/ASN.2012030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Berg AH, Drechsler C, Wenger J, et al. Carbamylation of serum albumin as a risk factor for mortality in patients with kidney failure. Sci Transl Med. 2013;5:175ra129. doi: 10.1126/scitranslmed.3005218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Holzer M, Zangger K, El-Gamal D, et al. Myeloperoxidase-derived chlorinating species induce protein carbamylation through decomposition of thiocyanate and urea: novel pathways generating dysfunctional high-density lipoprotein. Antioxid Redox Signal. 2012;17:1043–1052. doi: 10.1089/ars.2011.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.El-Gamal D, Rao SP, Holzer M, et al. The urea decomposition product cyanate promotes endothelial dysfunction. Kidney Int. 2014;86:923–931. doi: 10.1038/ki.2014.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Passarelli M, Tang C, McDonald TO, et al. Advanced glycation end product precursors impair ABCA1-dependent cholesterol removal from cells. Diabetes. 2005;54:2198–2205. doi: 10.2337/diabetes.54.7.2198. [DOI] [PubMed] [Google Scholar]
- 136.Brown BE, Nobecourt E, Zeng J, et al. Apolipoprotein A-I glycation by glucose and reactive aldehydes alters phospholipid affinity but not cholesterol export from lipid-laden macrophages. PLoS One. 2013;8:e65430. doi: 10.1371/journal.pone.0065430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57:1446–1454. doi: 10.2337/db08-0057. [DOI] [PubMed] [Google Scholar]
- 138.Rye KA, Barter PJ. Regulation of high-density lipoprotein metabolism. Circ Res. 2014;114:143–156. doi: 10.1161/CIRCRESAHA.114.300632. [DOI] [PubMed] [Google Scholar]
- 139.Bacchetti T, Masciangelo S, Armeni T, et al. Glycation of human high density lipoprotein by methylglyoxal: effect on HDL-paraoxonase activity. Metabolism. 2014;63:307–311. doi: 10.1016/j.metabol.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 140.Matafome P, Sena C, Seica R. Methylglyoxal, obesity, and diabetes. Endocrine. 2013;43:472–484. doi: 10.1007/s12020-012-9795-8. [DOI] [PubMed] [Google Scholar]
- 141.Annema W, von Eckardstein A. High-density lipoproteins. Multifunctional but vulnerable protections from atherosclerosis. Circ J. 2013;77:2432–2448. doi: 10.1253/circj.cj-13-1025. [DOI] [PubMed] [Google Scholar]
- 142.Yamamoto S, Yancey PG, Ikizler TA, et al. Dysfunctional high-density lipoprotein in patients on chronic hemodialysis. J Am Coll Cardiol. 2012;60:2372–2379. doi: 10.1016/j.jacc.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shroff R, Speer T, Colin S, et al. HDL in children with CKD promotes endothelial dysfunction and an abnormal vascular phenotype. J Am Soc Nephrol. 2014;25:2658–2668. doi: 10.1681/ASN.2013111212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhu X, Lee JY, Timmins JM, et al. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances proinflammatory response of macrophages. J Biol Chem. 2008;283:22930–22941. doi: 10.1074/jbc.M801408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Baragetti A, Norata GD, Sarcina C, et al. High density lipoprotein cholesterol levels are an independent predictor of the progression of chronic kidney disease. J Intern Med. 2013;274:252–262. doi: 10.1111/joim.12081. [DOI] [PubMed] [Google Scholar]
- 146.Kaseda R, Fazio S, Kon V. Chronic kidney disease (CKD) disruption of macrophage lipid handling and inflammation and effect of liver X receptor (LXR) agonism. ASN Kidney Week. 2014;25:869. [Google Scholar]
- 147.Kaseda R, Jabs K, Hunley TE, et al. Dysfunctional high-density lipoproteins in children with chronic kidney disease. Metabolism. 2015;64:263–273. doi: 10.1016/j.metabol.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Speer T, Rohrer L, Blyszczuk P, et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity. 2013;38:754–768. doi: 10.1016/j.immuni.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 149.Li XM, Tang WH, Mosior MK, et al. Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler Thromb Vasc Biol. 2013;7:1696–1705. doi: 10.1161/ATVBAHA.113.301373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tolle M, Pawlak A, Schuchardt M, et al. HDL-associated lysosphin-golipids inhibit NAD(P)H oxidase-dependent monocyte chemoattractant protein-1 production. Arterioscler Thromb Vasc Biol. 2008;28:1542–1548. doi: 10.1161/ATVBAHA.107.161042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Navab M, Anantharamaiah GM, Fogelman AM. The effect of apolipoprotein mimetic peptides in inflammatory disorders other than atherosclerosis. Trends Cardiovasc Med. 2008;18:61–66. doi: 10.1016/j.tcm.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Morena M, Cristol JP, Dantoine T, et al. Protective effects of high-density lipoprotein against oxidative stress are impaired in haemodialysis patients. Nephrol Dial Transplant. 2000;15:389–395. doi: 10.1093/ndt/15.3.389. [DOI] [PubMed] [Google Scholar]
- 153.Holzer M, Gauster M, Pfeifer T, et al. Protein carbamylation renders high-density lipoprotein dysfunctional. Antioxid Redox Signal. 2011;14:2337–2346. doi: 10.1089/ars.2010.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Honda H, Ueda M, Kojima S, et al. Oxidized high-density lipoprotein as a risk factor for cardiovascular events in prevalent hemodialysis patients. Atherosclerosis. 2012;220:493–501. doi: 10.1016/j.atherosclerosis.2011.10.038. [DOI] [PubMed] [Google Scholar]
- 155.Kennedy DJ, Tang WH, Fan Y, et al. Diminished antioxidant activity of high-density lipoprotein-associated proteins in chronic kidney disease. J Am Heart Assoc. 2013;2:e000104. doi: 10.1161/JAHA.112.000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Van Lenten BJ, Hama SY, de Beer FC, et al. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J Clin Invest. 1995;96:2758–2767. doi: 10.1172/JCI118345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Artl A, Marsche G, Lestavel S, et al. Role of serum amyloid A during metabolism of acute-phase HDL by macrophages. Arterioscler Thromb Vasc Biol. 2000;20:763–772. doi: 10.1161/01.atv.20.3.763. [DOI] [PubMed] [Google Scholar]
- 158.Marsche G, Frank S, Raynes JG, et al. The lipidation status of acute-phase protein serum amyloid A determines cholesterol mobilization via scavenger receptor class B, type I. Biochem J. 2007;402:117–124. doi: 10.1042/BJ20061406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.van der Westhuyzen DR, Cai L, de Beer MC, et al. Serum amyloid A promotes cholesterol efflux mediated by scavenger receptor B-I. J Biol Chem. 2005;280:35890–35895. doi: 10.1074/jbc.M505685200. [DOI] [PubMed] [Google Scholar]
- 160.Banka CL, Yuan T, de Beer MC, et al. Serum amyloid A (SAA): influence on HDL-mediated cellular cholesterol efflux. J Lipid Res. 1995;36:1058–1065. [PubMed] [Google Scholar]
- 161.De Nardo D, Labzin LI, Kono H, et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat Immunol. 2014;15:152–160. doi: 10.1038/ni.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Moody WE, Edwards NC, Madhani M, et al. Endothelial dysfunction and cardiovascular disease in early-stage chronic kidney disease: cause or association? Atherosclerosis. 2012;223:86–94. doi: 10.1016/j.atherosclerosis.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 163.Besler C, Heinrich K, Rohrer L, et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J Clin Invest. 2011;121:2693–2708. doi: 10.1172/JCI42946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Riwanto M, Landmesser U. High density lipoproteins and endothelial functions: mechanistic insights and alterations in cardiovascular disease. J Lipid Res. 2013;54:3227–3243. doi: 10.1194/jlr.R037762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chitalia VC, Murikipudi S, Indolfi L, et al. Matrix-embedded endothelial cells are protected from the uremic milieu. Nephrol Dial Transplant. 2011;26:3858–3865. doi: 10.1093/ndt/gfr337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Serradell M, Diaz-Ricart M, Cases A, et al. Uraemic medium accelerates proliferation but does not induce apoptosis of endothelial cells in culture. Nephrol Dial Transplant. 2003;18:1079–1085. doi: 10.1093/ndt/gfg161. [DOI] [PubMed] [Google Scholar]
- 167.Bode-Boger SM, Scalera F, Kielstein JT, et al. Symmetrical dimethylarginine: a new combined parameter for renal function and extent of coronary artery disease. J Am Soc Nephrol. 2006;17:1128–1134. doi: 10.1681/ASN.2005101119. [DOI] [PubMed] [Google Scholar]
- 168.Kielstein JT, Veldink H, Martens-Lobenhoffer J, et al. SDMA is an early marker of change in GFR after living-related kidney donation. Nephrol Dial Transplant. 2011;26:324–328. doi: 10.1093/ndt/gfq395. [DOI] [PubMed] [Google Scholar]