

In this issue of Science Signaling, Hongbo Chi’s group describes the role of the p38α-MAPK pathway in driving inflammation in experimental autoimmune encephalomyelitis (EAE), the murine model for multiple sclerosis and the immunologist’s favorite go-to tool for defining cellular mechanisms of autoimmunity. Mitogen activated protein kinases (MAPK) are activated by a myriad ligands and stimuli, including inflammatory cytokines, chemokines, pattern recognition receptors, etc. There are 14 mammalian MAPKs that act primarily in three major signaling cascades, known commonly as the p38, ERK and Jun kinase (JNK) pathways [1]. The p38 MAPK pathway, in particular, drives many of the acute inflammatory events that occur after infection or injury, leading to expression of early genes involved in the innate immune response. Consequently, numerous drugs designed to block this pathway are in use or under development to treat autoimmunity, immunopathologies such as COPD, or cancer [1]. However, since p38-MAPKs are activated by a myriad of stimuli, delineating their roles in specific diseases in a particular sequence of events is challenging.
The Chi lab previously demonstrated that p38α was crucial in DC, but not T cells or macrophages, during the initiation phase of EAE [2]. DC p38α signaling induced by PRR and/or T cell costimulation enhanced IL-6 and inhibited IL-27 production to promote the generation of Th17 cells. In the current paper, the authors use several elegant genetic approaches to further decipher the role of the p38α pathway in EAE and the IL-17 effector cytokine response in the CNS. Using a tamoxifen-inducible knockout system (p38CreER mice), they demonstrate that deletion of p38α at the onset of clinical disease alleviates symptoms. When EAE was induced by passive transfer of MOG-reactive Th17 cells, p38α was required in recipient mice, again pointing to a role for p38α in the effector phase. To demonstrate a role for CNS-resident cells, they created conditional knockout mice lacking p38α in Nestin+ cells. The Nestin-CRE transgene is specific to CNS-resident cells such as neurons, astrocytes and oligodendrocytes. Consistently, the p38αNesCre mice showed delayed onset and reduced severity of EAE, correlating with reduced immune cell infiltration. In both the p38αNesCre mice and the tamoxifen-targeted p38CreER mice, generation of antigen-specific Th1 and Th17 cells was not impaired, nor was their activity affected ex vivo. Rather, the numbers of CNS-infiltrating cells were suppressed, correlating with reduced expression of chemokines such as Ccl5, Cxcl2 and Cxcl1, thus suggesting a defect in leukocyte cell recruitment rather than generation.
The reduced expression of various chemokines and inflammatory genes seen in these mice was reminiscent of signals characteristically associated with IL-17 [3]. IL-17 is well established as a critical cytokine for EAE induction. Mice made deficient in IL-17A or IL-17RA show varying degrees of resistance to EAE induction ([4], authors’ unpublished data). Deletion of another IL-17 signaling intermediate, Act1, in CNS-resident cells also results in resistance to EAE induction, with similarly reduced chemokine production by astrocytes [5]. One note of caution is that nestin exhibits a rather ubiquitous expression in the CNS, so nestin-Cre deletes p38α in several cell types. Indeed, while Kang et al found significant defects in astrocyte responses using Actfl/flnestinCre mice, subsequent analysis using more restricted Cre promoters revealed that oligodendrocytes were the more dominant CNS target of IL-17 pathogenesis [6]. In the current study, Huang et al. demonstrate a clear role for p38α in astrocytes, but whether it is required in neurons or oligodendrocytes remains uncertain.
IL-17 is known to activate all three major MAPK pathways, though which pathway is dominantly activated varies by cell type [7]. Accordingly, the authors used specific anti-IL-17A Abs to confirm a mechanistic connection between p38α and IL-17 in EAE pathogenesis. Anti-IL-17A Abs partially, though not fully, reversed the phenotype. Thus, a p38α deficiency leads to reduced disease in EAE due at least in part to reduced IL-17 signal transduction in CNS-resident cells.
To demonstrate the link between p38α and IL-17 more directly, the authors cultured MEFs and astrocytes derived from p38CreER mice. IL-17-driven gene expression was indeed impaired in both cell types, indicating that there is an IL-17 signaling defect due to p38α-deficiency. CREB and MAPK-activated protein kinase 2 (MK2) are direct targets of p38α, and not surprisingly MK2 activation was impaired in response to IL-17 signaling in p38CreER cells. In contrast, IL-17-induced activation of CREB, NF-κB and ERK and were not affected. How does the p38α/MK2 axis control IL-17-induced gene expression? A major activity of MK2 is to phophorylate tristetraprolin (TTP), an RNA binding protein that destabilizes certain cytokine mRNA transcripts [1]. In this regard, IL-17 mediates stabilization of Cxcl1 mRNA and other chemokine transcripts, although interestingly in a TTP-independent manner [8]. In this setting the authors observed no effect of mRNA stability, at least for selected chemokines (Cxcl1, Cxcl2).
Finally, activation of inflammation must be kept in check in order to limit potential collateral tissue damage. In the case of the p38α pathway, the dual specificity phosphatase MKP1 (also known as DUSP1) is activated by MK1 and dephosphorylates p38α, thereby serving as a feedback inhibitor [1]. As would be expected, MKP1−/− mice (reconstituted with WT bone marrow) had the opposite phenotype of p38α-deficient mice, with exacerbated EAE symptoms and increased leukocyte infiltration. Consistent with IL-17 involvement, MKP-1-deficient MEF cells exhibited enhanced IL-17-dependent signaling. Therefore, both positive and negative regulation of the p38α signaling pathway contributes to immunoregulation during CNS inflammation.
Like any good study, this work raises intriguing new questions. Although IL-17A blockade reduced the impact of the p38α deletion during EAE, disease was not fully reversed, suggesting that other factors might be involved. An obvious candidate is GM-CSF, but this was not defined [9, 10]. The role of MKP-1 as a negative regulator is intriguing, but it would have been nice to see if this is a direct negative feedback loop initiated by the IL-17 pathway; in other words, is MKP-1 induced directly by IL-17, or regulated by crosstalk from other stimuli in the environment? Another open question relates to how IL-17 regulates downstream target gene expression through p38α. Since the effect of p38α seems to be independent of mRNA stability, which transcription factors are the targets of p38α? The AP1 complex is likely to be a target, although the AP1 binding site is dispensable for IL-17-dependent activation of the proximal promoter of at least one relevant target gene (IL-6) [11]. Finally, is p38α involved in IL-17 signaling in oligodendrocytes or is its activity truly specific to astrocytes? In summary, while activation and regulation of MAPK pathway is highly complex, this work convincingly limits its functional importance to a specific phase and cell type during CNS inflammation.
Figure. Role of p38α in IL-17-mediated signaling during EAE.
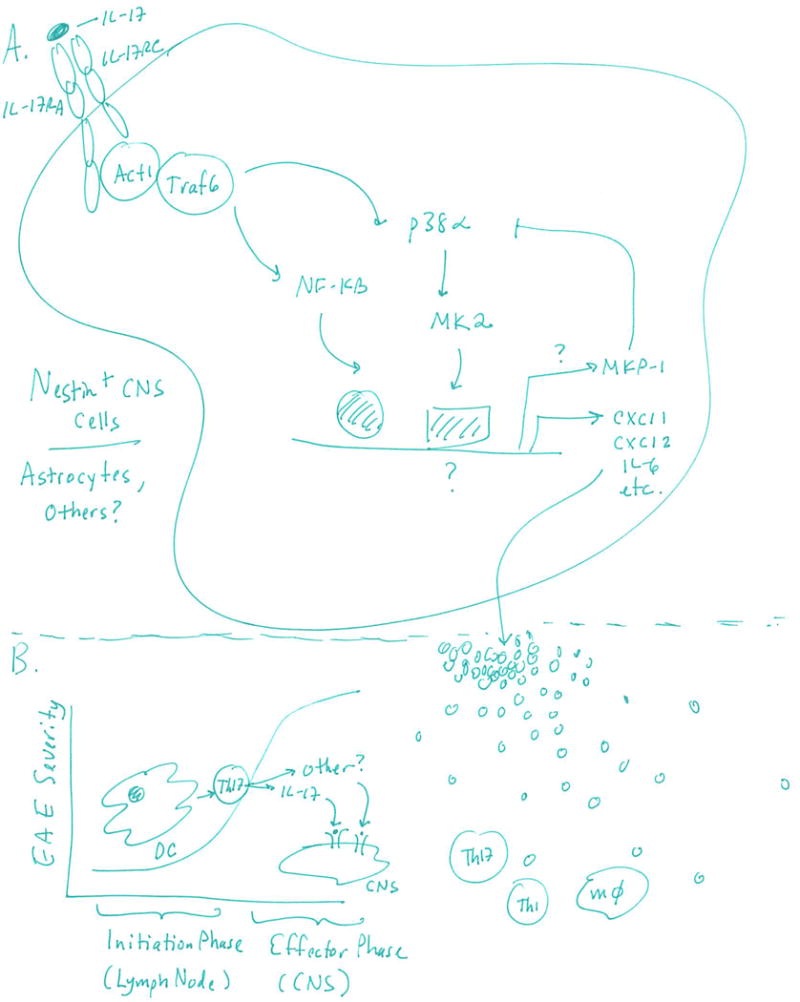
A. IL-17 binds to its receptor (IL-17RA, IL-17RC) on Nestin+ CNS-resident cells to activate Act1 and TRAF6, which in turn lead to activation of NF-κB and p38α. MK2 is a direct target of p38α phosphorylation and is essential for target gene expression. B. Chemokines and cytokines induced by the IL-17/p38α axis lead to recruitment of immune cells, particularly macrophages, Th17 and Th1 cells, which exert pathogenic effects in EAE through expression of IL-17 and probably additional cytokines. P38α expression in DCs was previously shown to be important during the initiation phase of EAE (Huang et al., Nature Immunol 2012), whereas this report shows that p38α expression in Nestin+ CNS-resident cells is important for IL-17 responsiveness during the effector phase of the disease.
References
- 1.Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13(9):679–92. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- 2.Huang G, et al. Signaling via the kinase p38alpha programs dendritic cells to drive TH17 differentiation and autoimmune inflammation. Nat Immunol. 2012;13(2):152–61. doi: 10.1038/ni.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Onishi R, Gaffen SL. IL-17 and its Target Genes: Mechanisms ofIL-17 Function in Disease. Immunology. 2010;129:311–321. doi: 10.1111/j.1365-2567.2009.03240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwakura Y, et al. Functional specialization of interleukin-17 family members. Immunity. 2011;34:149–162. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 5.Kang Z, et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32(3):414–25. doi: 10.1016/j.immuni.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang Z, et al. Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. NatNeurosci. 2013;16(10):1401–8. doi: 10.1038/nn.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaffen SL, et al. IL-23-IL-17 immune axis: Discovery, mechanistic understanding and clinical therapy. Nat Rev Immunol. 2014;14(9):585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Datta S, et al. IL-17 regulates CXCL1 mRNA stability via an AUUUA/tristetraprolin-independent sequence. J Immunol. 2010;184(3):1484–91. doi: 10.4049/jimmunol.0902423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Behi M, et al. The encephalitogenicity ofT(H)1 7 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12(6):568–75. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Codarri L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12(6):560–7. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 11.Ruddy MJ, et al. Functional cooperation between interleukin-1 7 and tumor necrosis factor-a is mediated by CCAAT/enhancer binding protein family members. J Biol Chem. 2004;279(4):2559–2567. doi: 10.1074/jbc.M308809200. [DOI] [PubMed] [Google Scholar]