

Abstract
Proximity enhancement is a central chemical tenet underpinning an exciting suite of small-molecule toolsets that have allowed us to unravel many biological complexities. The leitmotif of this opus is “tethering”—a strategy in which a multifunctional small molecule serves as a template to bring proteins/biomolecules together. Scaffolding approaches have been powerfully applied to control diverse biological outcomes such as protein–protein association, protein stability, activity, and improve imaging capabilities. A new twist on this strategy has recently appeared, in which the small-molecule probe is engineered to unleash controlled amounts of reactive chemical signals within the microenvironment of a target protein. Modification of a specific target elicits a precisely timed and spatially controlled gain-of-function (or dominant loss-of-function) signaling response. Presented herein is a unique personal outlook conceptualizing the powerful proximity-enhanced chemical biology toolsets into two paradigms: “multifunctional scaffolding” versus “on-demand targeting”. By addressing the latest advances and challenges in the established yet constantly evolving multifunctional scaffolding strategies as well as in the emerging on-demand precision targeting (and related) systems, this Perspective is aimed at choosing when it is best to employ each of the two strategies, with an emphasis toward further promoting novel applications and discoveries stemming from these innovative chemical biology platforms.
Better Together? Remodeling Proximity-Targeted Organic Chemistry
In the post-genomic era, emphasis is placed on understanding how various biological macromolecules specifically interact and assemble, either through protein–protein or protein–ligand associations, to orchestrate dynamic responses that influence physiologic decision-making.1,2 This impetus has sparked new methods to interrogate how these seemingly minor alterations drive phenotypic responses.3−7 The challenges in this field are manifold: there are over 20 000 protein-coding genes in humans8—and even more macromolecule or small-molecule potential binding partners. All these components can modulate the functions and dynamics of the interactome specific to individual gene products. Thus, identifying target(s), binding sites, and the nature of the input signal, and ultimately forging a precise link between these myriad upstream events and a phenotypic output is no easy task. Arguably, the go-to chemical biology method to engender specificity in these highly interconnected networks—space, time, gene, or pathway—is proximity enhancement. This fundamental tenet has been employed to resolve complex biological quandaries. This Perspective conceptualizes the latest proximity-enhanced design strategies with an eye toward stimulating further innovation and highlighting areas where improvements are required. We split chemical tethering into two classes (Figure 1) and systematically discuss the merits and drawbacks of both. Through this organization, we hope to help the researcher decide the ideal strategy to solve a specific biological problem, and to further seed new inspirations and spur on next-generation chemical biology innovations.
Figure 1.
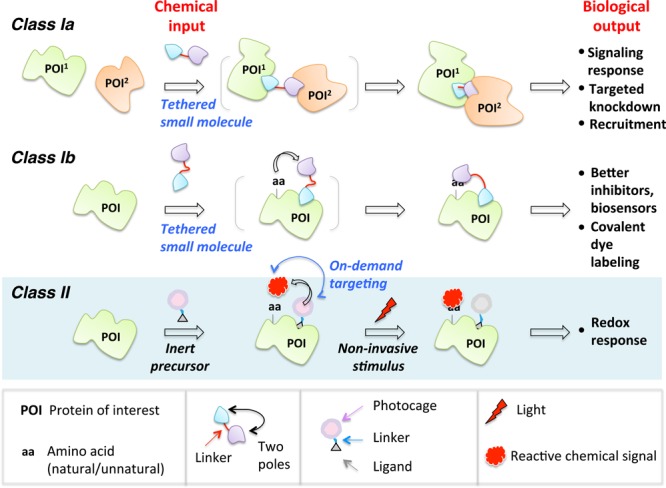
In this Perspective, the chemical biology toolsets built upon proximity enhancement are classified into two general categories, Class I and Class II. Class I is further broken into two subclasses: in Subclass Ia, the bifunctional probe enables recruitment of two or more distinct biological entities (proteins/cells), thereby promoting a response; and in Subclass Ib, one pole of the small molecule serves as an anchor to the POI and the other pole reacts intramolecularly with the same POI. In Class II, on-demand precision targeting enables the reactive entity to be unmasked in situ and targeted to (or the microenvironment of) POI. In all cases, in the absence of ligands known to bind POI, ligands generic to various protein- and peptide-based tags (Halo, CLIP, SNAP, PRIME, etc.) that can be fused to POIs may be integrated into the probes.
Tethering is the blueprint of established proximity-augmented heterofunctional small-molecule probes. We define Class I proximity enhancement to be a system in which the chemical linker between the two poles of a bifunctional small-molecule probe is unbroken throughout an experiment (Figure 1). There is widespread use and diverse applications of the Class I strategy in biology, yet due to the rapid expansion of the field, it is timely to re-stress the parallels of all these approaches at the chemical level while tipping our hat to the huge diversity they can engender at the biological level.6,7,9−16 The second act of this Perspective broaches Class II proximity enhancement. Instead of employing permanently tethered small-molecule scaffolds, Class II uses a bifunctional small molecule that contains a recognition unit at one pole but at the other a latent warhead (such as a sensitizer or a photocaged precursor) that can be activated in situ (Figure 1). Early incarnations of this new concept have interrogated specific redox response by targeted redox signaling on demand.17−19 Related small-molecule-directed in situ unmasking ideas of reactive entities have also been powerfully applied in characterizing nascent interactomes such as mitochondrial proteome mapping20−23 and ribosome profiling.24,25 We focus on the common concepts that drive and ultimately limit these emerging strategies. Astoundingly, because the core chemical concepts are similar, the respective classes show similar strengths and limitations, implying that there may be cross-cutting solutions to some of these problems. In most cases, we have limited ourselves to examples from the past 2–3 years.
Class I. Two Types of Multicomponent Tethers
The seminal proof of concept for the power of tethered small-molecule probes in biology was “chemical inducers of protein dimerization” (CID).14,26 CID uses a bifunctional small molecule that interacts with two separate proteins of interest (POIs) to induce the formation of an active complex or localization-specific recruited state. The CID-induced protein dimerization event was shown to be sufficient to prompt tripping of signaling nodes, eliciting selective pathway activation. CID was first introduced in 199326 and proved to be a game-changer in the field that spawned many tethered small-molecule modulators. These bifunctional tethered probes were successfully put to task to help understand diverse biological quandaries.13,27,28 Herein we subcategorize the established scaffolding systems into two subtypes: Class Ia, in which bifunctional small molecules forge new intermolecular interactions, such as heterodimerization, that in turn elicits a downstream biological response (typically a gain of function or increase in a background rate); and Class Ib, where two termini of the bifunctional small molecule interact with the same target POI, such as in site-specific irreversible dye labeling or covalent enzyme inactivation (Figure 1). We stress that although applications that stem from both subclasses are diverse and far-reaching, they are unified by the elegant simplicity of a design based upon proximity promoting a response/interaction. Beyond CID-based implementation of tethered probes,13,27,28 recent examples in the first subclass include proteasome targeting,29,30 T cell activity control,31 antibody recruitment,9,32 and development of hybrid pharmaceuticals.33,34 The second subclass is represented by multifunctional small-molecule-derived conjugates that function as potent and specific inhibitors,35,36 biosensors,37,38 reporters,10,39,40 and protein-activity modulators.41,42
Class Ia. Intermolecular Recruitment by Small-Molecule Tethers
We focus on the following latest developments exploiting small-molecule scaffolding to elicit gain-of-function responses. We draw the reader’s attention to recent excellent and comprehensive accounts of CID-driven biological applications13 and other approaches utilizing small-molecule-based tethering platforms.7
Targeted Protein Degradation by Non-Peptide-Based PROTACs
Proteolysis targeting chimeras (PROTACs) are bifunctional small molecules that induce degradation of their target POI.29 PROTACS work by a validated mechanism in which one pole of the PROTAC binds to the POI while the other PROTAC pole is a recognition unit that recruits a specific E3 ligase. The proximity of the ligase and target POI is sufficient to elicit polyubiquitination of the POI, marking the target out for proteasomal degradation (Figure 2). The original E3 ligase recognition pole was a functionalized peptide that specifically recruits the von Hippel–Lindau (VHL) E3 ligase. The reliance on peptide-based ligands was a significant stumbling block to the broad applicability of PROTACs because peptides suffer from low cell permeability43 and susceptibility to proteases,44 among other drawbacks. Recently, VHL-specific nonpeptide small-molecule ligands have been shown to function as the E3 ligase recognition pole.45 These next-generation PROTACS are active at subnanomolar concentrations. Most impressively, each heterobifunctional all-small-molecule next-generation PROTAC can facilitate multiple protein-degradation turnovers, resulting in >90% knockdown—a result on par with complementary genetic methods such as shRNA- and CRISPR/Cas9-assisted gene knockdowns and knockouts.46,47 Although many strategies for selective protein degradation of artificial POIs have been described,29 PROTACs remain one of the most promising for drug discovery because they have been proven to work on a variety of endogenous target POIs.
Figure 2.
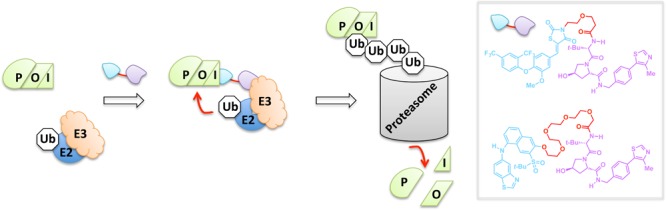
Endogenous protein degradation by PROTACs. The tethered bifunctional PROTAC unites the POI with an E3 ubiquitin ligase, allowing for an E2-mediated transfer of ubiquitin (Ub). The PROTAC thus enables targeted polyubiquitination and degradation by the 26S proteasome. Inset: nonpeptide-derived PROTACs that can induce targeted degradation of the nuclear estrogen-related receptor alpha (top) and the serine/threonine kinase RIPK2 (bottom).
T Cell Activity Control by On-Switch CARs
T cells engineered to express chimeric antigen receptors (CARs) have proven effective against refractory B cell cancers in clinical trials.48 Despite this promise, sporadic toxicity has been observed due to off-target killing of healthy cells. Thus, to design precision-controlled therapeutic T cells, a “split” CAR—in which the intracellular portion is expressed as two separate polypeptides that can coalesce in the presence of a bifunctional small molecule—was envisioned (Figure 3).31 In the absence of the small molecule, the antigen receptor is defunct, and the T cells cannot function effectively, even in the presence of their cognate antigen. However, addition of the small-molecule dimerizers allows the chimeric receptor to regain function, and only in the presence of the antigen, are T cells activated. This promising binary input system, coined “on-switch CARs”, was shown to be able to elicit small-molecule-titratable tumor cell killing and was tolerated by mice.
Figure 3.
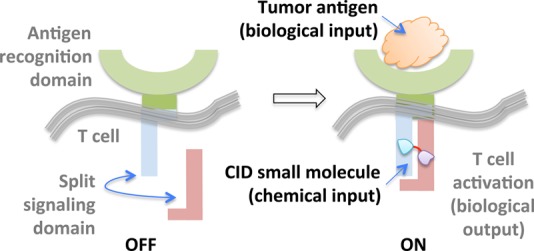
On-switch CARs are built upon a binary logic gate system in which simultaneous presence of tumor antigen and CID-based small molecule dimerizer (e.g., rapamycin analogue AP21967) triggers T cell activation.
Antibody Commissioning by SyAMs
Our next example shows how tethering can be equally effective at engendering a gain of function by bringing two different cell types together.32 Importantly, although the arena in which the events play out is vastly different from those above, the foundational chemical principles remain the same. Synthetic antibody mimics (SyAMs) use a bifunctional small-molecule that can bind to both a cell-surface membrane antigen, selectively overexpressed in prostate cancer cells, and an effector domain that is present in immune cells, Fc gamma receptors (FcγRs) (Figure 4).49 Thus, SyAMs initiate selective phagocytosis of the target cancer cells while avoiding noncancerous cells. The advantages of SyAMs over antibody therapies illustrate the need for contextually relevant comparisons: for instance, although SyAMs are much larger than conventional small-molecule therapeutics, they are only 5% of the molecular weight of an antibody (many of which are also used or in trials to treat various tumors), allowing for better solid tumor penetration. Many cell types also coexpress both activating and inhibitory FcγRs, both of which can be bound by antibodies. On the other hand, SyAMs only bind Type I (activator) FcγR, and hence are capable of selectively initiating immune-mediated toxicity.
Figure 4.
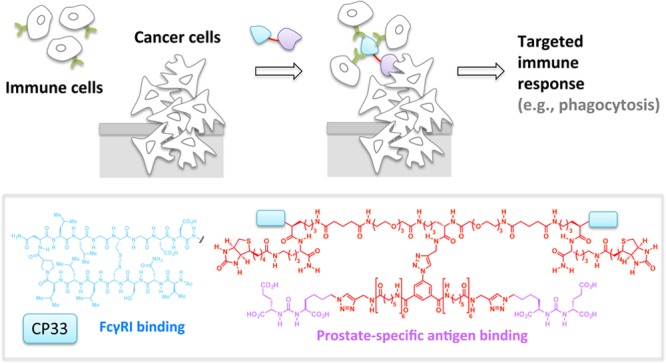
SyAMs. The tethered bifunctional SyAMs unite disease-relevant targets (cancer cells/viral and bacteria pathogens) and immune cells by binding simultaneously to a specific membrane antigen and Fc gamma receptor I. This prompts immune clearance of the targets. Inset, the third-generation SyAM-P3 comprising a pair of CP33, the FcgRI binding motif, and a pair of prostate-specific antigen binding motifs.
Interestingly, on-switch CARs and SyAMs show how a similar problem—how to harness immune cells for targeted therapeutic value—can be tackled in two different ways while still using Class Ia tethering as a core concept. In on-switch CARs, intracellular tethering relays signals in an engineered immune cell, whereas in SyAMs, extracellular tethering choreographs the physical interaction between the host’s immune cells and the intended target cell. This comparison thus serves as testament to the versatility of bifunctional small molecules in terms of their mechanism and applicability across various locales and contexts.
Most Class Ia systems have no Class II counterpart because the small molecule must behave as a scaffold for a sustained period to allow a response to occur. The recently demonstrated recyclability of PROTACs45 allows for low dosage while maintaining high efficacy, a benefit which is also conferred by Class II strategies (vide infra). In the case of PROTACs, high efficacy at low dosage is possible because the PROTAC proceeds through noncovalently linked intermediates and elicits a gain of function (degradation). The latter highlights an important “win” for bifunctional molecules over single-site binders (e.g., inhibitors), because small-molecule inhibitors typically require 50% or more target-site occupancy to be effective.50 On the down side, given the logistical issues with marshaling complex organizations, intricate design is required to achieve success of Class Ia systems. Furthermore, how such complex small molecules behave in vivo in terms of stability, tolerance, and off-target spectrum—which is likely a function of both the poles and the linkers—still needs to be fully evaluated. Even if there is little problem with chemical stability, the pathways targeted are intrinsically complex51−55 and future work must also seek to establish a deeper understanding of the generality, scope, and shortcomings in various cell and tissue types. Encouragingly, PROTACs have recently shown efficacy in various mouse tissues45 and early CID molecules have also proven effective in mouse models. Hopefully, the coming years will see more data evaluating utility of bifunctional small-molecule ligands in animals.
Class Ib. Sensing, Activity Modulation, and Imaging by Protein–Ligand Intramolecular Tethers
Reversible Binding Strategies
1. Biosensing by LUCIDs
The pressing need to monitor changes in the abundance as well as spatiotemporal patterns of small-molecule analytes in a wealth of applications has spurred on the development of numerous sophisticated biosensors.56−59 Several such biosensors exploiting tethered heterotrifunctional small molecules have emerged over the past decade. In a recent invention termed luciferase-based indicators of drugs (LUCIDs), a low-cost point-of-care drug sensor is patterned using a three-component fusion protein:38 SNAP Tag (a ∼20 kDa polypeptide that binds covalently to small molecules functionalized with a benzylguanine unit60), nanoluciferase (which engages in a chemiluminescent reaction with its substrate furimazine61), and the POI (which binds to the drug in question). Tethered trifunctional small molecules are custom-designed to recognize each piece of the three-component fusion protein: a benzylguanine motif that covalently binds to the SNAP Tag, a fluorophore to permit bioluminescence resonance energy transfer (BRET)62 with nanoluciferase, and a noncovalent ligand specific to the POI. Covalent binding of SNAP Tag to the trifunctional small molecule and ensuing proximity-assisted noncovalent association of the specific ligand on the opposite pole of the trifunctional small molecule to the POI facilitates BRET between the fluorophore and nanoluciferase (Figure 5). Upon addition of a free test ligand that competes with the tethered small molecule for binding to the POI, the noncovalent interaction between the tethered pole and the POI breaks, lowering the BRET ratio. This system renders the BRET ratio dependent upon test ligand concentration and presents a useful platform for real-time interrogations into dose-dependent fluxes of therapeutics in blood samples. The ratiometric measurement sidesteps some of the generic issues common to biosensors,36 affording the method an elegant simplicity of use as a portable system for field testing. However, masterminding these kinds of biosensors demands a great deal of protein engineering to solve a complex three-component problem. Additionally, this method is currently limited to POIs for which established reversible ligands exist that, despite being chemically modified to accommodate a linker, can bind a site on the POI that is regulated by binding of the test ligand, e.g., trimethoprim (TMP) to dihydrofolate reductase (DHFR),63 cyclosporin A to cyclophilin A,64 and benzenesulfonamide derivatives to human carbonic anhydrase II (HCA).65 This strategy will only be most accurate to titer concentrations of ligands that bind reversibly to the target site (arguably the most prevalent class of ligands available66), and careful considerations must be taken if the test ligand has long residence times67−69 on its target POI to ensure complete equilibration.
Figure 5.
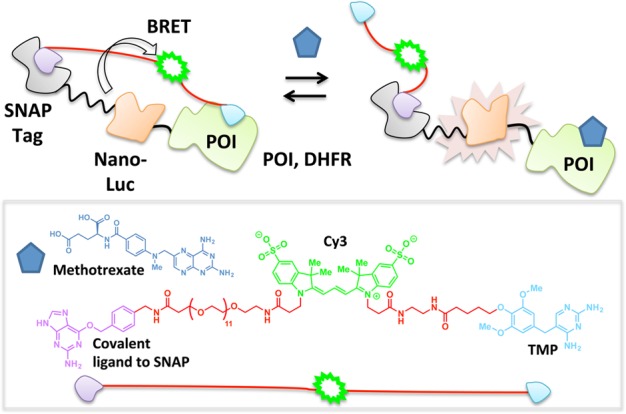
LUCIDs. Tethered TMP binding to DHFR (POI) enables BRET between the nano-Luc enzyme and Cy3 dye. Titration with methotrexate (blue pentagon) displaces TMP, thus preventing BRET.
2. Protein Activity Modulation by CLASH
Molecular switches have been reported in which a freely diffusible effector protein sterically regulates binding of a functionalized ligand to its POI.70 The setup of this platform is similar to LUCIDs38 in that a covalent interaction between SNAP tag and a benzylguanine motif orchestrates noncovalent assembly on a sophisticated fusion protein. In this instance, however, the engineered small-molecule probe contains binding sites for the fusion protein as well as a binding site for an exogenous protein (Figure 6a). The ligand to the exogenous protein and the ligand to the POI within the fusion protein system are juxtaposed such that addition of the exogenous protein and accompanying formation of a new protein–ligand complex prevents the POI from binding to the tethered ligand. This intervention provides a dynamic molecular switch that is controlled by the concentration of the effector protein. In a proof-of-principle example of this chemical ligand-associated steric hindrance (CLASH) method,42 proximity-directed control of the activity of luciferase fused to SNAP Tag was demonstrated (Figure 6a). In the ground state, a multifunctional conglomerate is created, in which luciferase is covalently linked to coelenteramide (a reversible inhibitor)71 via conjugation to the SNAP Tag. The coelenteramide pole is also functionalized with biotin.72 In the absence of streptavidin (a tetrameric protein which binds to biotin with high affinity), the coelenteramide pole efficiently binds and inhibits luciferase. Upon addition of streptavidin, the coelenteramide inhibitor can no longer bind luciferase, triggering luciferase bioluminescence73 (Figure 6a). This trifunctional tethered manifold was further extended to incorporate a Cy3 fluorophore74 between the SNAP Tag and the biotin which enables BRET with nanoluciferase in the ground state (Figure 6b). In this instance, the fluorophore was brought into contact with luciferase using HCA and benzenesulfonamide. By also appending biotin to complete the tetra-functional tethered small-molecule arrangement made up of benzylguanine, Cy3, benzenesulfonamide, and biotin, it was found that the interaction between HCA and benzenesulfonamide was prevented by streptavidin in a dose-dependent manner.
Figure 6.
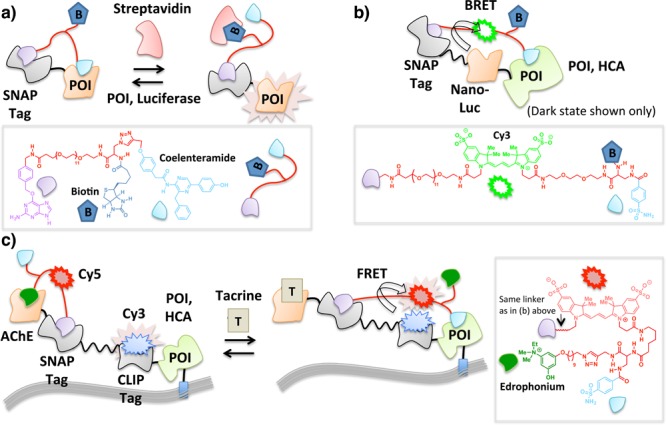
CLASH enables on/off switching of POI activity by making use of two ligands with mutually exclusive binding to POI. Tethering results in intramolecular protein–ligand interaction that is disrupted by an effector biomolecule [streptavidin in (a) and (b)] or small molecule [tacrine in (c)] that competes with the ligand within the tethered array that initially binds POI. In (b), only the ground state in the absence of streptavidin is shown. POI is exemplified by nanoluciferase (a) and HCA (b,c).
A potential drawback of the above strategy is that an exogenous protein (e.g., streptavidin) is required to effect a response. This set up is arguably more amenable to the detection of a particular protein rather than serving as a tunable molecular switch since protein expression level is less easily controlled and can be changed with less precision in terms of both timing and concentration than small-molecule ligand addition, especially in cells and whole organisms. To enable more precise control of the response, a four-component fusion protein comprising an acetylcholine esterase (AChE),75 SNAP,60 and CLIP (a ∼20 kDa protein tag that reacts with benzyl cytosine76) domains, and a membrane-bound HCA was conceived (Figure 6c). A tethered small-molecule array was constructed in which edrophonium (ligand to AChE) and benzenesulfonamide (ligand to HCA) were joined to a benzylguanine motif (irreversible binder to SNAP) via a linker containing a Cy5 fluorophore.74 The CLIP domain of the four-component fusion protein expressed at the cell surface was prelabeled with a Cy3 fluorophore through the use of a benzylcytosine (irreversible ligand to CLIP)-functionalized Cy3 probe (Figure 6c). In the absence of tacrine (an AChE inhibitor formerly used to treat Alzheimer’s disease77), intramolecular binding of the tethered edrophonium to AChE keeps the system in a low-FRET state while the SNAP domain functions as a hinge point through which covalent ligand–protein conjugation is achieved. When edrophonium is displaced from AChE by tacrine, the system allows the tethered benzenesulfonamide to interact with HCA (Figure 6c), switching to a high-FRET state as a result of donor (Cy3) to acceptor (Cy5) interaction. Tacrine induction and removal, respectively, enables the tethered ligand to reversibly toggle between the binding of tacrine and edrophonium to AChE, which corresponds to high- vs low-FRET states, respectively. This elegant strategy demonstrates the power of multiple intramolecular tethers that can be fabricated in situ at the cell surface and controlled by a simple change in conditions. However, the level of complexity of the system is high, and successful examples have only been demonstrated with extracellular and cell-free systems.
Covalent Capture Strategies
1. Proximity-Directed Ligation
Glycan alterations at cell surfaces are responsible for many essential biological functions, such as antigen presentation, cell-to-cell communication, and cell differentiation and migration.78−80 It is thus critically important to understand the makeup of the cell-surface glycome. Many methods to visualize global glycosylation at the plasma membrane have been reported;80,81 however, chemical tethering has recently allowed individual membrane protein glycosylation events to be identified.82 The approach uses a DNA aptamer83,84 that can bind specific cell-surface targets. The aptamer is modified with biotin (or a small-molecule fluorophore) at one terminus and cyclooctyne at the other. Cells are first pretreated with peracetylated N-azidoacetylneuraminic acid (SiaNAz), which is incorporated into glycoproteins by endogenous glycosylation pathways. When the aptamer binds to its target receptor at the cell surface, provided the target itself has been sialylated with SiaNAz, accompanying enhanced local concentration primes rapid covalent conjugation of the cyclooctyne and the azide group via copper-free click coupling85,86 (Figure 7). Using this approach, PTK7, an important receptor in the Wnt signaling pathway,87 was identified as a novel sialylated receptor. Although this method promises exciting future expansion and applications, the authors provide some evidence to suggest that in certain applications, the cyclooctyne ligand can undergo reaction with nucleophilic residues88 leading to relatively high background.
Figure 7.
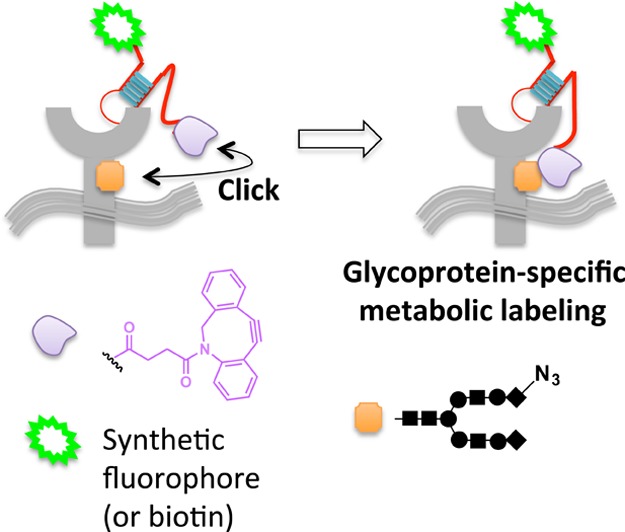
Scaffolding via an aptamer allows glycoprotein-specific fluorophore targeting against the backdrop of metabolically SiaNAz-labeled cell-surface glycans. Proximity enables strain-promoted click coupling between cyclooctyne tethered to aptamer and the azidosugar-labeled receptor.
2. Isozyme-Selective Regulation by BOLT
The demand for targeted small-molecule inhibitors or ligands that can selectively alter POI activity without affecting the entire proteome has continued to grow over the years.89,90 This need is magnified because there are many protein targets with high homology, but distinct differences in their biological functions.91 Identification of specific ligands for these homologous targets remains a significant challenge.92,93 By incorporating a genetically encoded unnatural amino acid94 on the POI, a new bioorthogonal ligand tethering (BOLT) approach was designed which is capable of regulating the desired POI with unparalleled rapidity and selectivity. The unnatural amino acid within the POI reacts with one pole of a bifunctional inhibitor, while the second pole within the tether functions as a low-affinity ligand. Many proteins may be able to bind the low-affinity ligand, yet only the engineered mutant POI can be inactivated irreversibly due to formation of a stable ligand-POI complex. The binding enhancement conferred by the covalent interaction is most pronounced when a ligand concentration below KD is used, such that the ligand has low nontemplated occupancy, but irreversible chemistry drives saturation of the target POI. The utility of this method was illustrated by rapid and specific inhibition of specific MEK isozymes95 featuring complementary unnatural amino acids.
A further development uses photochemistry to allow toggling between the inhibited and uninhibited states. This work builds upon prior art in which photochromic tethered ligands (PTLs) have been used to elicit optogenetic control of living systems. A recent review thoughtfully summarizes the design and applications of tethered small-molecule photoswitches in live cells and animals.96 The resulting on/off approach called “photo-BOLT” uses a cis–trans photoisomerizable linker between the two poles of the ligand, affording dynamic optochemical control (Figure 8). In the photo-BOLT platform,41 when the linker is in the trans conformation,97 one pole of the bifunctional small molecule is covalently conjugated to the unnatural amino acid, and the other pole noncovalently binds to the active site of the POI, resulting in inhibition. Inhibition is reversed by illumination of the cells at a wavelength that initiates trans to cis isomerization of the linker. In the cis isomer, the spatial arrangement of the poles is changed such that the noncovalent-binding pole of the ligand is no longer able to interact with the POI and activity is restored. Initial proof-of-principle work has demonstrated that BOLT and its derivatives are effective in targeting a specific enzyme over a number of closely related analogues. The reliance on unnatural amino acids is liberating as there is no need for large domain fusions that may affect protein function, but also potentially restrictive, because a readily functionalizable residue must be found in proximity to the target ligand binding site that must preserve activity even (in the case of photo-BOLT) when functionalized with one geometrical isomer of the BOLT ligand. One interesting question is how distance from the target binding site and linker length affect the benefit conferred by intermolecular binding. Depending on the importance of these parameters, one could envision using a separate binding domain (e.g., SNAP) to anchor the ligand to the POI in a similar way to iBOLT.
Figure 8.
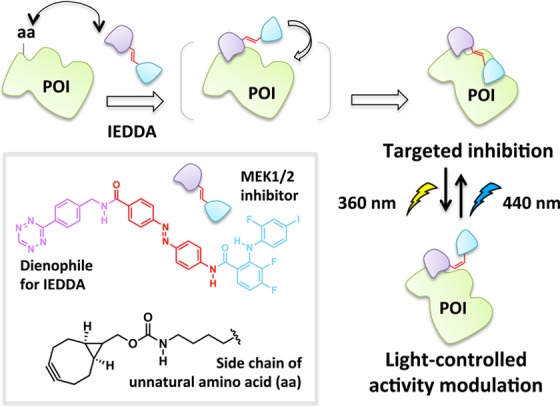
iBOLT and photoBOLT. The bifunctional small-molecule probe undergoes an inverse-electron demand Diels–Alder (IEDDA)/retro Diels–Alder reaction with the unnatural amino acid on the POI to form a covalent linkage. Light-driven cis–trans isomerization enables reversible activity modulation.
Both proximity-directed ligation and BOLT use enhanced local concentrations of a semispecific probe to direct a specific interaction to a desired point of origin. Clearly for many scenarios this strategy is highly effective. However, it remains to be seen how effective systems like BOLT will be in more complex settings. For instance, when the target POI can form hetero-oligomers with other isozymes that can interact with the noncovalent-binding pole of the bifunctional probe, specificity will likely be diminished. In the event that interprotein interactions dominate, inhibition of the undesired isoform/protein may result. Judicious choice of a linker length/unnatural amino acid position that favors intramolecular interactions may circumvent this issue.
3. Stable Fluorophore Integration via Proximal Cysteines
The site-specific noncovalent labeling of DHFR-fusion proteins with TMP-conjugated fluorescent dyes and analogous small-molecule dye labeling methods are widely used.98,99 In the case of DHFR and TMP, the assembly exploits the selective, noncovalent binding of the TMP ligand to Escherichia coli as opposed to human DHFR, which results in a protein–ligand complex with a dissociative half-life on the order of 20 min.100 To increase the residence time of the ligand on the target enzyme, this platform has been recently amended such that a covalent (essentially permanent) bond between the TMP and DHFR is formed (Figure 9).39,41,101 This required single modifications to the ligand and protein: synthetic derivatization of the TMP-conjugated dye to incorporate an acrylamide appendage, and a point mutation (K28C) within DHFR. Binding affinity of the K28C-DHFR to TMP is similar to that of the wild type, indicating that the mutant enzyme retains its affinity for TMP; however, when the acrylamide-functionalized ligand is used, a proximity-induced Michael addition covalently links the nucleophilic C28 residue on the mutant DHFR with the ligand. A related development utilizing intramolecular tethering for covalent-dye labeling makes use of fluorophore-binding peptides, called fluorettes (Figure 10).40 In this case, a reactive cysteine residue is built into the linker region of the fusion protein construct made up of a fluorette fused to the POI. The tethered small molecule is designed with an α-halocarbonyl function appended to a TexasRed-derived fluorophore known to bind reversibly to the specific fluorette.102 The rapid noncovalent interaction between the fluorophore and the fluorette templates a covalent reaction between the α-halocarbonyl motif on the tether and the proximal cysteine on the linker attached to the target POI.
Figure 9.
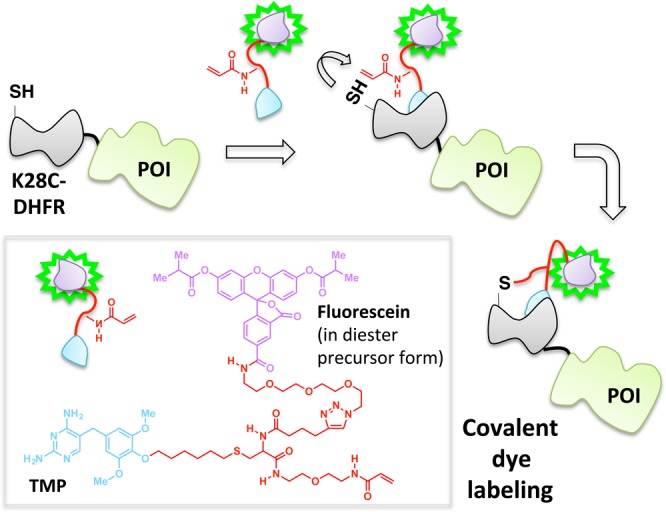
Covalent capture enables stable dye incorporation through an acylamide appendage that can conjugate with the engineered cysteine K28C-DHFR mutant fused to POI.
Figure 10.
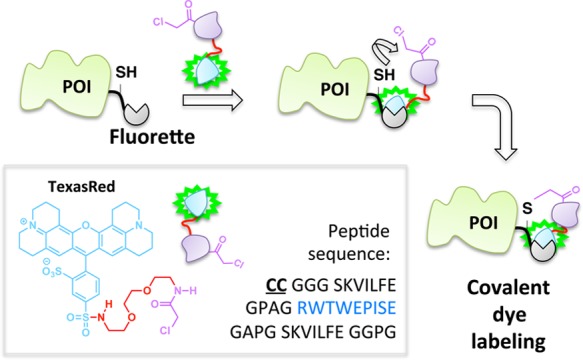
Covalent capture concept in stable dye incorporation using a fusion peptide that bears a Texas Red fluorophore-binding sequence (blue) and reactive Cys residues.
The crux of the method is that typically covalent interactions occur slower than noncovalent associations. Should a relatively rapid noncovalent association facilitate covalent bond formation, the rate enhancement relative to nonspecific covalent bond formation will allow a specific covalent bond forming event to occur. Thus, the best systems will have low intrinsic reactivity of the reactive pole, or “warhead”. Obviously, for this process to occur a nucleophilic residue (usually cysteine) must be in close enough proximity to the ligand when bound to its target to form a covalent bond to the acrylamide moiety. Similar considerations have been echoed by the pharmaceutical communities as attempts are made to develop irreversible inhibitors from known reversible binders.66,103−105 The acrylamide warhead demonstrated for the DHFR/TMP system is of relatively low intrinsic reactivity/promiscuity, so its use will limit off-target binding. It is thus ideally set to pair with trimethoprim, which is a high-affinity, reversible, yet slow onset inhibitor of DHFR. It is possible to increase the promiscuity of the reactive unit appended to the ligand [such as the α-halocarbonyl in the fluorette system (Figure 10)] to drive covalent attachment; however, increasing reactivity will typically elevate off-target binding, and will ultimately limit the versatility of the method. Clearly, such promiscuous reactive ligands may not be kinetically compatible with slow onset inhibitor manifolds either. Similar ways to engender specificity have been achieved using unnatural amino acids as in BOLT for instance, and cyclooctyne such as in proximity-enhanced aptamer ligation. One alternative solution applicable to ligands with relatively long residence times on their target is on-demand activation of a reactive functional group once the recognition element has been bound to the POI and the excess washed away. In this context, recently developed on-demand targeting or in situ unmasking ideas of proximity enhancement have appeared in the literature (vide infra).
Current Challenges
The pioneering lines of method development within the Class I framework over the recent decade have also highlighted several general as well as specific challenges unique to individual subclasses. We here summarize these salient points of consideration.
1. Covalent vs Non-Covalent Ligand-Binding Modalities
Both covalent and noncovalent modes of ligand binding are evident in the examples illustrated above, but the decision as to which strategy is better integrated in developing a new toolset is often not obvious. Each tactic has limitations and the following general points must be considered. Key aspects are the following: (1) Reversible binders can function catalytically allowing low ligand load while maintaining high efficacy, whereas irreversible binders cannot. This aspect applies most readily to Class Ia. (2) Although typically assumed to be rapid, binding equilibria of noncovalent ligands may be relatively slowly established and occur in a complex, multistep process involving enzyme conformational changes, especially for high-affinity binders. Crystal structures of protein/ligand complexes will typically show end-point conformations, and thus careful consideration of proximal residues must be made especially if a Class Ib strategy is under consideration. (3) Off-rates (residence times) of high-affinity reversible ligands can also be prolonged, making washout of ligand prior to downstream analysis, or sensing of analyte concentrations, require careful optimization. (4) Covalent bond formation is typically slow (detection of a binding event may take minutes to hours),66,106 leading to time-dependent associations that need to be addressed carefully to account for off-target effects and characterization of ligand–protein interactions. (5) Although not all off-target interactions of irreversible target binders are necessarily covalent, those that are will increase as a function of time. Unlike for noncovalent off-target effects, the covalent off-targets, once formed, necessitate new protein synthesis to regain activity if target inactivation has occurred. Alternatively, if gain of function has occurred, degradation coupled with new protein synthesis is required. Given the above, we finally stress that conversion of noncovalent binders to covalent binders in a bid to assess on- and/or off-target binding should be approached with caution.66,103−107
2. Exploiting Functional Constraints
One trivial, but nonetheless common issue is that for Classes Ia and b, care must be taken to ensure that tethered macromolecules forged by the multifunctional small-molecule scaffolds (either via intermolecular protein–protein tethering enabled by the probes, or intramolecular protein–ligand tethering) are able to maintain functionality. An arduous series of chemical syntheses (or permuted polypeptides), optimizations of linkers and ordering of individual functional groups along the tethered multifunctional small-molecule array are often prerequisites of many successful Class I methods discussed above. Currently there are no clear predictive tools for achieving the optimal design a priori. However, what has been less well recognized is the fact that these considerations can also be used to the researcher’s advantage. In CLASH42 (Figure 6), problems associated with steric repulsion have been turned on their head by designing techniques based on steric-strain-directed ligand binding and toggling. Photo-BOLT41 also utilizes rigidity-enhanced steric and stereoelectronic requirements of intramolecular bond formation to its advantage (Figure 8).
3. Auto-inhibition—The Hook Effect
One generally less attractive feature of systems in which a bifunctional ligand must interact with two independent binding sites is the Prozone or hook effect,108 wherein a drop in ternary complex formation occurs at high ligand concentrations. This behavior counteracts one of the key benefits of small molecules—their predictable dose response. The hook effect occurs because once the ligand is in excess relative to its target protein partners, there is sufficient free ligand available for each of the two poles to bind its respective protein target separately.109 The result is that these systems are often autoinhibitory. Although this behavior has been helpful to model binding mechanisms in vitro, this complex operation confounds downstream biology, especially in cells and whole organisms in which available small-molecule ligand concentrations are not a simple parameter to control but often dependent on pleiotropic factors. This problem is most applicable to cases where both poles of the bifunctional probe interact reversibly as in Class Ia systems discussed above. If one pole behaves as an irreversible binder, the hook effect can be reduced, principally because probe concentrations can be more readily controlled and excess can be removed. On the other hand, the hook effect is less of an issue if the ligand can behave catalytically as in next-generation PROTACs because ligand concentrations can be minimized such that competition for binding sites does not occur. Finally, the hook effect may be almost completely obviated in platforms such as BOLT in which one pole of the bifunctional probe reacts irreversibly and essentially exclusively with its target site and the second binding site is also on the same protein (Class Ib systems).
4. The Extent of On-Target Specificity
In terms of downstream applications, the majority of applications using proximity-induced chemistry are limited by the complexity of the molecules required to achieve their ultimate goal. Thus, most multicomponent tethered probes (Classes Ia and Ib)—even those currently at the cutting edge of this field—work well in cultured cells, but much less information is available on how well they are adaptable to applications in animal models or for human disease treatments.110 One of the most obvious roadblocks is a question of size and complexity. The molecular design inherently calls for two poles that are each sufficiently complex to bind selectively to a specific target POI, and a linker separating the poles. These prerequisites render it challenging to create bifunctional small molecules that adhere to basic drug design rules to optimize ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties,111 such as Lipinski’s rule of five.112
Beyond the above considerations, increased size/complexity of chimeric ligands may engender lower target specificity compared to single-site binders. However, this proposition is not necessarily true because the downstream application of single-site-binding ligands and bifunctional molecules can be very different. Single-site binders often act as inhibitors of a specific enzyme, but dimerizers (i.e., Class Ia) induce gain of function through forcing protein–protein associations. It is thus possible that by setting up low-occupancy binding conditions (i.e., less than stoichiometric target-binding), even chimeras derived from promiscuous ligands can have highly specific effects. This is because formation of the desired protein complex will cause a large gain in activity even at low ligand occupancy, whereas unintentional off-target binding will be low occupancy and is unlikely to usher a gain of function.
5. Modularity and Non-Invasive Functionalization
The multicomponent scaffolding is the centerpiece of both Classes Ia and Ib probe design in which each of the components is typically derived from known ligands. Unfortunately, not all protein ligands are amenable to functionalization, either because they lack a suitable functionalizable appendage, or because chemical modification perturbs function. Thus, there are relatively few bona fide ligand–protein pairs directly amenable to Class I approaches. Furthermore, most of the functionalizable protein ligands are actually inhibitors of a specific enzyme, principally because protein binders have traditionally been identified from activity modulation screens. This caveat often limits downstream chemical biology applications because protein targets are mostly restricted to those with an enzymatic function,113 but that activity will be inhibited as consequence of ligand binding. Various methods have been used to combat this deficiency, including the use of artificial domains (Classes Ia and Ib) such as DHFR. These considerations are not applicable if a pole can be found that interacts with a site that is not functionally coupled to the target enzyme’s activity; e.g., the target protein is part of a complex. For instance, in the case of PROTACS, catalytic chemistry (ubiquitination) is carried out by the E2 ligase, which is complexed to the target of the PROTAC, an E3 ligase.
More recently, drug-screening methods that append macromolecular barcodes onto small-molecule ligands have become widespread.114 These innovations provide a goldmine of potential small-molecule ligands (not necessarily inhibitors) selective for a particular POI (not necessarily those with enzymatic functions) that must be tolerable to functionalization to appear as a hit. On the bright side, for proof of principle, genetically encodable protein tags—such as SNAP,60,76 CLIP,76 DHFR,115 Halo,116 and PRIME117—can greatly extend the scope and generality of all methodologies, provided the researcher is prepared to study ectopic proteins and evaluate that the tag does not perturb protein function drastically. The ease with which knockdowns/knockouts or inducible systems can now be generated46,47 additionally provides opportunities to study these fusion POIs against the null background or with minimal perturbation to the endogenous organizations. With the advent of engineered immune cells for targeted therapy,48,118 there is also a distinct possibility that these artificial domains could be used in “real-life” applications.
Class II. Proximity-Directed On-Demand Targeting
The most recent years have witnessed the emergence of proximity-directed controlled release/delivery of reactive small-molecules in situ.17−19,70,119 This method can obviate some of the problems associated with conventional multicomponent tethering (Class I). Instead of linking multiple small-molecule modules through a chemical tether that remains unbroken throughout the course of the assay (Figure 1, Classes Ia and Ib), a latent warhead is linked to a recognition element for a specific POI. Activation of the warhead can be achieved (a) by sensitization (generation of a reactive intermediate upon contact with one pole of the ligand) or (b) through photo-uncaging of a protected, inert precursor of the reactive signal built into one pole of the ligand, breaking the tether in the process (Figure 1, Class II) thereby enabling proximity targeting to the POI. The basic design of Class II—particularly the sensitization strategy—is not new; chromophore/fluorophore-assisted laser activation (CALI/FALI) methods were introduced in 1988.120−122 In these systems, reactive oxygen species (ROS) are overproduced in cells by excitation of photosensitizers—engineered fluorescent proteins or dye molecules. POI-specific targeting is achieved either via tethering organic chromophores/fluorophore—such as malachite green or fluorescein—to POI-binding antibodies (delivered into cells via microinjection) or small-molecule ligands. Generality was subsequently shown through the use of tagging domains fused to the POI. These early proof-of-concept studies required high irradiation doses with focused laser beams and arc lamps that have deleterious effects on cells.123,124 A further limitation of the early method was off-target photodamage during irradiation, stemming from the requirement for excess quantities of toxic dyes that cannot be easily washed away.121 Thus, high-affinity binding of the chromophore/fluorophore to the POI is often a necessary factor.
To overcome some of these shortcomings, a more effective visible-light-driven singlet oxygen-generating CALI reagent was recently developed by tethering a [RuII(bpy)3]2+ photocatalyst (an oxygen sensitizer) to cell-permeable peptoids125 that are selective binders of POIs (Figure 11).119 This reagent was field tested with both intracellular and integral membrane POIs. The hyperpotency of these CALI-based peptoid–ruthenium conjugate inhibitors was demonstrated in direct comparison with their parent compounds in light-triggered inactivation of the vascular endothelial growth factor receptor 2 (VEGFR2)126 and the chymotrypsin-like activity of the 26S proteasome (Figure 11).127
Figure 11.
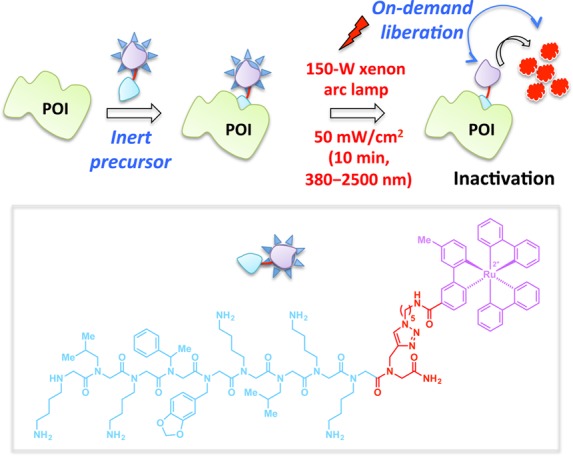
In this CALI-based strategy, Ru(II)(tris-bipyridyl)2+ (purple motif, inset) functions as a photosensitizer for the generation of singlet oxygen (1O2). Binding of the peptoid (inset, blue motif) to a specific POI (e.g., VEGFR, 26S proteasome) allows targeted inactivation of POI by excess 1O2 produced by the Ru catalyst upon irradiation with the visible light of indicated parameters.
On-Demand Flipping of Redox Switches by T-REX
In the recent decades, many reports have indicated that endogenous electrophiles can exert a specific redox-linked signaling role in cells.128−132 Electrophile signaling is presumed to occur through modification of specific redox sensor proteins. Until recently, researchers used bolus dosing with the reactive signal of choice to model these endogenous signaling events. Unfortunately, because of the promiscuity of these reactive signals [e.g., >400–800 functional targets have been profiled for the reactive signaling molecule 4-hydroxynonenal (HNE)],130,133−135 it is challenging to pinpoint gain-of-function signaling responses that require only modest levels of modifications on a single sensor protein.131,133,135 Conditions of global stimulation and the associated off-target effects can also lead to generation of secondary signaling metabolites/oxidative stress that further confound analysis and data interpretation. Motivated to provide a gateway to track specific gain-of-function responses brought about by non-enzyme-dependent redox-linked protein modifications, our laboratory has pioneered the concept of targetable reactive electrophiles and oxidants (T-REX).17−19 A bifunctional small-molecule ligand was designed with one pole featuring the HaloTag116-recognition unit, a chloroalkane, and the other end bearing a photocaged precursor to a specific reactive signal. Photouncaging releases the signal with temporal precision and in substoichiometric quantity with respect to the target POI fused to Halo (Figure 11). The released entity can either react with the target POI, or can diffuse away from the solvent cage, where it is likely intercepted by reactive small-molecule thiols in the cell such as glutathione. The engineered bacterial dehalogenase domain, HaloTag (∼33 kDa protein),116 ensures target POI specificity, limits the amount of electrophile to be at most stoichiometric to POI, and was shown to be inert to most reactive electrophiles.19 T-REX has recently shown that single-protein modifications by HNE are sufficient to elicit pathway activation. The commercial availability of the human and mouse ORFeomes as halo-tagged libraries also enables identification of novel HNE-sensing proteins using the Halo-ORFeome library in combination with T-REX.
CALI/FALI and T-REX embody the Class II proximity-directed regimen and a comparison of these systems with Classes Ia and Ib tethering systems reveals interesting advantages and disadvantages. The key advantage of the Class II is that the small molecule that would constitute the second pole in Class I is free to diffuse and is unaffected by chemical modification. By contrast, Class I requires the second pole—while still tethered to the first pole—to be effective in POI binding. Because of specific mechanistic differences, the Class I approach is compatible with irreversible and reversible ligands, whereas the Class II approach only works for long residence (most likely irreversible) binders. In contrast to Class I in which high-selectivity ligands are typically preferred, the aim of Class II is to endow a nonspecific ligand with high specificity for a particular POI [or for proteins in a specific locale (vide infra)] through proximity enhancement. There is little to be gained from release of high selectivity ligands unless its targeted delivery to one subcellular locale is required.
On the other hand, the differences between CALI/FALI119−122 and T-REX17−19 lie principally in their release stoichiometry. In CALI/FALI, a sensitization event repeatedly allows activation of cellular oxygen for the duration of the laser irradiation. In T-REX, a controlled amount of a reactive entity to a maximum level of stoichiometry is unleashed. Thus, CALI/FALI protocols can generate a large quantity of reactive signals; T-REX, on the other hand, guarantees a substoichiometric release of the reactive electrophile, mimicking substoichiometric redox-linked modifications and signaling. Thus, T-REX works best on reactive sensor proteins, often rich in reactive Cys that rapidly capture highly reactive electrophiles: whereas, CALI/FALI can affect proteins that are less reactive because reactive oxygen species can be generated until photobleaching occurs. By similar logic, T-REX is best used to study a small-molecule-initiated gain of function (or dominant loss of function) or a new downstream signaling event, rather than small-molecule-induced inhibition or loss of function. Because CALI/FALI is a “targeted overload” method, it works well to inhibit target proteins because 100% target protein labeling is possible, although precision control of target POI labeling stoichiometry may be more challenging.
It is possible that spatial positioning of the chromophore or caged precursor, respectively, relative to the tethered protein will affect targeted delivery in both CALI/FALI and T-REX. Although this weakness would be expected to apply most to T-REX, for the proof-of-concept target POI, Keap1, the position of the HaloTag affects neither delivery nor Cys-residue specificity. This result is consistent with T-REX targeting the most reactive cysteine(s) in the target protein, which are likely to be the ones that sense freely diffusive endogenous electrophilic signals. Finally, T-REX requires UV photo-uncaging (Figure 12), which may limit utility to cell culture and transparent organisms. Successful integration of two-photon uncaging techniques may surmount this problem.
Figure 12.
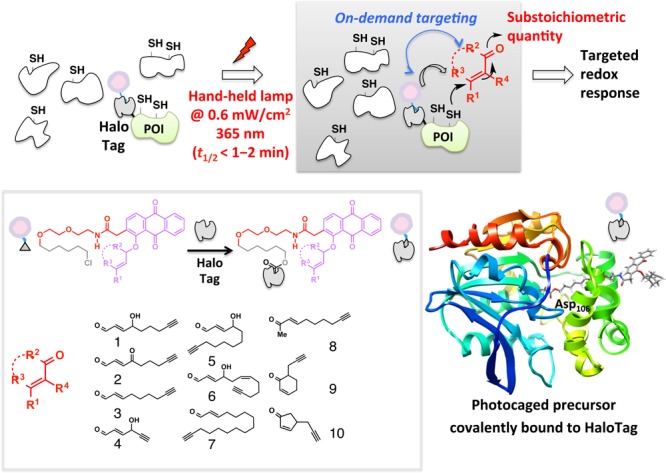
T-REX exploits the on-demand precision targeting concept. Inert photocaged precursors delivered to cells selectively and covalently bind HaloTag fusion proteins (inset) and excess is washed away. Photouncaging driven by 365 nm light rapidly unleashes up to stoichiometric amounts of reactive lipid-derived signaling electrophiles (LDEs) from the HaloTag as a point source (inset, representative LDEs). Targeted LDE modifications on a specific POI in turn elicit gene-specific redox signaling on demand in the backdrop of an unperturbed cell. Reproduced from ref (19). Copyright 2015 American Chemical Society.
Methods Analogous to Class II On-Demand Targeting
Some methods closely related to CALI/FALI and T-REX have recently been reported. These use proximity targeting through in situ unmasking of reactive entities with a goal of understanding interacting proteins, or which proteins are present in a particular locale.
Biotin-dependent proximity-driven small-molecule chemistry has afforded powerful insights into biological systems. This general strategy is most readily exemplified by proximity-dependent biotin identification also known as BioID.136 The heart of the BioID platform is a reactive, adenosine monophosphate (AMP)-activated biotin generated in situ. An engineered promiscuous biotin ligase (BirA*) is fused to the POI (Figure 13). Upon biotin stimulation, BirA* creates activated biotin that is subsequently released within the local environment of the POI, enabling biotinylation of lysine residues within spatially proximate (10 nm radius)137 proteins. Subsequent streptavidin enrichment, peptidic digest, and MS proteomics permit identification of unknown targets or validation of hypothesis-driven binding partners to POI. A recent case of this innovative tool was demonstrated with functional characterizations of large protein assemblies in nuclear pore complex organization.137
Figure 13.
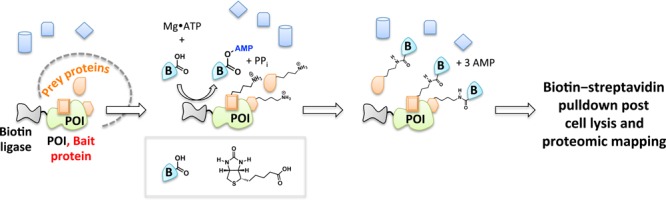
BioID. BioID enables identification of proximate proteins for a candidate POI fused to biotin ligase BirA* upon stimulation of cells with biotin. The practical labeling radius in cells is ∼10 nm.
Another biotin-dependent proximity capture system has also emerged aimed at ribosome profiling (Figure 14).24,25,138 A spatially restricted biotin ligase (BirA) fused to, for instance, an endoplasmic reticulum (ER) or mitochondrial-specific protein, is coexpressed with ribosomes that are engineered to bear AviTags, substrates for BirA. Whole-cell stimulation with biotin causes proximity-targeted biotinylation, enabling the elucidation of specific subgroups of ribosomes at defined locations or with specific interacting factors. The method was successfully applied to paint a detailed spatiotemporal picture of local protein synthesis and posttranslational translocation at the ER as well as at ER-associated ribosomes.
Figure 14.
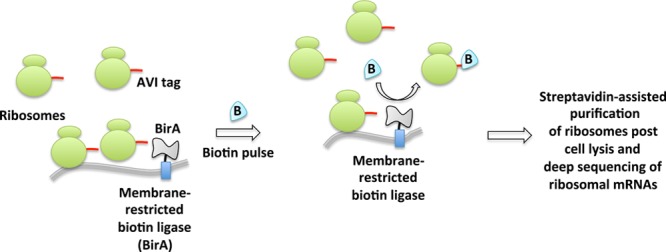
Proximity-specific ribosome profiling. This deep sequencing-based method requires coexpression of spatially restricted (e.g., ER or mitochondrial membrane) biotin ligase BirA and ribosomes fused to its substrate, AVI tag. Pulsing of live cells with biotin enables biotinylation of proximal ribosomes. Enrichment of biotinylated ribosomes and subsequent deep sequencing of ribosome-protected mRNAs inform spatiotemporally precise message translation.
In another innovation dubbed “APEX”, an engineered class I cytosolic pea or soybean ascorbate peroxidase (28-kDa) is specifically targeted to a particular organelle using a localization sequence.20−23 The peroxidase catalyzes H2O2-mediated oxidation of biotin phenol introduced to cells. Temporal control is achieved by 1 min H2O2 stimulation of the cells (Figure 15). The nascent short-lived radical thus formed covalently attacks electron-rich amino acids on nearby protein targets. These endogenous biotinylated targets are enriched with streptavidin beads, and identified by MS sequencing. This method has proven successful in answering key questions underlying mitochondrial compartmentalization.
Figure 15.
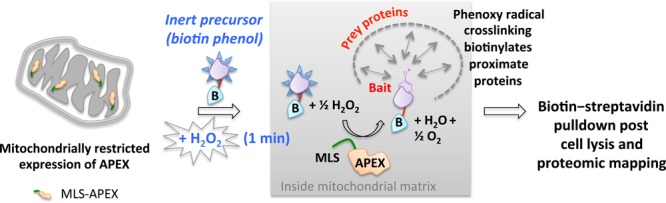
APEX. The ectopically expressed APEX peroxidase targeted to a specific organelle (in this case mitochondria) catalyzes the oxidation of biotinphenol by exogenously added H2O2. The reactive biotinphenoxy radical covalently labels proximate mitochondrial matrix proteins, enabling proteomics profiling of biotinylatated proteins from defined locations. MLS, mitochondrial localization sequence.
The proximity-dependent biotin-derived capture methods above have proven powerful as discovery-based tools. The choice of capture method used is dependent on many factors. Since, in principle, any Class II and related approaches can be used to profile associated or proximal proteins, we catalog the key points to consider: (1) Half-life of the reactive molecule generated. This parameter defines the “radius of influence” or distance over which a reactive small molecule can “label” prior to decay. Consensus second-order rate constants for the reaction of probes with reduced glutathione, a biologically relevant reference reacting partner, are shown in Table 1. (2) Reactivity spectrum of the activated probe. Ideally, an affinity capture agent will react nondiscriminately with any protein. This parameter will scale roughly with bimolecular rate constants in Table 1, but is also a function of amount of probe produced, and duration of experiment. (3) Stability of the complex formed after reaction. This variable defines whether the signal can be relayed to another protein post capture, thereby potentially diluting the signal among secondary players. For instance, acyl phosphates, like those generated during the course of BioID, can in principle react with any nucleophilic residue including those bearing amines139 and thiols.140 The latter creates a thioester, which can react with other nucleophilic residues (potentially on other proteins), whereas the former makes a stable amide bond. (4) Reaction context/conditions. Acyl phosphates, for instance, vary in their stability and reactivity from having second-order kinetics ranging from slow (undetectable) to relatively fast as a function of metal ion/Lewis acid activation141 and pH of the microenvironment.142 Radicals, on the other hand, are intrinsically more reactive and of more transient nature, making them more generally applicable.
Table 1. Rate Constants for Reaction with GSH.
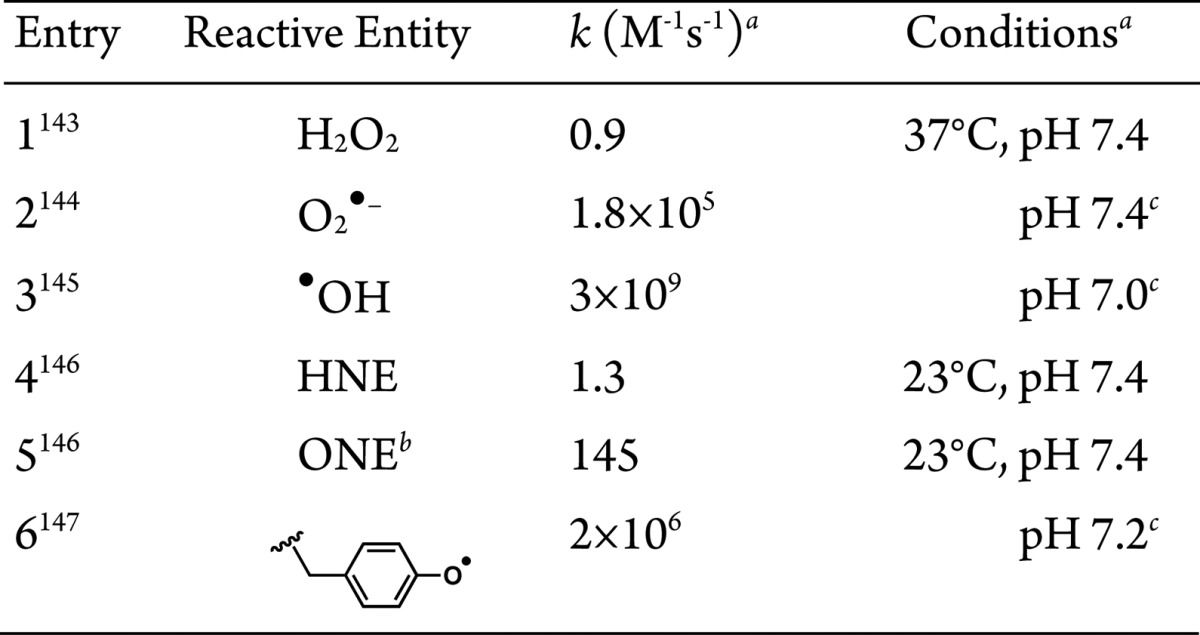
See references for details.
4-Oxononenal.
Temperature not reported.
At present, all biotin-dependent capture methods rely on exogenous small-molecule stimulation. In the case where the stimulant is a high concentration of H2O2 such as in APEX, even though this is only for 1 min, it remains to be addressed how the approach may affect the very proteins intended to be profiled. This question is critical because exogenous H2O2 challenge is known to perturb signal transduction pathways148,149 and organelle dynamics.150 For instance, 10-s pulsing with 1 μM H2O2 reportedly activates cellular redox relay chains151 and 5 min exposure to 1.5 mM H2O2 has been shown to elicit Ca2+-release channel activation.152 It is possible that a combination of a colocalized point source of H2O2 (generated on-demand in a controlled manner) and BirA in conjunction with global biotin-phenol administration could remedy this issue. Using such a dual delivery system, locale/protein specificity will likely be enhanced because any aberrant APEX or H2O2 generation would be unlikely to colocalize. It should further be noted that APEX and its derivatives, as well as BirA mutants, are the products of extensive genetic engineering in which the desired activity is honed through a combination of directed evolution, computational analysis, and rational mutagenesis based on structural knowledge of the tagging enzyme.
Summary and Outlook
Given the complexity of the systems chemical biologists study, there is no panacea to universally solve all problems. However, as our discussion shows, there is a considerable amount of overlap between all tethered processes, because they share common physiochemical principles. We thus recommend a thorough survey of tethered chemistry prior to undertaking/designing experiments to try to anticipate problems, and afford preemptive solutions. It is also paramount going forward that we have as many tools available to study biology from a chemical perspective as possible. For this reason, both Class I and Class II approaches should be considered when planning an experiment or developing a method. The tethered approach can endow low affinity/selectivity ligands/proteins with laser-guided binding accuracy, and it can further force reactive associations, which otherwise would require an unknown stimulus or cue. It is thus often the case that the Class I approaches require that an outcome/association/end point be known or presumed; for example, a specific POI is inhibited, such as in BOLT, or a specific POI forms a tether through a specific sugar as in aptamer ligation, or a specific POI is degraded through a specific pathway, such as with PROTACS. This requirement may be restrictive, however the power of small-molecule tethering to promote an atypical cellular process will remain an important arrow in the quiver of life scientists. This is principally because gain of function is not a common mechanism for compounds that bind single sites on enzymes, such as therapeutics.153,154 Thus, there remains a huge scope to explore phenotypic output using substoichiometric modification through gain-of-function-induced proximity targeting.
T-REX shows that on-demand targeting through provisional or momentary tethering can afford levels of specificity similar to those obtained with permanent tethering. However, there is no evidence that T-REX induces electrophilic modification of proteins that are not intrinsically electrophile reactive, thus the target POI and reactive signal likely should be chosen to be a “matched” pair. Assuming that the players are known, T-REX is another method to elicit gain-of-function events in cells, and thus elicit “cellular mind control”. On-going experiments show that T-REX is a useful screening tool and can identify the most reactive proteins in a panel of postulated sensor proteins. Beyond eliciting redox responses through on-demand redox targeting, T-REX currently stands as a test case for future development of synthetic systems that would enable precision-controlled gain-of-function posttranslational modifications through, for instance, low-stoichiometry acetylation155,156 and methylation events157 on a specific POI that in turn drive specific biological outcomes. Aside from CALI, the other Class II-related approaches are arguably best used to identify new interactions. This is because Class II and related ideas are, by their nature, unbiased: only one ligand pole is bound to a specific protein, leaving the other pole free to react. The power of BioID and APEX testifies to the efficacy of this strategy. It remains to be shown whether systems like T-REX can be used in similar applications.
A huge body of evidence indicates that the proximity-dependent approach has potential to be used in a wide array of circumstances. However, current approaches are directed mostly at the basic science level. We anticipate future efforts will also be devoted to extending these proof-of-concept design principles to testing in whole organisms and ultimately in humans.
Acknowledgments
We thank Profs. Jeremy Baskin and Rick Cerione of Cornell University for their comments on the manuscript and overall feedback, respectively, and Mr. Saba Parvez of the Aye laboratory for helpful suggestions. We acknowledge an NIH Director’s New Innovator award (1DP2GM114850), an NSF CAREER award (CHE-1351400), a Beckman Young Investigator award, a Burroughs Wellcome Fund CRTG, a Sloan Research Fellowship, and an intercampus seed grant from Cornell University and Weill Cornell Medicine (to Y.A). J.R.P. acknowledges the NIH CBI training grant (NIGMS T32GM008500, PI Prof. Hening Lin, Cornell University). Cornell university is the owner of the trademark for T-REX platform conceived and developed by the Aye laboratory.
The authors declare no competing financial interest.
References
- Thorner J.; Hunter T.; Cantley L. C.; Sever R. Cold Spring Harbor Perspect. Biol. 2014, 6, a022913. 10.1101/cshperspect.a022913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes N. E.; Ingham P. W.; Lim W. A.; Marshall C. J.; Massague J.; Pawson T. Nat. Rev. Mol. Cell Biol. 2013, 14, 393. 10.1038/nrm3581. [DOI] [PubMed] [Google Scholar]
- Schreiber S. L.; Kotz J. D.; Li M.; Aube J.; Austin C. P.; Reed J. C.; Rosen H.; White E. L.; Sklar L. A.; Lindsley C. W.; Alexander B. R.; Bittker J. A.; Clemons P. A.; de Souza A.; Foley M. A.; Palmer M.; Shamji A. F.; Wawer M. J.; McManus O.; Wu M.; Zou B.; Yu H.; Golden J. E.; Schoenen F. J.; Simeonov A.; Jadhav A.; Jackson M. R.; Pinkerton A. B.; Chung T. D.; Griffin P. R.; Cravatt B. F.; Hodder P. S.; Roush W. R.; Roberts E.; Chung D. H.; Jonsson C. B.; Noah J. W.; Severson W. E.; Ananthan S.; Edwards B.; Oprea T. I.; Conn P. J.; Hopkins C. R.; Wood M. R.; Stauffer S. R.; Emmitte K. A. Cell 2015, 161, 1252. 10.1016/j.cell.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Cole P. A. Curr. Opin. Chem. Biol. 2015, 28, 115. 10.1016/j.cbpa.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghatelian A.; Cravatt B. F. Nat. Chem. Biol. 2005, 1, 130. 10.1038/nchembio0805-130. [DOI] [PubMed] [Google Scholar]
- Rakhit R.; Navarro R.; Wandless T. J. Chem. Biol. 2014, 21, 1238. 10.1016/j.chembiol.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corson T. W.; Aberle N.; Crews C. M. ACS Chem. Biol. 2008, 3, 677. 10.1021/cb8001792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nature 2004, 431, 931. 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- Kiessling L. L.; Gestwicki J. E.; Strong L. E. Angew. Chem., Int. Ed. 2006, 45, 2348. 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean K. M.; Palmer A. E. Nat. Chem. Biol. 2014, 10, 512. 10.1038/nchembio.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W.; Gao L.; Chen X. Curr. Opin. Chem. Biol. 2015, 28, 156. 10.1016/j.cbpa.2015.07.020. [DOI] [PubMed] [Google Scholar]
- Lowder M. A.; Appelbaum J. S.; Hobert E. M.; Schepartz A. Curr. Opin. Chem. Biol. 2011, 15, 781. 10.1016/j.cbpa.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRose R.; Miyamoto T.; Inoue T. Pfluegers Arch. 2013, 465, 409. 10.1007/s00424-012-1208-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree G. R.; Schreiber S. L. Trends Biochem. Sci. 1996, 21, 418. 10.1016/S0968-0004(96)20027-1. [DOI] [PubMed] [Google Scholar]
- Lang K.; Chin J. W. ACS Chem. Biol. 2014, 9, 16. 10.1021/cb4009292. [DOI] [PubMed] [Google Scholar]
- Xue L.; Karpenko I. A.; Hiblot J.; Johnsson K. Nat. Chem. Biol. 2015, 11, 917. 10.1038/nchembio.1959. [DOI] [PubMed] [Google Scholar]
- Fang X.; Fu Y.; Long M. J.; Haegele J. A.; Ge E. J.; Parvez S.; Aye Y. J. Am. Chem. Soc. 2013, 135, 14496. 10.1021/ja405400k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez S.; Fu Y.; Li J.; Long M. J.; Lin H. Y.; Lee D. K.; Hu G. S.; Aye Y. J. Am. Chem. Soc. 2015, 137, 10. 10.1021/ja5084249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H. Y.; Haegele J. A.; Disare M. T.; Lin Q.; Aye Y. J. Am. Chem. Soc. 2015, 137, 6232. 10.1021/ja5132648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam S. S.; Martell J. D.; Kamer K. J.; Deerinck T. J.; Ellisman M. H.; Mootha V. K.; Ting A. Y. Nat. Methods 2015, 12, 51. 10.1038/nmeth.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martell J. D.; Deerinck T. J.; Sancak Y.; Poulos T. L.; Mootha V. K.; Sosinsky G. E.; Ellisman M. H.; Ting A. Y. Nat. Biotechnol. 2012, 30, 1143. 10.1038/nbt.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee H.-W.; Zou P.; Udeshi N. D.; Martell J. D.; Mootha V. K.; Carr S. A.; Ting A. Y. Science 2013, 339, 1328. 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. L.; Hu Y.; Udeshi N. D.; Lau T. Y.; Wirtz-Peitz F.; He L.; Ting A. Y.; Carr S. A.; Perrimon N. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 12093. 10.1073/pnas.1515623112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C. C.; Jan C. H.; Weissman J. S. Science 2014, 346, 748. 10.1126/science.1257522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan C. H.; Williams C. C.; Weissman J. S. Science 2014, 346, 716. 10.1126/science.1257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer D.; Wandless T. J.; Crabtree G. R.; Schreiber S. L. Science 1993, 262, 1019. 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- Kley N. Chem. Biol. 2004, 11, 599. 10.1016/j.chembiol.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Gestwicki J. E.; Marinec P. S. Comb. Chem. High Throughput Screening 2007, 10, 667. 10.2174/138620707782507296. [DOI] [PubMed] [Google Scholar]
- Buckley D. L.; Crews C. M. Angew. Chem., Int. Ed. 2014, 53, 2312. 10.1002/anie.201307761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y.; Ishikawa M.; Naito M.; Hashimoto Y. J. Am. Chem. Soc. 2010, 132, 5820. 10.1021/ja100691p. [DOI] [PubMed] [Google Scholar]
- Wu C. Y.; Roybal K. T.; Puchner E. M.; Onuffer J.; Lim W. A. Science 2015, 350, aab4077. 10.1126/science.aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEnaney P. J.; Parker C. G.; Zhang A. X.; Spiegel D. A. ACS Chem. Biol. 2012, 7, 1139. 10.1021/cb300119g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery E. L.; Chatterjee A. K.; Winzeler E. A. Nat. Rev. Microbiol. 2013, 11, 849. 10.1038/nrmicro3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees D. C.; Congreve M.; Murray C. W.; Carr R. Nat. Rev. Drug Discovery 2004, 3, 660. 10.1038/nrd1467. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A.; Wells J. A.; Braisted A. C. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 199. 10.1146/annurev.biophys.33.110502.140409. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A.; Lam J. W.; Wiesmann C.; Luong T. N.; Simmons R. L.; DeLano W. L.; Choong I. C.; Burdett M. T.; Flanagan W. M.; Lee D.; Gordon E. M.; O’Brien T. Nat. Biotechnol. 2003, 21, 308. 10.1038/nbt786. [DOI] [PubMed] [Google Scholar]
- Cornell B. A.; Braach-Maksvytis V. L. B.; King L. G.; Osman P. D. J.; Raguse B.; Wieczorek L.; Pace R. J. Nature 1997, 387, 580. 10.1038/42432. [DOI] [PubMed] [Google Scholar]
- Griss R.; Schena A.; Reymond L.; Patiny L.; Werner D.; Tinberg C. E.; Baker D.; Johnsson K. Nat. Chem. Biol. 2014, 10, 598. 10.1038/nchembio.1554. [DOI] [PubMed] [Google Scholar]
- Gallagher S. S.; Sable J. E.; Sheetz M. P.; Cornish V. W. ACS Chem. Biol. 2009, 4, 547. 10.1021/cb900062k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunbul M.; Nacheva L.; Jaschke A. Bioconjugate Chem. 2015, 26, 1466. 10.1021/acs.bioconjchem.5b00304. [DOI] [PubMed] [Google Scholar]
- Tsai Y.-H.; Essig S.; James J. R.; Lang K.; Chin J. W. Nat. Chem. 2015, 7, 554. 10.1038/nchem.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena A.; Griss R.; Johnsson K. Nat. Commun. 2015, 6, 7830. 10.1038/ncomms8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craik D. J.; Fairlie D. P.; Liras S.; Price D. Chem. Biol. Drug Des. 2013, 81, 136. 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- Werner H. M.; Cabalteja C. C.; Horne W. S. ChemBioChem 2015, 10.1002/cbic.201500312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K. K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Nat. Chem. Biol. 2015, 11, 611. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C. L.; Scott W. Nat. Cell Biol. 2014, 16, 10. 10.1038/ncb2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna J. A.; Charpentier E. Science 2014, 346, 1258096. 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Kochenderfer J. N.; Rosenberg S. A. Nat. Rev. Clin. Oncol. 2013, 10, 267. 10.1038/nrclinonc.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEnaney P. J.; Fitzgerald K. J.; Zhang A. X.; Douglass E. F. Jr.; Shan W.; Balog A.; Kolesnikova M. D.; Spiegel D. A. J. Am. Chem. Soc. 2014, 136, 18034. 10.1021/ja509513c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreotti G.; Pedone E.; Giordano A.; Cubellis M. V. Mol. Genet. Genomic Med. 2013, 1, 32. 10.1002/mgg3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn F.; Gordan S.; Lux A. Trends Immunol. 2015, 36, 325. 10.1016/j.it.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. Curr. Opin. Cell Biol. 1992, 4, 1024. 10.1016/0955-0674(92)90135-Y. [DOI] [PubMed] [Google Scholar]
- Komander D.; Rape M. Annu. Rev. Biochem. 2012, 81, 203. 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Nalepa G.; Rolfe M.; Harper J. W. Nat. Rev. Drug Discovery 2006, 5, 596. 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- Geng F.; Wenzel S.; Tansey W. P. Annu. Rev. Biochem. 2012, 81, 177. 10.1146/annurev-biochem-052110-120012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Jensen M. K.; Keasling J. D. Curr. Opin. Chem. Biol. 2015, 28, 1. 10.1016/j.cbpa.2015.05.013. [DOI] [PubMed] [Google Scholar]
- Bruchez M. P. Curr. Opin. Chem. Biol. 2015, 27, 18. 10.1016/j.cbpa.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggeling L.; Bott M.; Marienhagen J. Curr. Opin. Biotechnol. 2015, 35C, 30. 10.1016/j.copbio.2014.12.021. [DOI] [PubMed] [Google Scholar]
- Howes P. D.; Chandrawati R.; Stevens M. M. Science 2014, 346, 1247390. 10.1126/science.1247390. [DOI] [PubMed] [Google Scholar]
- Juillerat A.; Gronemeyer T.; Keppler A.; Gendreizig S.; Pick H.; Vogel H.; Johnsson K. Chem. Biol. 2003, 10, 313. 10.1016/S1074-5521(03)00068-1. [DOI] [PubMed] [Google Scholar]
- Hall M. P.; Unch J.; Binkowski B. F.; Valley M. P.; Butler B. L.; Wood M. G.; Otto P.; Zimmerman K.; Vidugiris G.; Machleidt T.; Robers M. B.; Benink H. A.; Eggers C. T.; Slater M. R.; Meisenheimer P. L.; Klaubert D. H.; Fan F.; Encell L. P.; Wood K. V. ACS Chem. Biol. 2012, 7, 1848. 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z.; Rao J. Curr. Opin. Biotechnol. 2009, 20, 37. 10.1016/j.copbio.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Cornish V. W.; Min W. Curr. Opin. Chem. Biol. 2013, 17, 637. 10.1016/j.cbpa.2013.05.018. [DOI] [PubMed] [Google Scholar]
- Schreiber S. L. Science 1991, 251, 283. 10.1126/science.1702904. [DOI] [PubMed] [Google Scholar]
- Sethi K. K.; Verma S. M.; Tanc M.; Purper G.; Calafato G.; Carta F.; Supuran C. T. Bioorg. Med. Chem. 2014, 22, 1586. 10.1016/j.bmc.2014.01.031. [DOI] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. Nat. Rev. Drug Discovery 2011, 10, 307. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Tummino P. J.; Copeland R. A. Biochemistry 2008, 47, 5481. 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- Lu H.; Tonge P. J. Curr. Opin. Chem. Biol. 2010, 14, 467. 10.1016/j.cbpa.2010.06.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A.; Pompliano D. L.; Meek T. D. Nat. Rev. Drug Discovery 2007, 5, 730. 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Buskirk A. R.; Liu D. R. Chem. Biol. 2005, 12, 151. 10.1016/j.chembiol.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Head J. F.; Inouye S.; Teranishi K.; Shimomura O. Nature 2000, 405, 372. 10.1038/35012659. [DOI] [PubMed] [Google Scholar]
- Dundas C. M.; Demonte D.; Park S. Appl. Microbiol. Biotechnol. 2013, 97, 9343. 10.1007/s00253-013-5232-z. [DOI] [PubMed] [Google Scholar]
- Dunlap P. In Bioluminescence: Fundamentals and Applications in Biotechnology; Springer: Berlin/Heidelberg, 2014; p 37. [Google Scholar]
- Lavis L. D.; Raines R. T. ACS Chem. Biol. 2014, 9, 855. 10.1021/cb500078u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soreq H.; Seidman S. Nat. Rev. Neurosci 2001, 2, 294. 10.1038/35067589. [DOI] [PubMed] [Google Scholar]
- Gautier A.; Juillerat A.; Heinis C.; Correa I. R. Jr.; Kindermann M.; Beaufils F.; Johnsson K. Chem. Biol. 2008, 15, 128. 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Qizilbash N.; Whitehead A.; Higgins J.; Wilcock G.; Schneider L.; Farlow M. JAMA 1998, 280, 1777. 10.1001/jama.280.20.1777. [DOI] [PubMed] [Google Scholar]
- Dalziel M.; Crispin M.; Scanlan C. N.; Zitzmann N.; Dwek R. A. Science 2014, 343, 1235681. 10.1126/science.1235681. [DOI] [PubMed] [Google Scholar]
- Freeze H. H. Nat. Rev. Genet. 2006, 7, 537. 10.1038/nrg1894. [DOI] [PubMed] [Google Scholar]
- Prescher J. A.; Bertozzi C. R. Cell 2006, 126, 851. 10.1016/j.cell.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Laughlin S. T.; Bertozzi C. R. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 12. 10.1073/pnas.0811481106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson P. V.; de Almeida-Escobedo G.; de Groot A. E.; McKechnie J. L.; Bertozzi C. R. J. Am. Chem. Soc. 2015, 137, 10452. 10.1021/jacs.5b04279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe A. D.; Pai S.; Ellington A. Nat. Rev. Drug Discovery 2010, 9, 537. 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P. V.; Chen X.; Smyrniotis C.; Xenakis A.; Hu T.; Bertozzi C. R.; Wu P. Angew. Chem., Int. Ed. 2009, 48, 4030. 10.1002/anie.200806319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescher J. A.; Bertozzi C. R. Nat. Chem. Biol. 2005, 1, 13. 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Angew. Chem., Int. Ed. 2001, 40, 2004. 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Peradziryi H.; Tolwinski N. S.; Borchers A. Arch. Biochem. Biophys. 2012, 524, 71. 10.1016/j.abb.2011.12.019. [DOI] [PubMed] [Google Scholar]
- Lo Conte M.; Staderini S.; Marra A.; Sanchez-Navarro M.; Davis B. G.; Dondoni A. Chem. Commun. 2011, 47, 11086. 10.1039/c1cc14402b. [DOI] [PubMed] [Google Scholar]
- Allen T. M. Nat. Rev. Cancer 2002, 2, 750. 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- Huang M.; Shen A.; Ding J.; Geng M. Trends Pharmacol. Sci. 2014, 35, 41. 10.1016/j.tips.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Koonin E. V.; Galperin M. Y. In Evolution–Function: Computational Approaches in Comparative Genomics; Kluwer Academic: Boston, 2003. [PubMed] [Google Scholar]
- Knight Z. A.; Shokat K. M. Chem. Biol. 2005, 12, 621. 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Crews C. M. Chem. Biol. 1996, 3, 961. 10.1016/S1074-5521(96)90162-3. [DOI] [PubMed] [Google Scholar]
- Liu C. C.; Schultz P. G. Annu. Rev. Biochem. 2010, 79, 413. 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- Skarpen E.; Flinder L. I.; Rosseland C. M.; Orstavik S.; Wierod L.; Oksvold M. P.; Skalhegg B. S.; Huitfeldt H. S. FASEB J. 2008, 22, 466. 10.1096/fj.07-8650com. [DOI] [PubMed] [Google Scholar]
- Broichhagen J.; Trauner D. Curr. Opin. Chem. Biol. 2014, 21, 121. 10.1016/j.cbpa.2014.07.008. [DOI] [PubMed] [Google Scholar]
- Knoll H. In CRC Handbook of organic photochemistry and photobiology, 2nd ed.; Horspool W., Lenci F., Eds.; CRC Press: Boca Raton, FL, 2004; p 1. [Google Scholar]
- Miller L. W.; Sable J.; Goelet P.; Sheetz M. P.; Cornish V. W. Angew. Chem., Int. Ed. 2004, 43, 1672. 10.1002/anie.200352852. [DOI] [PubMed] [Google Scholar]
- Hoskins A. A.; Friedman L. J.; Gallagher S. S.; Crawford D. J.; Anderson E. G.; Wombacher R.; Ramirez N.; Cornish V. W.; Gelles J.; Moore M. J. Science 2011, 331, 1289. 10.1126/science.1198830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin A. D.; Kiick K. L. Bioconjugate Chem. 2011, 22, 1946. 10.1021/bc200148v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing C.; Cornish V. W. ACS Chem. Biol. 2013, 8, 1704. 10.1021/cb300657r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks K. M.; Rosinov M.; Nolan G. P. Chem. Biol. 2004, 11, 347. 10.1016/j.chembiol.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Sabnis Y.; Zhao Z.; Zhang T.; Buhrlage S. J.; Jones L. H.; Gray N. S. Chem. Biol. 2013, 20, 146. 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah R.; Thomas J. R.; Shafer C. M. Bioorg. Med. Chem. Lett. 2014, 24, 33. 10.1016/j.bmcl.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Serafimova I. M.; Pufall M. A.; Krishnan S.; Duda K.; Cohen M. S.; Maglathlin R. L.; McFarland J. M.; Miller R. M.; Frodin M.; Taunton J. Nat. Chem. Biol. 2012, 8, 471. 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. T. Annu. Rev. Biochem. 1984, 53, 493. 10.1146/annurev.bi.53.070184.002425. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists, 2nd ed.; Wiley-Interscience: Weinheim, 2013. [PubMed] [Google Scholar]
- Weinstock C.; Schnaidt M. Int. J. Immunogenet. 2013, 40, 171. 10.1111/j.1744-313X.2012.01147.x. [DOI] [PubMed] [Google Scholar]
- Mack E. T.; Perez-Castillejos R.; Suo Z.; Whitesides G. M. Anal. Chem. 2008, 80, 5550. 10.1021/ac800578w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K. M. Pediatr. Res. 2010, 67, 505. 10.1203/PDR.0b013e3181d35017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson M. P.; Hersey A.; Montanari D.; Overington J. Nat. Rev. Drug Discovery 2011, 10, 197. 10.1038/nrd3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Adv. Drug Delivery Rev. 2001, 46, 3. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Imming P.; Sinning C.; Meyer A. Nat. Rev. Drug Discovery 2006, 5, 821. 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- Krutzik P. O.; Nolan G. P. Nat. Methods 2006, 3, 361. 10.1038/nmeth872. [DOI] [PubMed] [Google Scholar]
- Jing C.; Cornish V. W. Curr. Protoc. Chem. Biol. 2013, 5, 131. 10.1002/9780470559277.ch130019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los G. V.; Encell L. P.; McDougall M. G.; Hartzell D. D.; Karassina N.; Zimprich C.; Wood M. G.; Learish R.; Ohana R. F.; Urh M.; Simpson D.; Mendez J.; Zimmerman K.; Otto P.; Vidugiris G.; Zhu J.; Darzins A.; Klaubert D. H.; Bulleit R. F.; Wood K. V. ACS Chem. Biol. 2008, 3, 373. 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- Uttamapinant C.; Sanchez M. I.; Liu D. S.; Yao J. Z.; Ting A. Y. Nat. Protoc. 2013, 8, 1620. 10.1038/nprot.2013.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M.; Brentjens R.; Riviere I. Cancer Discovery 2013, 3, 388. 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Udugamasooriya D. G.; Lim H. S.; Kodadek T. Nat. Chem. Biol. 2010, 6, 258. 10.1038/nchembio.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman-Kim D.; Diefenbach T. J.; Eustace B. K.; Jay D. G. Methods Cell Biol. 2007, 82, 335. 10.1016/S0091-679X(06)82011-X. [DOI] [PubMed] [Google Scholar]
- Jacobson K.; Rajfur Z.; Vitriol E.; Hahn K. Trends Cell Biol. 2008, 18, 443. 10.1016/j.tcb.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay D. G. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 5454. 10.1073/pnas.85.15.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König K.; Liang H.; Berns M. W.; Tromberg B. J. Nature 1995, 377, 20. 10.1038/377020a0. [DOI] [PubMed] [Google Scholar]
- Tinevez J. Y.; Dragavon J.; Baba-Aissa L.; Roux P.; Perret E.; Canivet A.; Galy V.; Shorte S. Methods Enzymol. 2012, 506, 291. 10.1016/B978-0-12-391856-7.00039-1. [DOI] [PubMed] [Google Scholar]
- Simon R. J.; Kania R. S.; Zuckermann R. N.; Huebner V. D.; Jewell D. A.; Banville S.; Ng S.; Wang L.; Rosenberg S.; Marlowe C. K.; Spellmeyer D. C.; Tan R.; Frankel A. D.; Santi D. V.; Cohen F. E.; Bartlett P. A. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 9367. 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M. Cancer Sci. 2003, 94, 751. 10.1111/j.1349-7006.2003.tb01514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg A. L. Science 1995, 268, 522. 10.1126/science.7725095. [DOI] [PubMed] [Google Scholar]
- Delmastro-Greenwood M.; Freeman B. A.; Wendell S. G. Annu. Rev. Physiol. 2014, 76, 79. 10.1146/annurev-physiol-021113-170341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggar K. K.; Li S. S. Nat. Rev. Mol. Cell Biol. 2015, 16, 5. 10.1038/nrm3915. [DOI] [PubMed] [Google Scholar]
- Yang J.; Tallman K. A.; Porter N. A.; Liebler D. C. Anal. Chem. 2015, 87, 2535. 10.1021/ac504685y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs A. T.; Marnett L. J. Acc. Chem. Res. 2010, 43, 673. 10.1021/ar900286y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopfer F. J.; Cipollina C.; Freeman B. A. Chem. Rev. 2011, 111, 5997. 10.1021/cr200131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Weerapana E.; Blewett M. M.; Cravatt B. F. Nat. Methods 2014, 11, 79. 10.1038/nmeth.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codreanu S. G.; Ullery J. C.; Zhu J.; Tallman K. A.; Beavers W. N.; Porter N. A.; Marnett L. J.; Zhang B.; Liebler D. C. Mol. Cell. Proteomics 2014, 13, 849. 10.1074/mcp.M113.032953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E.; Wang C.; Simon G. M.; Richter F.; Khare S.; Dillon M. B.; Bachovchin D. A.; Mowen K.; Baker D.; Cravatt B. F. Nature 2010, 468, 790. 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux K. J. Cell. Mol. Life Sci. 2013, 70, 3657. 10.1007/s00018-013-1287-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. I.; Birendra K. C.; Zhu W.; Motamedchaboki K.; Doye V.; Roux K. J. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E2453. 10.1073/pnas.1406459111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan C. H.; Williams C. C.; Weissman J. S. Science 2015, 348, 1217. 10.1126/science.aaa8299. [DOI] [PubMed] [Google Scholar]
- Di Sabato G.; Jencks W. P. J. Am. Chem. Soc. 1961, 83, 4393. 10.1021/ja01482a024. [DOI] [Google Scholar]
- Pal M.; Bearne S. L. Org. Biomol. Chem. 2014, 12, 9760. 10.1039/C4OB02079K. [DOI] [PubMed] [Google Scholar]
- Gould R. F. In Reactions of Coordinated Ligands; Busch D. H., Ed.; American Chemical Society: Washington, DC: 1962; Vol. 37, p i. [Google Scholar]
- Wodzinska J.; Kluger R. J. Org. Chem. 2008, 73, 4753. 10.1021/jo800667b. [DOI] [PubMed] [Google Scholar]
- Winterbourn C. C.; Metodiewa D. Free Radical Biol. Med. 1999, 27, 322. 10.1016/S0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- Jones C. M.; Lawrence A.; Wardman P.; Burkitt M. J. Biochem. Soc. Trans. 2003, 31, 1337. 10.1042/bst0311337. [DOI] [PubMed] [Google Scholar]
- Mezyk S. P. J. Phys. Chem. 1996, 100, 8861. 10.1021/jp9535553. [DOI] [Google Scholar]
- McGrath C. E.; Tallman K. A.; Porter N. A.; Marnett L. J. Chem. Res. Toxicol. 2011, 24, 357. 10.1021/tx100323m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkes L. K.; Trujillo M.; Bartesaghi S.; Radi R.; Wardman P. Arch. Biochem. Biophys. 2011, 506, 242. 10.1016/j.abb.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Winterbourn C. C. Methods Enzymol. 2013, 528, 3. 10.1016/B978-0-12-405881-1.00001-X. [DOI] [PubMed] [Google Scholar]
- Murphy M. P.; Holmgren A.; Larsson N. G.; Halliwell B.; Chang C. J.; Kalyanaraman B.; Rhee S. G.; Thornalley P. J.; Partridge L.; Gems D.; Nystrom T.; Belousov V.; Schumacker P. T.; Winterbourn C. C. Cell Metab. 2011, 13, 361. 10.1016/j.cmet.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems P. H.; Rossignol R.; Dieteren C. E.; Murphy M. P.; Koopman W. J. Cell Metab. 2015, 22, 207. 10.1016/j.cmet.2015.06.006. [DOI] [PubMed] [Google Scholar]
- Sobotta M. C.; Liou W.; Stocker S.; Talwar D.; Oehler M.; Ruppert T.; Scharf A. N.; Dick T. P. Nat. Chem. Biol. 2015, 11, 64. 10.1038/nchembio.1695. [DOI] [PubMed] [Google Scholar]
- Oba T.; Ishikawa T.; Yamaguchi M. Am. J. Physiol. Cell. Physiol. 1998, 274, C914. [DOI] [PubMed] [Google Scholar]
- Howell A. Crit Rev. Oncol Hematol 2006, 57, 265. 10.1016/j.critrevonc.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Nasr R.; Lallemand-Breitenbach V.; Zhu J.; Guillemin M. C.; de The H. Clin. Cancer Res. 2009, 15, 6321. 10.1158/1078-0432.CCR-09-0209. [DOI] [PubMed] [Google Scholar]
- Verdin E.; Ott M. Nat. Rev. Mol. Cell Biol. 2015, 16, 258. 10.1038/nrm3931. [DOI] [PubMed] [Google Scholar]
- Choudhary C.; Weinert B. T.; Nishida Y.; Verdin E.; Mann M. Nat. Rev. Mol. Cell Biol. 2014, 15, 536. 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- Jensen O. N. Nat. Rev. Mol. Cell Biol. 2006, 7, 391. 10.1038/nrm1939. [DOI] [PubMed] [Google Scholar]