

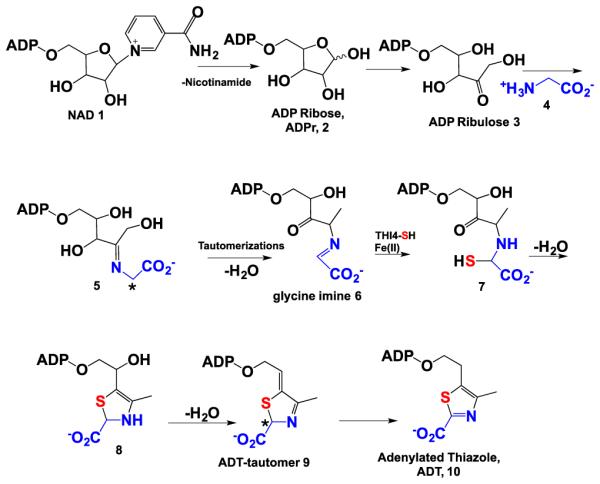
Abstract
Bacteria and yeast utilize different strategies for sulfur incorporation in the biosynthesis of the thiamin thiazole. Bacteria use thiocarboxylated proteins. In contrast, Saccharomyces cerevisiae thiazole synthase (THI4p) uses an active site cysteine as the sulfide source and is inactivated after a single turnover. Here, we demonstrate that the Thi4 ortholog from Methanococcus jannaschii uses exogenous sulfide and is catalytic. Structural and biochemical studies on this enzyme elucidate the mechanistic details of the sulfide transfer reactions.
Thiamin (Vitamin B1), an essential cofactor in all living systems, is required for key steps in carbohydrate and amino acid metabolism. Biosynthesis of thiamin is achieved by the separate synthesis of the pyrimidine and thiazole moieties and subsequent coupling of these two heterocyclic rings.1 In bacteria, the thiazole sulfur is derived from a protein thiocarboxylate and this sulfide transfer strategy has now been identified in the biosynthesis of methionine,2 cysteine,3,4 thioquinolobactin,5 molybdopterin,6 2-thiosugar,7 and thiopyrimidines.8-11 The eukaryotic pathway to the thiamin thiazole employs a different strategy involving sulfide transfer from an active site cysteine residue of the thiazole synthase (THI4p). This protein, isolated from Saccharomyces cerevisiae, catalyzes the conversion of NAD and glycine to the adenylated thiazole (ADT) 10 in a reaction where the thiazole sulfide is derived from C205 of THI4p (Scheme 1).12-14 This sulfur transfer reaction is iron-dependent and generates inactive enzyme through the formation of dehydroalanine, making THI4p a single turnover suicide enzyme. The mechanistic role of Fe(II) in the sulfide transfer and the molecular logic of using an active site cysteine as the sulfur source are poorly understood.
Scheme 1.
Methanococcus jannaschii live in sulfide rich habitats and contain millimolar concentrations of sulfide.15 Recent findings have shown that the biosynthesis of Fe/S clusters, methionine and thiouridine in methanogenic archaea utilize free sulfide.16,17 This suggested that these organisms might also use sulfide for the synthesis of the thiamin thiazole and thus overcome what appears to be an inefficient strategy in the case of the S. cerevisiae enzyme. In support of this, sequence analysis demonstrated that many Archaeal species contain an ortholog of the eukaryotic thiazole synthase (THI4p) and lack the bacterial thiazole biosynthesis genes. In the ortholog from Methanococcus jannaschii (Mj0601, MjThi4), the active site cysteine of the yeast enzyme, is replaced by a histidine residue. Here we report that MjThi4 catalyzes the formation of ADT 10 using exogenous sulfide and that this enzyme was particularly amenable to structural and mechanistic studies.
The Mj0601 gene was overexpressed in Escherichia coli BL21 (DE3) using a THT vector (pET28-based vector with a TEV protease site and an N-terminal His-tag) and purified by Ni-affinity chromatography. NAD 1, ADT 10, FMN (Figure S1), and smaller amounts of ADP-ribose 2 and ADP-ribulose 3 were found associated with the purified protein (data not shown). In assays run in the absence of sulfide, using NAD 1 (or ADP-ribose 2) and glycine 4 as substrates, MjThi4 generated the same sulfur-free intermediates observed in ScTHI4 (ADP-ribose 2, ADP-ribulose 3 , glycine imine 6).13 The rates of formation of these intermediates were not iron dependent (Figure S2) and ranged from 0.1 to 0.3 min−1 at 65 °C. As expected from the sequence analysis, neither ADT 10 nor its tautomer 9 was detected (Figure 1A and 1B), demonstrating that MjThi4 is missing the cysteine sulfide donor found in ScThi4p.
Figure 1.

(A) HPLC chromatogram showing formation of ADT 10 in the presence of different sulfur sources. Assays were carried out in the presence of 20 μM MjThi4, 500 μM ADP-ribose (ADPr) 2, 1 mM glycine, 400 μM ferrous ammonium sulfate (FeAS) and 0.8-1 mM of each of the sulfur compounds. Assays were quenched with 8 M GuHCl after 1 hour anaerobic incubation at 85 °C. (B) HPLC chromatogram showing conversion of NAD into ADT in the presence 100 μM MjThi4, 300 μM NAD, 1 mM glycine, 400 μM FeAS and 800 μM Na2S at 25 °C. (C) Overlaid UV-Vis spectra of the reaction product shown in panel A and an authentic sample of ADT prepared using ScTHI4. (D) Production of ADP-ribulose (ADPrl) 3 from ADPr 2 by MjThi4. Conditions were as follows; 500 μM MjThi4, 200 μM ADPr, 1 mM glycine, 1 mM FeAS and 800 μM Na2S inside a glovebox at 25 °C.
Several possible sulfur sources for thiazole formation were tested in the presence of Fe(II) (sodium sulfide, cysteine, thioacetate, thiosulfate and glutathione). Assays were carried out at 65 to 85 °C because M. jannaschii is a hyperthermophilic organism.18 Of these, only sodium sulfide and thioacetate led to the formation of a new product. This product had an identical UV-Vis spectrum and retention time to an authentic sample of ADT 10 purified from the ScTHI4p reaction (Figure 1B and 1C). ADT formed in the presence of thioacetate was later determined to be due to hydrolysis of thioacetate to sulfide. Addition of FMN and reductants (DTT and dithionite) to the assay mixture did not affect the activity, demonstrating that the copurified FMN is not required for ADT formation. ADT was not formed in the absence of iron (Figure 1B). Unlike the ScThi4, MjThi4 can catalyze multiple turnovers because the sulfide is not enzyme derived. Up to five turnovers were observed in a period of 2 h at 85 °C (0.04 min−1). Time dependent formation of ADT shows the presence of a burst phase at the first turnover with a rate of 0.15 min−1 followed by a slower linear phase of 0.005 min−1 (Figure 2). The burst amplitude is close to 1 (0.89), indicating that product dissociation rather than formation is almost completely rate limiting.
Figure 2.
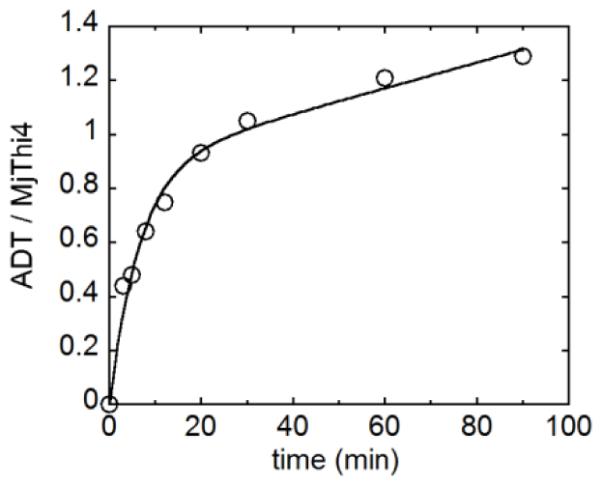
Time-dependent formation of ADT 10. Enzyme (10 μM) was reacted with 500 μM ADPr 2, 1 mM glycine, 400 μM FeAS and 800 μM Na2S at 80 °C anaerobically and quenched with 8 M GuHCl at various time points. ADT concentrations were determined by HPLC analysis.
The Fe(II) content of the enzyme was determined using the ferrozine reagent.19 MjThi4 samples expressed in M9 or LB medium lacked bound iron after purification (<1%). However, when MjThi4 was treated with excess ferrous ammonium sulfate and the weakly bound iron removed, the iron content was approximately one iron per monomer. Although enzyme reconstituted with Fe(II) had a much faster rate compared to the enzyme with Fe(III) for ADT formation during the first turnover (0.0056 min−1 and 0.0014 min−1 respectively, Figure S3), the total amount of ADT formed is almost the same under multiple turnover conditions for both Fe(II) and Fe(III) (Figure S4). In addition to Fe(II) and Fe(III), which showed the highest levels of activity, Mn(II), Co(II), Zn(II), Ni(II), Mg(II) and Ca(II) but not Cu(I) and Cu(II) also supported activity (Figure S4). In addition, ESI-MS analysis of MjThi4 showed that the protein is intact in contrast to the ScTHI4 protein, which purifies from LB medium with a significant portion having a mass loss of 32 Da (loss of H2S, Figure S5).12
To gain more insight into the nature of the active site iron, EPR spectra of the enzyme in the Fe(II) and Fe(III) reconstituted forms were measured at 10 K (Figure 3). As expected, the Fe(II) form of the enzyme was EPR silent.20 The Fe(III) form of the enzyme exhibited a peak centered at g ~ 4.3, characteristic of a high-spin mononuclear Fe(III) site with S = 5/2.21,22 Furthermore, the nitric oxide complex of the ferrous enzyme showed peaks at g = 4.05 and g = 3.98 indicative of an Fe-NO system with a spin of S =3/2, similar to the EPR spectra of other nitric oxide complexes of mononuclear nonheme iron enzymes.20,23,24
Figure 3.
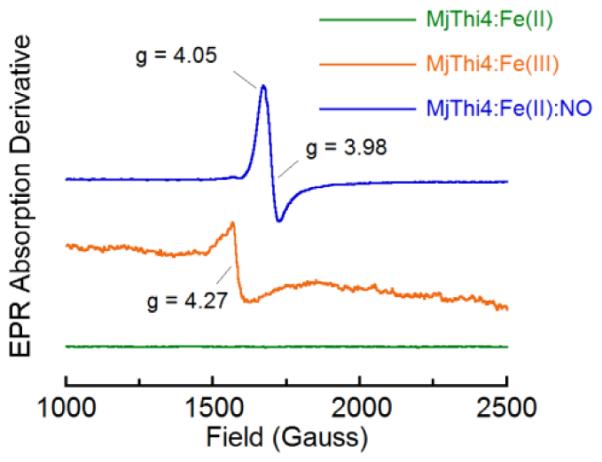
EPR spectra of various forms of MjThi4. Green trace: Purified MjThi4 (250 μM) reconstituted with ferrous iron; Orange trace: Ferric MjThi4 (250 μM), generated by air oxidation of the green trace sample; Blue trace: Nitric oxide treated (375 μM) ferrous MjThi4 (250 μM). Spectra were measured at 9.46 GHz at 10 K with field a modulation of 100 kHz and a microwave power of 0.2 mW.
We were able to study the structure and kinetics of intermediates in the MjThi4 reaction by sequential addition of the substrates to the enzyme. Upon addition of NAD 1 or ADP-ribose 2, in the presence of Fe(II), an ADP-ribulose complex 3 was obtained (Figure 1D). A chromophore absorbing at ~380 nm was formed upon addition of glycine to this complex (Figure S6). Treatment of this chromophore with sodium sulfide, under anaerobic conditions, resulted in its conversion to a new species with an absorbance at 305 nm characteristic of ADT tautomer, 9 (Figures S7A and S7B). The rate of this reaction matches the rate of ADT tautomer formation, suggesting that the loss of the 380 nm signal is due to the conversion of the Fe/glycine imine complex 6 to the ADT tautomer 9.
Primary deuterium isotope effects were measured to evaluate the kinetic significance of the loss of the two glycine protons (5 to 6 and 9 to 10, labeled with * in Scheme 1). No observable kinetic isotope effect was detected on the rate of formation of the 380 nm chromophore, demonstrating that the glycine deprotonation, involved in the conversion of 5 to 6, is not rate limiting (Figure S8B). An isotope effect on the rate of decay of the 380 nm absorbing intermediate was also not observed demonstrating that the second glycine deprotonation is not limiting this decay (Figure S8C). The formation of ADT 10 from the ADT tautomer 9 exhibits a primary deuterium kinetic isotope effect of 2.5 (Figure S9). However, this intrinsic isotope effect is not reflected in the overall rate of ADT formation, suggesting that slower earlier steps mask this isotope effect. In addition, this isotope effect was observed with R-glycine-2-d but not with S-glycine-2-d demonstrating that the pro-S hydrogen of glycine is removed in the conversion of 5 to 6 and the pro-R hydrogen is removed in the conversion of 9 to 10.
The crystal structure of the closely related Methanococcus igneus Thi4 shows the glycine imine 6 bound to the active site iron via the C3 ketone and the glycine imine nitrogen and carboxy groups (ribose numbering). In addition, the iron is also liganded to Asp161 and His176, and to a water molecule (Figure 4, manuscript in preparation). The formation of the 380 nm absorbing intermediate requires the bound metal ion (Figure S6B). The intensity and peak position of the absorbance changed when Fe(II) was replaced with Co(II) or with Fe(III), further supporting the dependence of the 380 nm peak on the active site metal (Figure S6B). The wavelength of the absorbance and its relatively high extinction coefficient (~5 mM−1cm−1) suggest that it is due to a charge-transfer transition between iron and the glycine imine intermediate 6. Other nonheme enzymes also exhibit similar charge transfer bands.25
Figure 4.
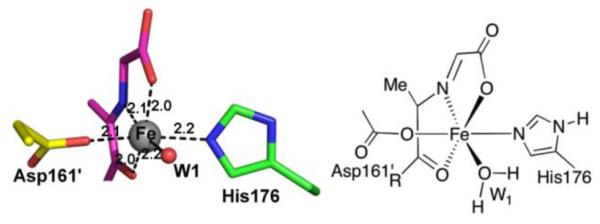
The iron-binding site of MjThi4. Active site residues His176 (green) and Asp161′ from the adjacent protomer (yellow), and the iron-bound glycine imine intermediate (11, purple) are shown as stick representations. The six metal bonds are shown as black dashes and labeled with bond lengths.
The structure of the iron-bound glycine imine 11 (Figure 4) suggests a mechanism for the iron mediated sulfide transfer steps involved in thiazole formation (Figure 5). In this mechanism, sulfide displaces the C3 carbonyl group of 11 to give 12. This allows the carbonyl group to rotate so that the iron-ligated sulfide can add to give 13. Loss of water gives thioketone 14. In complex 14, the sulfur cannot react with the glycine imine without major ligand rearrangement. We therefore propose that the iron dissociates from 14 to give 15, which then undergoes the necessary C-N bond rotation to allow formation of the second carbon sulfur bond (16 to 17), at which point Fe is lost. As the stereochemistry at C2 of 9 is known (R), the sulfur addition to the imine of 16 occurs from the re face.26 Confirmation of the thioketone will require further biophysical characterization.
Figure 5.
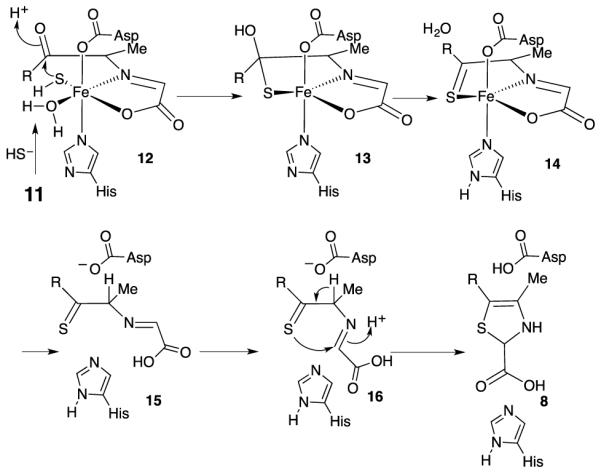
Mechanistic proposal for the role of iron in mediating the sulfur transfer chemistry required for thiamin thiazole formation.
Unlike its eukaryotic ortholog, MjThi4 uses free sulfide as the thiamin sulfur source and can catalyze multiple turnovers. Since the active sites of MjThi4 and ScTHI4 are quite similar, the C205A variant of ScTHI4p was assayed for ADT formation and found to be active (Figure S10) using sulfide as the sulfur source. This stands in contrast to the inactivity of the dehydroalanine form of ScTHI4 in the presence of sulfide and raises the question as to why the yeast enzyme is a suicidal enzyme when there appears to be no chemical necessity to use Cys 205 as a sulfur donor. One possibility is that the “inactive” form of ScTHI4 has additional functions in yeast that are not needed in archaea and that yeast thiamin thiazole biosynthesis represents a sophisticated post translational modification in which the posttranslational modification chemistry is coupled to the production of an essential metabolite.
Supplementary Material
ACKNOWLEDGMENT
We would like to thank Dr. Mrinmoy Chakrabarti for assistance with EPR experiments.
Funding Sources
This work was supported by NIH grants DK67081 and GM103403 (to SEE) and DK44083 (to TPB) and by the Robert A. Welch Foundation (A-0034 to TPB).
Footnotes
Supporting Information
Procedures for protein preparation and enzymatic assays, supplementary Figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interests.
REFERENCES
- (1).Begley TP, Ealick SE, McLafferty FW. Biochem Soc. Trans. 2012;40:555–60. doi: 10.1042/BST20120084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Krishnamoorthy K, Begley TP. J. Am. Chem. Soc. 2010;133:379–386. doi: 10.1021/ja107424t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Burns KE, Baumgart S, Dorrestein PC, Zhai H, McLafferty FW, Begley TP. J. Am. Chem. Soc. 2005;127:11602–11603. doi: 10.1021/ja053476x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jurgenson CT, Burns KE, Begley TP, Ealick SE. Biochemistry. 2008;47:10354–10364. doi: 10.1021/bi800915j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Godert AM, Jin M, McLafferty FW, Begley TP. J. Bacteriol. 2007;189:2941–2944. doi: 10.1128/JB.01200-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Leimkuehler S. Adv. Biol. 2014:1–22. [Google Scholar]
- (7).Sasaki E, Zhang X, Sun HG, Lu M-YJ, Liu T.-l., Ou A, Li J.-y., Chen Y.-h., Ealick SE, Liu H.-w. Nature. 2014;510:427. doi: 10.1038/nature13256. DOI: 10.1038/nature13256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Numata T, Fukai S, Ikeuchi Y, Suzuki T, Nureki O. Structure (Cambridge, MA, U. S.) 2006;14:357–366. doi: 10.1016/j.str.2005.11.009. [DOI] [PubMed] [Google Scholar]
- (9).Black KA, Dos Santos PC. J. Bacteriol. 2015;197:1952–1962. doi: 10.1128/JB.02625-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ikeuchi Y, Shigi N, Kato J.-i., Nishimura A, Suzuki T. Mol. Cell. 2006;21:97–108. doi: 10.1016/j.molcel.2005.11.001. [DOI] [PubMed] [Google Scholar]
- (11).Noma A, Sakaguchi Y, Suzuki T. Nucleic Acids Res. 2009;37:1335–1352. doi: 10.1093/nar/gkn1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chatterjee A, Abeydeera ND, Bale S, Pai P-J, Dorrestein PC, Russell DH, Ealick SE, Begley TP. Nature. 2011;478:542–546. doi: 10.1038/nature10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chatterjee A, Jurgenson CT, Schroeder FC, Ealick SE, Begley TP. J. Am. Chem. Soc. 2007;129:2914–2922. doi: 10.1021/ja067606t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jurgenson CT, Chatterjee A, Begley TP, Ealick SE. Biochemistry. 2006;45:11061–11070. doi: 10.1021/bi061025z. [DOI] [PubMed] [Google Scholar]
- (15).Liu Y, Beer LL, Whitman WB. Trends Microbiol. 2012;20:251–258. doi: 10.1016/j.tim.2012.02.002. [DOI] [PubMed] [Google Scholar]
- (16).Liu Y, Sieprawska-Lupa M, Whitman WB, White RH. J. Biol.Chem. 2010;285:31923–31929. doi: 10.1074/jbc.M110.152447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Liu Y, Zhu X, Nakamura A, Orlando R, Söll D, Whitman WB. J. Biol. Chem. 2012;287:36683–36692. doi: 10.1074/jbc.M112.405688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jones W, Leigh J, Mayer F, Woese C, Wolfe R. Arch. Microbiol. 1983;136:254–261. [Google Scholar]
- (19).Stookey LL. Anal. Chem. 1970;42:779–781. [Google Scholar]
- (20).Brown CA, Pavlosky MA, Westre TE, Zhang Y, Hedman B, Hodgson KO, Solomon EI. J. Am. Chem. Soc. 1995;117:715–732. [Google Scholar]
- (21).Zhang Y, Gan QF, Pavel EG, Sigal E, Solomon EI. J. Am. Chem. Soc. 1995;117:7422–7427. [Google Scholar]
- (22).Neidig ML, Wecksler AT, Schenk G, Holman TR, Solomon EI. J. Am. Chem. Soc. 2007;129:7531–7537. doi: 10.1021/ja068503d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hegg EL, Whiting AK, Saari RE, McCracken J, Hausinger RP, Que L. Biochemistry. 1999;38:16714–16726. doi: 10.1021/bi991796l. DOI: 10.1021/bi991796l. [DOI] [PubMed] [Google Scholar]
- (24).Han AY, Lee AQ, Abu-Omar MM. Inorg. Chem. 2006;45:4277–4283. doi: 10.1021/ic060478p. DOI: 10.1021/ic060478p. [DOI] [PubMed] [Google Scholar]
- (25).Que L, Jr., Ho RY. Chem Rev. 1996;96:2607–2624. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- (26).Hazra AB, Han Y, Chatterjee A, Zhang Y, Lai R-Y, Ealick SE, Begley TP. J. Am. Chem. Soc. 2011;133:9311–9319. doi: 10.1021/ja1110514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.