

Abstract
Reduced dietary sodium intake (sodium reduction) increases heart rate in some studies of animals and humans. As heart rate is independently associated with the development of heart failure and increased risk of premature death a potential increase in heart rate could be a harmful side-effect of sodium reduction. The purpose of the present meta-analysis was to investigate the effect of sodium reduction on heart rate. Relevant studies were retrieved from an updated pool of 176 randomized controlled trials (RCTs) published in the period 1973–2014. Sixty-three of the RCTs including 72 study populations reported data on heart rate. In a meta-analysis of these data sodium reduction increased heart rate with 1.65 beats per minute [95% CI: 1.19, 2.11], p < 0.00001, corresponding to 2.4% of the baseline heart rate. This effect was independent of baseline blood pressure. In conclusion sodium reduction increases heart rate by as much (2.4%) as it decreases blood pressure (2.5%). This side-effect, which may cause harmful health effects, contributes to the need for a revision of the present dietary guidelines.
Keywords: dietary sodium, heart rate, blood pressure, side-effect, meta-analysis
Introduction
About 0.5 g salt per day is sufficient to maintain vital physiological effects, but a daily habitual salt intake up to 40 g has been described (Battarbee, 1978). Sixty-five percent of the World's populations eat salt in the interval 8–10 g and 95% in the interval 6–12 g (McCarron et al., 2013). Consequently, world-wide the salt intake is very tightly regulated (Geerling and Loewy, 2008) in the low end of the tolerable interval of 0.5–40 g. Still, some health institutions consider this salt intake to be unhealthy due to an assumed association of sodium with blood pressure (BP) (WHO, 2012; US Department of Health and Human Services, 2015), and therefore recommend lowering sodium intake below 100 mmol (2300 mg, 5.8 g salt) (Box 1). However, meta-analyses question this assumption, as the effect of sodium reduction (SR) on BP is only 1.27/0.05 mmHg in individuals with a normal BP (Graudal et al., 2011) without indication of a dose-response relationship (Graudal et al., 2015). A moderate effect in individuals with hypertension (5.5/2.75 mmHg) (Graudal et al., 2011) does not justify SR for the whole population.
Box 1.
1000 mg of sodium (Na) corresponds to 2542 mg salt (NaCl) (43 mmol). 100 mmol NaCl corresponds to 2299 mg of sodium and 5844 mg of salt. In this article we use the term “sodium” and the unit “mmol.”
Compensating physiological mechanisms, like increases in renin and aldosterone (Brunner et al., 1972; Graudal et al., 1998) and noradrenalin and adrenalin (Graudal et al., 1998, 2011) during reduced sodium intake may contribute not only to maintain BP, but also to increase heart rate (HR). Animal experiments have shown that sodium reduction to one tenth of normal intake increase HR with 25% in rats (Ely et al., 1985). Folkow et al. concluded that “when it comes to the potential impact of the salt intake level which, when altered, in most subjects change BP and HR in opposite directions, these two parameters should be carefully and jointly analyzed” and “in studies of the effects of salt intake on blood pressure, influences on heart rate are usually neglected even though the long-term load on both left ventricle and systemic arteries is better related to the product of HR × BP than to pressure alone” (Folkow and Ely, 1998). This indication of the clinical importance of HR is further emphasized by prospective observational studies, which have shown that HR is independently associated with mortality (Jensen et al., 2012; Ho et al., 2014), just as BP (Lewington et al., 2002). Such an effect on HR could contribute to the finding of an association of low sodium intake with increased mortality in several population studies (Graudal et al., 2014; O'Donnell et al., 2014; Pfister et al., 2014).
The purpose of the present meta-analysis was to investigate the effect of SR on HR at different blood pressure levels. We also intended to identify longitudinal studies to define a time point for maximal efficacy of SR on HR and studies investigating different doses of sodium intake to establish a dose-response relationship between sodium intake and HR.
Methods
Trial search
Studies to be included were selected from an updated pool of trials identified in a previous review (Graudal et al., 2011), in which the details of the search procedure are described. Using this procedure the literature search was updated in April 2015.
Types of studies
Only randomized controlled trials (RCTs) were included.
Participants
Studies of healthy persons or persons with hypertension irrespective of age and race were included. Studies systematically investigating patients with diseases other than elevated BP (e.g., diabetes or heart failure), were excluded.
Intervention
The intervention is reduced sodium chloride intake (sodium reduction: SR). Studies, in which reduced sodium intake was controlled by means of 24-h urine sodium excretion or 8-h urine sodium excretion, were included. Studies not designed to control for the confounding effect of co-interventions were excluded, i.e., studies treating persons with a concomitant intervention such as an antihypertensive medication, potassium supplementation or weight reduction were only included if the concomitant intervention was identical in both the reduced and the usual sodium diet groups.
Primary outcome
Effect on HR calculated as the difference between the change during intervention in the low and usual sodium intake dietary groups. HR is measured as beats per minute (bpm) on the first and the last day of the intervention.
Supplementary analysis
The effect of SR on HR in subgroups defined by the 25, 50, and 75% BP-distribution percentiles of the American population (Wright et al., 2011) were investigated.
Secondary outcomes
The within study effect of SR on HR at different time points and at different doses of sodium reduction.
Data extraction
Two authors independently recorded the following data from each trial: HR (SD) before and after intervention for each measurement time and for each dose of sodium reduction; the sample size (N); the mean age of participants; SR measured as the difference between 24-h urinary sodium excretion during low-sodium and usual-sodium diets and standard deviation (SD); SBP (SD) and DBP (SD) before and after intervention. If there were discrepancies between reviewers they looked at the data together and came to an agreement.
Risk of bias (quality) assessment
This was performed using the Cochrane Risk of bias tool, including recording of allocation, blinding, incomplete outcome data, and selective reporting (Higgins and Green, 2011). Reporting bias was evaluated by an estimate of asymmetry in Funnel plots (Cochrane Collaboration, 2011; Higgins and Green, 2011).
Heterogeneity
A chi-squared test included in the forest plot was used to assess whether observed differences in results are compatible with chance alone. A low P value (or a large chi-squared statistic relative to its degree of freedom) provides evidence of heterogeneity of intervention effects (variation in effect estimates beyond chance) (Higgins and Green, 2011).
Data synthesis
The HR outcome was defined as the weighted mean difference (MD) between the changes from baseline to end of treatment during low and usual sodium diets. The combined effect measures were compared by means of the inverse variance method in Review Manager (Cochrane Collaboration, 2011). As we accumulated data from a series of studies that had been performed by researchers operating independently, and as the goal of the analysis was to extrapolate to other populations, we used a random effect model in our primary analysis to estimate the summary measure as the mean of a distribution of effects. In a secondary analysis we used a fixed-effect model assuming that the true effect size for all studies is identical. As we move from random effect to fixed effect, extreme studies will gain influence if they are large, and will lose influence if they are small. If there is no heterogeneity (tau2 = 0 and I2 = 0), the two models are identical.
Results
Trial identification
The last update of the 2011 search was performed April 15 2015. During the update the de-duplicated results from the searches revealed 728 articles. On the basis of headlines 645 were excluded. Eighty-three abstracts were read and 33 full articles obtained of which 12 fulfilled the inclusion criteria. During the process we identified 3 duplicates in the 2011 review (Graudal et al., 2011), which were eliminated. A total of 176 references (164 from the 2011 review plus 12 new references) were thus included in the present updated version. These 176 references included 198 study populations. Information on HR was available in 72 of these study populations, described in 63 references. Figure 1 shows a flow diagram of the trial identification.
Figure 1.
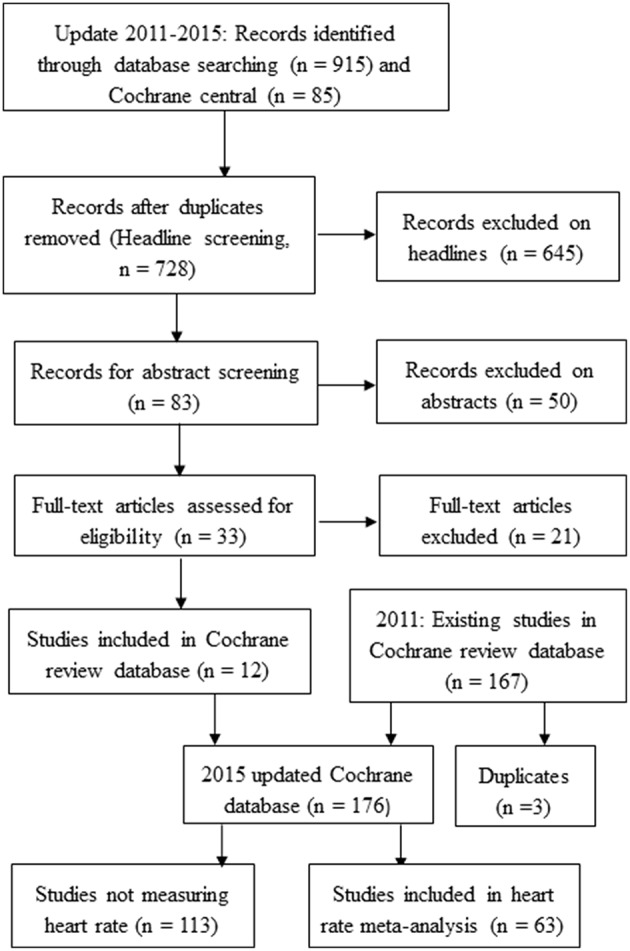
Flow diagram of the trial identification.
Trial characteristics
The 72 study populations included in the present meta-analysis are shown in Table 1.
Table 1.
Study characteristics and references of included studies.
| No. | Study ID | Dur. Days | N | Age (years) | Sodium reduction mmol | HR (beats/min) | References |
|---|---|---|---|---|---|---|---|
| 1 | Paulsen | 4 | 22 | 24 | 47 | 54.0 | Scand J Clin Lab Invest 2009;69:323–9 |
| 2 | Seals | 90 | 35 | 63.5 | 46 | 68.0 | J Am Coll Cardiol 2001;38:506–13 |
| 3 | Morgan | 14 | 16 | 63 | 50 | 77.0 | J Hum Hypertens 1988;1:311–5 |
| 4 | Grobee | 42 | 40 | 24 | 72 | 70.0 | BMJ 1987;293:27–9 |
| 5 | Dickinson | 42 | 25 | 35.1 | 42 | 68.0 | Atherosclerosis 2014;232:211–6 |
| 6 | Ishimitsu N | 7 | 7 | 53 | 195 | 64.0 | Clin Sci 1996;91:293–8 |
| 7 | Fotherby | 35 | 17 | 73 | 79 | 75.0 | J Hypertens 1993;11:657–63 |
| 8 | He B | 42 | 69 | 50 | 44 | 68.0 | Hypertension 2009;54:482–8 |
| 9 | ANHMRCDSSMC | 48 | 103 | 58.4 | 63 | 75.7 | Lancet 1989;1:399–402 |
| 10 | Doig | 4 | 8 | 25 | 117 | J Cardiovasc Pharm 1995;25:511–17 | |
| 11 | Sharma | 6 | 23 | 25 | 246 | 64.3 | J Hypertens 1991;9:329–35 |
| 12 | Gillies | 42 | 24 | 56.7 | 77 | 67.2 | Clin Exp Pharm Phys 1984;11:395–8 |
| 13 | Starmans-Kool | 14 | 10 | 32 | 97 | 56.0 | J Appl Physiol 2011;110:468–71 |
| 14 | Jessani | 7 | 184 | 49.5 | 81 | 82.0 | Am J Hypertens 2008;21:1238–1244 |
| 15 | Beeks | 7 | 117 | 53.4 | 99 | 63.5 | Hypertension 2004;44:419–23 |
| 16 | Wing | 42 | 17 | 61 | 59 | 80.0 | Blood Pressure 1998;7:299–307 |
| 17 | Schorr | 28 | 16 | 64.1 | 61 | 78.0 | J Hypertens 1996;14:131–5 |
| 18 | Fliser | 7 | 7 | 25.5 | 180 | 59.0 | Eur J Clin Invest 1995;25:39–43 |
| 19 | Bruun H | 4 | 12 | 47 | 331 | 73.0 | J Hypertens 1990;8:219–27 |
| 20 | Chiolero H | 7 | 38 | 43 | 183 | 76.0 | J Hypertens 2000;36:631–7 |
| 21 | Benetos | 28 | 20 | 41.5 | 78 | 75.3 | J Hypertens 1992;10:355–60 |
| 22 | Skrabal | 14 | 52 | 23 | 156 | 69.3 | Hypertension 1984;6:152–8 |
| 23 | Ferri | 14 | 61 | 47.1 | 264 | 73.2 | J Am Soc Nephrol 1996;7:443–53 |
| 24 | Sharma | 7 | 40 | 25 | 214 | 61.0 | Hypertension 1990;16:407–13 |
| 25 | Davrath | 5 | 8 | 25.1 | 96 | 63.5 | Aviat Space Env Med 1999;70:577–82 |
| 26 | Miller | 14 | 36 | 23.4 | 58 | 60.0 | Psychosom Med 1995;57:381–9 |
| 27 | Ishimitsu H | 7 | 23 | 55 | 193 | 59.7 | Clin Sci 1996;91:293–8 |
| 28 | Cooper | 24 | 113 | 16.3 | 64 | 81.0 | J Hypertens 1984;2:361–6 |
| 29 | Bonfils N | 5 | 12 | 39 | 140 | 64.0 | J Hypertens 2013;31:2220–9 |
| 30 | Graffe | 4 | 21 | 26 | 172 | 65.0 | Am J Physiol Renal Physiol 2012;15:F264–75 |
| 31 | Tzemos | 5 | 16 | 27 | 149 | 67.0 | Hypertension 2008;51:1525–35 |
| 32 | Dishy | 6 | 25 | 34 | 300 | 67.0 | J Hypertens 2003;21:153–7 |
| 33 | Zanchi | 7 | 9 | 25 | 250 | 66.0 | J Clin Endocrin Metab 2004;89:1140–5 |
| 34 | Burnier | 7 | 15 | 22.7 | 188 | 71.0 | J Hypertens 2000;18:1657–64 |
| 35 | Bech | 5 | 12 | 23.8 | 235 | 56.0 | Am J Physiol 1998;274:914–23 |
| 36 | Herlitz | 4 | 6 | 46 | 98 | 73.0 | Blood Press 1998;7:47–52 |
| 37 | He W | 42 | 71 | 52 | 55 | 65.0 | Hypertension 2009;54:482–8 |
| 38 | Hyperpath C2 | 7 | 211 | 49.2 | 211 | 63.5 | Hypertension 2012;60:1359–66 |
| 39 | Draaijer | 7 | 10 | 41 | 259 | 69.8 | J Hum Hypertens 1995;9:263–9 |
| 40 | Schorr | 7 | 187 | 25 | 206 | 56.6 | J Hypertens 1999;17:475–9 |
| 41 | Ambrosioni | 42 | 25 | 23 | 60 | 76.0 | Hypertension 1982;4:789–94 |
| 42 | He A | 42 | 29 | 47 | 68 | 67.0 | Hypertension 2009;54:482–8 |
| 43 | Chiolero N | 7 | 12 | 40 | 201 | 67.0 | J Hypertens 2000;36:631–7 |
| 44 | Sullivan N | 4 | 27 | 28.8 | 146 | 61.0 | Hypertension 1980;2:506–14 |
| 45 | Fuchs | 9 | 17 | 20 | 229 | 64.7 | Brazi J Med Biol Res 1987;20:25–34 |
| 46 | Sullivan H | 4 | 19 | 27 | 153 | 64.0 | Hypertension 1980;2:506–14 |
| 47 | Johnson | 14 | 40 | 68.8 | 73 | J Hypertens 2001;19:1053–60 | |
| 48 | Cuzzola | 14 | 19 | 47 | 161 | 71.0 | Am J Hypertens 2001;14:224–30 |
| 49 | Stein | 5 | 7 | 33.7 | 183 | 61.2 | Clin Pharm Ther 1995;58:425–33 |
| 50 | Parker | 28 | 59 | 52 | 73 | 74.0 | Hypertension 1990;16:398–406 |
| 51 | Uzu | 7 | 70 | 50.5 | 173 | 64.8 | Am J Hypertens 1999;12:35–9 |
| 52 | Mallamaci | 14 | 32 | 48 | 165 | 70.0 | J Hypertens 2013;31:1424–30 |
| 53 | Bonfils OW | 5 | 12 | 39 | 157 | 68.0 | J Hypertens 2013;31:2220–9 |
| 54 | Damgaard | 7 | 12 | 56.8 | 129 | 64.0 | Am J Physiol - Reg Integ Comp Physiol 2006;290:R1294-R1301 |
| 55 | Bruun N | 4 | 10 | 46 | 341 | 60.0 | J Hypertens 1990;8:219–27 |
| 56 | Mark | 10 | 6 | 28 | 305 | 63.3 | Circ Res 1975;(Suppl 1):36–7: I194–I198 |
| 57 | Overlack | 7 | 46 | 45.3 | 245 | 64.7 | Am J Hypertens 1995;8:829–36 |
| 58 | Ruppert | 7 | 163 | 38 | 274 | 63.2 | Hypertens 1993;11:743–9 |
| 59 | Fliser | 16 | 16 | 25 | 187 | 77.1 | J Hypertens 1993;6:320–4 |
| 60 | Sciarrone | 56 | 91 | 53.5 | 82 | J Hypertens 1992;10:287–98 | |
| 61 | Allen | 5 | 70 | 24 | 83 | 72.0 | J Hypertens 2014;32:374–82 |
| 62 | Mak | 7 | 13 | 24 | 190 | 56.0 | Eur Heart Journal Cardiovascular Imaging 2013;14:1092–8 |
| 63 | Inoue | 7 | 14 | 46 | 293 | 71.5 | J Hum Hypertens 1996;10:523–9 |
| 64 | Townsend | 6 | 20 | 30 | 171 | 67.0 | Clin Sci (London) 2007;113:141–8 |
| 65 | Boero | 14 | 13 | 51 | 209 | 76.0 | Min Urol Nefrol 2000;52:13–6 |
| 66 | Hargreaves | 14 | 8 | 23.4 | 106 | 59.0 | Clin Sci 1989;76:553–7 |
| 67 | Skrabal | 14 | 20 | 23 | 150 | 62.2 | Lancet 1981; II:895–900 |
| 68 | Carey C1 | 7 | 185 | 47 | 203 | 68.1 | Hypertension 2012;60:1359–66 |
| 69 | Bonfils H | 5 | 12 | 43 | 131 | 73.0 | J Hypertens 2013;31:2220–9 |
| 70 | Sudhir | 12 | 6 | 34.7 | 133 | 61.0 | Clin Sci 1989;77:605–10 |
| 71 | Melander | 28 | 39 | 53 | 89 | 67.7 | J Hypertens 2007;25:619–27 |
| 72 | Friberg | 13 | 10 | 33.3 | 117 | 63.0 | Hypertension 1990;16:121–30 |
Effect of SR on HR
Figure 2 shows that the effect of SR is highly significant, although clinically small: 1.65 bpm [95%CI: 1.19, 2.11], p < 0.00001, random effect, or 1.40 bpm [95% CI: 1.05, 1.75], fixed effect. As the mean baseline HR for the whole population was 67.8 bpm, this corresponds to an effect of 2.4% (1.65/67.8). Table 2 shows the effect of SR on HR in BP-distribution quartiles defined by quartiles of the American population (Wright et al., 2011). The effect of SR on HR was significant in all four quartiles indicating that the effect of SR on HR is independent of the baseline BP.
Figure 2.

Forrest plot showing effect of sodium reduction on heart rate in 72 study populations. The overall effect of sodium reduction is 1.65 b.p.m. The studies are ordered according to the effect size and correspond to the order in Table 1.
Table 2.
Mean heart rate effect of sodium reduction stratified by blood pressure quartiles of the American population.
| Systolic BP percentile, mmHg | Number of studies (participants) | Heart rate, MD (95%CI) (beats/min) | Z (p) |
|---|---|---|---|
| 0–25 %, −110 | 3 (304) | 0.91 [0.37, 1.45] | 3.28 (0.001) |
| 25–50 %, 110–119 | 18 (1204) | 2.18 [1.26, 3.11] | 4.62 (0.00001) |
| 50–75 %, 119–131 | 20 (1472) | 1.99 [0.72, 3.27] | 3.06 (0.002) |
| 75–100 %, 131 – | 31 (2452) | 1.41 [0.75, 2.07] | 4.21 (0.0001) |
The most contrasting risk of bias element was blinding (24 double-blind vs. 48 open studies). In 24 double-blind studies (n = 1386) the effect of SR was 1.34 bpm [0.54, 2.15], p < 0.001, random and fixed effect (I2 = 0%). In 48 open studies (n = 4040) the effect of SR was 1.83 bpm [1.24, 2.43], p < 0.00001, random effect, or 1.42 bpm [1.03, 1.81], p < 0.00001, fixed effect (I2 = 18%).
Longitudinal effect of SR on HR
Only 2 studies gave such results. In the study by Ruppert et al. (1993) the effect of SR on HR was 1.6 bpm at week 1 and 1.1 bpm at week 4. In the study by Fuchs et al. (1987) the outcome varied at 3, 6, and 9 days but without an obvious trend (Table 3).
Table 3.
Changes in mean heart rate in studies investigating different doses of sodium.
| References | Minor sodium reduction (b/m) | Moderate sodium reduction | Extreme sodium reduction | Study Duration, days |
|---|---|---|---|---|
| 45 Fuchs (1987) | NA | 4.6 | 3.3 | 3 |
| 45 Fuchs (1987) | NA | 1.5 | 1.5 | 6 |
| 45 Fuchs (1987) | NA | −0.9 | 4.5 | 9 |
| 19 Bruun (1990) (N) | NA | 1 | 2 | 4 |
| 19 Bruun (1990) (H) | NA | 0 | 1 | 4 |
| 34 Burnier (2000) | NA | 1 | 2 | 7 |
| 47 Johnson (2001) | −1 | 1.2 | 1.8 | 14 |
Dose-response relationship of SR vs. HR
Table 3 shows the outcome of seven dose-response analyses in four RCTs (Fuchs et al., 1987; Bruun et al., 1990; Burnier et al., 2000; Johnson et al., 2001). The trend is that HR increases with increasing SR but the number of investigations is few and the differences are small.
Discussion
This meta-analysis shows that SR increases HR. The increase is statistically highly significant (Z = 7.0, p < 0.00001), but the clinical significance of the size of the increase, which is 1.65 bpm corresponding to 2.4%, is debatable. The effect size matches quantitatively the effect of SR on SBP in the investigated RCTs, which is 3.4 mmHg (mean baseline SBP = 135 mmHg), corresponding to 2.5% (3.4/135). However, in contrast to the BP effect, which is moderate in hypertensives and low in normotensives, the HR effect is independent of baseline BP. This means that individuals with a BP in the lower 50% percentile of the BP distribution may get an increase in HR without a reduction in BP. Study blinding had no significant influence on the effect size. Two longitudinal studies were too few to determine the time of maximal efficacy (Fuchs et al., 1987; Ruppert et al., 1993). Seven dose-response relationships from 4 RCTs (Fuchs et al., 1987; Bruun et al., 1990; Burnier et al., 2000; Johnson et al., 2001) indicate that a dose-response relationship does exist, but the data are not sufficient for a reliable conclusion. Previously we have shown that ethnicity only has a minor impact on BP (Graudal and Jürgens, 2015), but we could not determine whether ethnicity has an impact on the effect of SR on HR, as only 2 Black and 4 Asian study populations were identified in the present meta-analysis.
A series of population studies have not been able to demonstrate a beneficial association between low sodium intake and health outcomes. A recent IOM report [Institute of Medicine (IOM), 2013] concluded “Science was insufficient and inadequate to establish whether reducing sodium intake below 2300 mg/d (100 mmol) either decreases or increases CVD risk in the general population.” A later meta-analysis of these population studies found that a sodium intake below 115 mmol was associated with increased mortality, as was a sodium intake above 215 mmol (Graudal et al., 2014). This U-shaped relation between sodium intake and mortality has been identified in several individual population studies (Thomas et al., 2011; O'Donnell et al., 2014; Pfister et al., 2014). The reason for this U-shape could be that SR has side effects as indicated in our previous meta-analysis, which shows that SR significantly increases plasma renin, plasma aldosterone, plasma adrenaline, plasma noradrenaline, plasma cholesterol, and plasma triglyceride (Graudal et al., 2011). In addition, the present finding of an increase in HR can be considered a side-effect, because HR has been associated with mortality (Jensen et al., 2012; Ho et al., 2014) and development of heart insufficiency (Pfister et al., 2012) in observational studies. In the Copenhagen City Study a 10 bpm increase in resting HR was associated with increased cardiovascular and all-cause mortality in both univariate (about 20%) and multivariate (about 10%) models (Jensen et al., 2012), and in the Framingham study an 11 bpm increase in resting HR was also associated with increased cardiovascular (18%) and all-cause (17%) mortality (Ho et al., 2014). A 20 mmHg increase in SBP leads to about 100% increase in mortality (Lewington et al., 2002). Assuming that these associations are linear, it can be estimated that an increase in HR of about 3 bpm outbalances the effect of a reduction in SBP of 1 mmHg. As mentioned the clinical significance of a 2.4% increase in HR may be questionable. However, the SR recommendations of the health institutions are based on the assumption that a small BP reduction in the whole population will lead to a significant reduction in mortality (WHO, 2012; US Department of Health and Human Services, 2015). Assuming that this concept is correct a 2.4% increase in HR should have the opposite effect especially when combined with increases in other factors know to be predictive of mortality, such as renin, noradrenalin and cholesterol. Indeed, observational population studies indicate that side effects of SR trump the BP effect (Thomas et al., 2011; Graudal et al., 2014; O'Donnell et al., 2014; Pfister et al., 2014). This seems to be the case, not only in healthy normotensive individuals, but also in heart failure patients. In a recent study of symptomatic patients with chronic heart failure sodium restriction was associated with significantly higher risk of death or HF-hospitalization (hazard rate = 1.85; 95% confidence interval: 1.21 to 2.84; p < 0.004) (Doukky et al., 2016). These findings indicate that health institutions in their Dietary Guidelines (US Department of Health and Human Services, 2015; DeSalvo et al., 2016) to a greater extent should recognize the lack of RCTs on the effect of SR on health outcomes, the possible side effects of SR and the association of low sodium intake with increased mortality in observational studies (Graudal, 2016). In the light of the ongoing debate, the process of preparing guidelines should include “full transparency, a lack of bias, and the inclusion and consideration of all of the latest available research and scientific evidence, even that which challenges current dietary recommendations” (Whoriskey, 2016). The present sodium recommendations do not fulfill these criteria. In conclusion SR increases HR. This should be considered a side-effect, which may have a harmful effect on the general population. This effect contributes to the need for a revision of the present dietary guidelines, which now has been initiated by the American Congress (Whoriskey, 2016).
Author contributions
NG: Substantial contributions to the conception, design of the work; and the acquisition, analysis, or interpretation of data for the work, Drafting the work, and Final approval of the version to be published. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. TH: Substantial contributions to the acquisition, analysis, and interpretation of data for the work, Revising it critically for important intellectual content, and Final approval of the version to be published. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. GJ: Substantial contributions to the acquisition, analysis, and interpretation of data for the work, Revising it critically for important intellectual content, and Final approval of the version to be published. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Battarbee H. D. (1978). The toxicity of salt. CRC Crit. Rev. Toxicol. 5, 355–376. 10.3109/10408447809081011 [DOI] [PubMed] [Google Scholar]
- Brunner H. R., Laragh J. H., Baer L., Newton M. A., Goodwin F. T., Krakoff L. R., et al. (1972). Essential hypertension: renin and aldosterone, heart attack and stroke. N. Engl. J. Med. 286, 441–449. 10.1056/NEJM197203022860901 [DOI] [PubMed] [Google Scholar]
- Bruun N. E., Skøtt P., Nielsen M. D., Rasmussen S., Schütten H. J., Leth A., et al. (1990). Normal renal tubular response to changes of sodium intake in hypertensive man. J. Hypertens 8, 219–227. [PubMed] [Google Scholar]
- Burnier M., Monod M., Chiolero A., Maillard M., Nussberger J., Brunner H. R. (2000). Renal sodium handling in acute and chronic salt loading/depletion protocols: the confounding influence of acute water loading. J. Hypertens 18, 1657–1664. 10.1097/00004872-200018110-00018 [DOI] [PubMed] [Google Scholar]
- Cochrane Collaboration (2011). Review Manager (RevMan) (Computer program) Version 5.1. Copenhagen: The Nordic Cochrane Centre. [Google Scholar]
- DeSalvo K. B., Olson R., Casavale K. O. (2016). Dietary guidelines for Americans. JAMA 315, 457–458. 10.1001/jama.2015.18396 [DOI] [PubMed] [Google Scholar]
- Doukky R., Avery E., Mangla A., Collado F. M., Ibrahim Z., Poulin M. F., et al. (2016). Impact of dietary sodium restriction on heart failure outcomes. JACC Heart Fail. 4, 24–35. 10.1016/jfchf.2015.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ely D. L., Friberg P., Nilsson H., Folkow B. (1985). Blood pressure and heart rate responses to mental stress in spontaneously hypertensive (SHB) and normotensive (WKY) rats on various sodium diets. Acta Physiol. Scand. 123, 159–169. 10.1111/j.1748-1716.1985.tb07573.x [DOI] [PubMed] [Google Scholar]
- Folkow B., Ely D. (1998). Importance of the blood pressure-heart rate relationship. Blood Press. 7, 133–138. 10.1080/080370598437321 [DOI] [PubMed] [Google Scholar]
- Fuchs F. D., Wannmacher C. M., Wannmacher L., Guimarães F. S., Rosito G. A., Gastaldo G., et al. (1987). Effect of sodium intake on blood pressure, serum levels and renal excretion of sodium and potassium in normotensives with and without familial predisposition to hypertension. Braz. J. Med. Biol. Res. 20, 25–34. [PubMed] [Google Scholar]
- Geerling J. C., Loewy A. D. (2008). Central regulation of sodium appetite. Exp. Physiol. 93, 177–209. 10.1113/expphysiol.2007.039891 [DOI] [PubMed] [Google Scholar]
- Graudal N. (2016). A radical sodium reduction policy is not supported by randomized controlled trials or observational studies: grading the evidence. Am. J. Hypertens. [Epub ahead of print]. 10.1093/ajh/hpw006 [DOI] [PubMed] [Google Scholar]
- Graudal N. A., Galløe A. M., Garred P. (1998). Effects of sodium restriction on blood pressure, renin, aldosterone, catecholamines, cholesterols, and triglyceride. JAMA 279, 1383–1391. [DOI] [PubMed] [Google Scholar]
- Graudal N. A., Hubeck-Graudal T., Jurgens G. (2011). Effects of low sodium diet versus high sodium diet on blood pressure, renin, aldosterone, catecholamines, cholesterol, and triglyceride. Cochrane Database Syst. Rev. 11:CD004022. 10.1002/14651858.cd004022.pub3 [DOI] [PubMed] [Google Scholar]
- Graudal N. A., Hubeck-Graudal T., Jurgens G., McCarron D. A. (2015). The significance of duration and dose of sodium reduction intervention in normotensive and hypertensive individuals. A meta-analysis. Adv. Nutr. 6, 169–177. 10.3945/an.114.007708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graudal N., Jürgens G. (2015). The blood pressure sensitivity to changes in sodium intake is similar in Asians, Blacks and Whites. An analysis of 92 randomized controlled trials. Front. Physiol. 6:157. 10.3389/fphys.2015.00157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graudal N., Jürgens G., Baslund B., Alderman M. H. (2014). Compared with usual sodium intake, low- and excessive-sodium diets are associated with increased mortality: a meta-analysis. Am. J. Hypertens 27, 1129–1137. 10.1093/ajh/hpu028 [DOI] [PubMed] [Google Scholar]
- Higgins J. P. T., Green S. (eds.). (2011). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration. Available online at: www.cochrane-handbook.org
- Ho J. E., Larson M. G., Ghorbani A., Cheng S., Coglianese E. E., Vasan R. S., et al. (2014). Long-term cardiovascular risks associated with an elevated heart rate: the Framingham Heart Study. J. Am. Heart Assoc. 3:e000668. 10.1161/JAHA.113.000668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Institute of Medicine (IOM) (2013). Sodium Intake in Populations: Assessment of Evidence. Washington, DC: National Academies Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M. T., Marott J. L., Allin K. H., Nordestgaard B. G., Jensen G. B. (2012). Resting heart rate is associated with cardiovascular and all-cause mortality after adjusting for inflammatory markers: the Copenhagen City Heart Study. Eur. J. Prev. Cardiol. 19, 102–108. 10.1177/1741826710394274 [DOI] [PubMed] [Google Scholar]
- Johnson A. G., Nguyen T. V., Davis D. (2001). Blood pressure is linked to salt intake and modulated by the angiotensinogen gene in normotensive and hypertensive elderly subjects. J. Hypertens. 19, 1053–1060. 10.1097/00004872-200106000-00009 [DOI] [PubMed] [Google Scholar]
- Lewington S., Clarke R., Qizilbash N., Peto R., Collins R. (2002). Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 360, 1903–1913. 10.1016/S0140-6736(02)11911-8 [DOI] [PubMed] [Google Scholar]
- McCarron D. A., Kazaks A. G., Geerling J. C., Stern J. S., Graudal N. A. (2013). Normal range of human dietary sodium intake: a perspective based on 24-hour urinary sodium excretion worldwide. Am. J. Hypertens 26, 1218–1223. 10.1093/ajh/hpt139 [DOI] [PubMed] [Google Scholar]
- O'Donnell M., Mente A., Rangarajan S., McQueen M. J., Wang X., Liu L., et al. (2014). PURE Investigators. Urinary sodium and potassium excretion, mortality, and cardiovascular events. N. Engl. J. Med. 371, 612–623. 10.1056/NEJMoa1311889 [DOI] [PubMed] [Google Scholar]
- Pfister R., Michels G., Sharp S. J., Luben R., Wareham N. J., Khaw K. T. (2012). Resting heart rate and incident heart failure in apparently healthy men and women in the EPIC-Norfolk study. Eur. J. Heart Fail. 14, 1163–1167. 10.1093/eurjhf/hfs104 [DOI] [PubMed] [Google Scholar]
- Pfister R., Michels G., Sharp S. J., Luben R., Wareham N. J., Khaw K. T. (2014). Estimated urinary sodium excretion and risk of heart failure in men and women in the EPIC-Norfolk study. Eur. J. Heart Fail. 16, 394–402. 10.1002/ejhf.56 [DOI] [PubMed] [Google Scholar]
- Ruppert M., Overlack A., Kolloch R., Kraft K., Göbel B., Stumpe K. O. (1993). Neurohormonal and metabolic effects of severe and moderate salt restriction in non-obese normotensive adults. Hypertens 11, 743–749. 10.1097/00004872-199307000-00010 [DOI] [PubMed] [Google Scholar]
- Thomas M. C., Moran J., Forsblom C., Harjutsalo V., Thorn L., Ahola A., et al. (2011). FinnDiane Study Group. The association between dietary sodium intake, ESRD, and all-cause mortality in patients with type 1 diabetes. Diabetes Care 34, 861–866. 10.2337/dc10-1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- US Department of Health Human Services (2015). US Department of Agriculture. 2015-2020 Dietary Guidelines for Americans, 8th Edn. Washington, DC: US Dept of Health and Human Services; Available online at: http://www.health.gov/DietaryGuidelines (Accessed December, 2015). [Google Scholar]
- WHO (2012). Sodium Intake for Adults and Children. Geneva: WHO. [Google Scholar]
- Whoriskey P. (2016). Congress: We Need to Review the Dietary Guidelines for Americans. Washington. Available online at: http://wpo.st/iQCy0
- Wright J. D., Hughes J. P., Ostchega Y., Yoon S. S., Nwankwo T. (2011). Mean systolic and diastolic blood pressure in adults aged 18 and over in the United States, 2001–2008. Natl. Health Stat. Rep. 35, 1–24. [PubMed] [Google Scholar]