

Abstract
Purpose
Prostate Cancer (PCa) is one of the most common cancers in men and its early detection can provide a high chance of cure. The detection of Vitamin D Receptor (VDR) gene polymorphisms may be useful as a molecular indicator of clinical outcome, once VDR is implicated in a wide variety of biological processes including modulation of the immune response and inhibition of cancer cell growth, angiogenesis and metastasis. In this study we explored the Single Nucleotide Polymorphisms (SNPs) FokI, BsmI, ApaI and TaqI, to evaluate the susceptibility locus for PCa and verify its correlation with clinical parameters.
Methods
VDR polymorphisms were detected by PCR followed by Restriction Fragment Length Polymorphism (PCR–RFLP). DNA samples were extracted from peripheral blood of 342 patients: 132 PCa, 41 Benign Prostatic Hyperplasia and 169 young healthy volunteers.
Results
Statistical analysis showed a noteworthy correlation among SNPs and clinical pathological features. CC genotype (TaqI) was correlated with the age at diagnosis (>58 years old), and GG (BsmI) was associated to lower Prostate-Specific Antigen (PSA) levels (<10 ng/mL). Moreover, when PCa patients were subgrouped, G allele (BsmI) significantly increased the estimated chance for PSA < 10 ng/mL, and GG/GG genotype (BsmI/ApaI) provided a 9.75 fold increased chance of patients with PCa to present lower PSA levels.
Conclusions
The polymorphisms of VDR gene showed a genotype-phenotype association and presented new correlations with different parameters as age and PSA levels.
Electronic supplementary material
The online version of this article (doi:10.1186/s40064-016-2009-8) contains supplementary material, which is available to authorized users.
Keywords: Vitamin D receptor, Polymorphisms, Prostate cancer, Prostate-specific antigen
Background
Prostate cancer (PCa) is the second most common type of cancer in American men. In PCa, structural genomic rearrangements resulting in mutation is associated with the activation of oncogenes leading to tumors development (Prostate Cancer 2015; Wyatt et al. 2013). Early diagnosis and consequently early treatment have resulted in decreased rate of mortality among PCa patients. One of the most common tests used to diagnose PCa was introduced in 1986, when FDA approved the Prostate-Specific Antigen (PSA) for evaluation of the disease progression. In 1994, FDA defined the PSA concentration of 4.0 ng/mlas the upper limit of normal prostate tissue (Barve et al. 2014). However, although it is clinically accepted, the low specificity makes its detection controversial (Cuzick et al. 2014). Therefore, it is highly necessary the development of specific methods that allow early diagnosis of the disease, contributing to the reduction of mortality and providing information on the prognosis before a certain treatment.
Prostate cell division is influenced by two steroid hormones: testosterone and vitamin D. The action of these hormones is mediated by their respective receptors: androgen receptor (AR) and Vitamin D Receptor (VDR) (Jingwi et al. 2015). Prostate epithelial cells express multiple members of nuclear receptor superfamily that regulate proliferation and differentiation of cells in the prostate gland. Their action is disturbed in PCa, presenting molecular alterations and mutations related to the diagnosis of the disease and response to therapy (Gommersall et al. 2004).
VDR is a member of the superfamily of nuclear hormone receptors that regulate gene transcription. The idea that the VDR gene may influence the occurrence of PCa and other diseases is mainly based on the notion that vitamin D is implicated in a wide variety of biological processes including modulation of the immune response and inhibition of cancer cell growth, angiogenesis and metastasis (Li et al. 2015).
The most frequently studied single nucleotide polymorphisms (SNP) in VDR are rs1544410, rs731236, rs2228570 and rs7975232, sites for the BsmI, TaqI, FokI and ApaI restriction enzymes, respectively (Gandini et al. 2014). Such polymorphisms are found in exon 2 (FokI), intron 8 (ApaI and BsmI) and exon 9 (TaqI). The FokI polymorphism occurs due to a different initiation site associated with a frameshift in the VDR protein (Xu et al. 2014; Mehta et al. 2013). The BsmI and ApaI polymorphisms are located into a noncoding region and thus do not affect the quantity, the structure or function of the VDR protein generated. Finally, the TaqI is a silent polymorphism caused by the substitution of a cytosine for thymine. As these variants are situated close to the 3ʹ region they can influence the stability of the messenger RNA, altering protein expression (Yang et al. 2014).
Genetic studies have provided excellent opportunities to link molecular insights to epidemiological data. The discovery of genetic variants linked to susceptibility of diseases, mainly the wide variety of tumors, may be the key to improve advances in preventive medicine. Polymorphisms in the VDR gene may be useful to detect individuals with higher risk of disease development, assisting in the early detection and therapy. Furthermore, analysis of haplotypes may be useful to identify groups of linked SNPs, simplifying association analysis and facilitating the understanding of these risk alleles. The aim of this study was to investigate the relationship between the polymorphisms FokI (g.27823C>T), BsmI (g.60890G>A), ApaI (g.61888G>T) and TaqI (g.61968T>C); alleles namely according to their genomic position (NCBI: #AY342401), and the susceptibility to prostate cancer development as well as their association with clinical parameters.
Methods
Study design and sample collection
This work was developed in the Laboratory of Nanobiotechnology of the Federal University of Uberlândia (UFU), approved by the UFU Research Ethics Committee under the approval number 005/2001, together with the Urology Service of the Clinical Hospital of UFU. Peripheral blood samples from Benign Prostatic Hyperplasia (BPH) and PCa were collected before surgery in a vacutainer™ tube containing K2 EDTA 7.2 mg, and maintained at 4 °C.
Peripheral blood samples from 342 patients were grouped into three classes: 132 PCa patients, 41 benign prostatic hyperplasia samples and 169 healthy volunteers. Patients were selected by using the following criteria: negative X-rays and bone scan analyses, and rectal examination compatible with organ-confined (i.e. limited to the prostate gland) cancer. Moreover, it was selected BPH patients who were submitted to Transurethral Resection of Prostate (TURP) and PCa patients who were submitted to radical prostatectomy.
PSA levels were obtained through the IMMULITE 1000 System for quantitative detection (Siemens Healthcare Diagnostics Inc.), considering normal values between 0 and 4.0 ng/mL. DNA was extracted from leukocytes according to protocol previously published elsewhere (Sambrook et al. 1989) and the concentration and quality were obtained spectrophotometrically by the absorbance readings at 260 and 280 nm.
Single nucleotide polymorphisms screening
The SNPs with restriction site for FokI, BsmI, ApaI e TaqI enzymes presented in VDR gene were detected by PCR followed by restriction fragment length polymorphism (PCR–RFLP) method (Fig. 1). To identify the FokI mutation, we amplified the 265-bp PCR fragment (Fig. 1a) and performed an endonuclease FokI digestion. In the absence of the mutation, cleaved fragments of 196 and 69-bp were detected (Fig. 1b). For the BsmI mutation, the 825-bp amplified fragment (Fig. 1c) was digested by BsmI, and in the absence of the mutation, cleaved fragments of 650 and 175-bp were detected (Fig. 1d). To detect the ApaI mutation, the 352-bp (Fig. 1e) amplified fragment was digested by ApaI. If the fragment had this mutation, cleaved fragments of 214 and 138-bp could be obtained (Fig. 1f). For the TaqI mutation, the 352-bp amplified fragment was digested by TaqI, and cleaved fragments of 293 and 59-bp could be obtained if the fragment had this mutation (Fig. 1g). Each fragment was detected in a 2.5 % agarose gel stained with ethidium bromide. Primers designed for fragments amplification are described in Table 1.
Fig. 1.
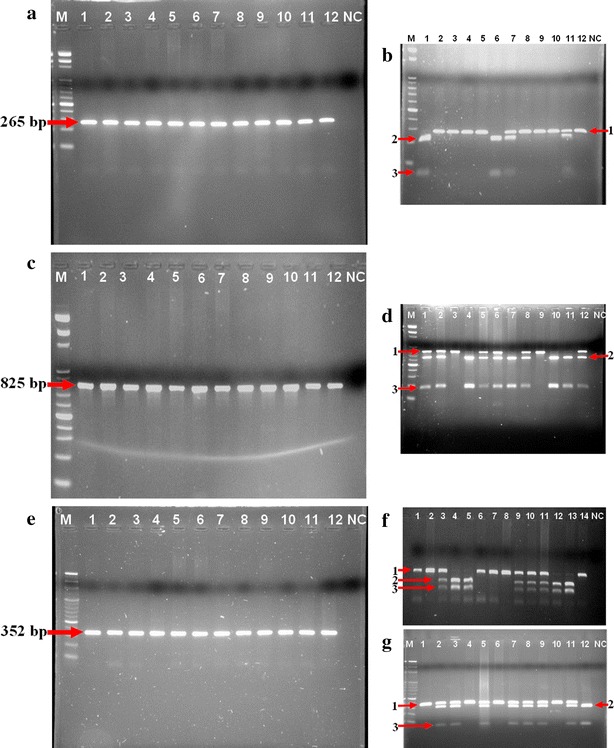
Amplification of VDR gene and restriction endonuclease digestion pattern for FokI, BsmI, ApaI and TaqI polymorphims. The 265 bp fragment (a) was digested by FokI enzyme (b) generating two fragments, 196 (2) and 69 bp (3) for T allele. C allele, containing 265 bp (1), represents the absence of restriction site. The 825 bp fragment (c) was digested by BsmI enzyme (d) presenting two fragments, 650 (2) and 175 bp (3) for G allele, and 825 bp (1) for A allele (no restriction site). The amplicon of 325 bp (e) was used for detection of ApaI and TaqI polymorphims. For ApaI, the 352 bp fragment (1) represents T allele, and the 214 bp (2) and 18 bp (3) fragments indicate the presence of G allele (f). Finally, after digestion with TaqI enzyme (g) it was verified two fragments, 293 (2) and 59 bp (3) for C allele and a unique fragment of 352 bp (1) for T allele. M: 100 bp DNA ladder. Lanes 1–4: represent PCa patients; Lanes 5–8: represent BPH patients; Lanes 9–12: represent healthy volunteers. NC negative control
Table 1.
Oligonucleotide sequences for DNA amplification
| SNPs | Primer sequence 5′–3′ (forward/reverse) | Position in database sequence | Amplicon (pb) |
|---|---|---|---|
| rs731236 (TaqI) | CAGAGCATGGACAGGGAGCAAG | exon 9 | 352 |
| GGTGGCGGCAGCGGATGTACGT | |||
| rs7975232 (ApaI) | CAGAGCATGGACAGGGAGCAAG | intron 8 | 352 |
| GGTGGCGGCAGCGGATGTACGT | |||
| rs15444410 (BsmI) | CAACCAAGACTCAAGTACCGCGTCAGTGA | intron 8 | 823 |
| AACCAGCGGAAGAGGTCAAGGG | |||
| rs2228570 (FokI) | AGCTGGCCCTGGCACTGACTCTGCTCT | exon 2 | 265 |
| ATGGAAACACCTTGCTTCTTCTCCCTC |
SNP single nucleotide polymorphism
Statistical analyses
Allele frequency analysis and tests of deviation from Hardy–Weinberg equilibrium were carried out using the GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA). Chi square analyses were performed to compare genotypic and allelic frequencies for the average of clinical parameters, such as: age, PSA serum levels, TNM (Tumor-Node-Metastasis), adenocarcinoma histopathological staging and Gleason score. The odds ratio (OR) was determined to verify the risk of prostate cancer development. P values <0.05 were considered statistically significant.
Associations between SNPs and clinical data were performed with the contingency coefficient C by using BioEstat 5.3 software.
Results
Table 2 shows VDR polymorphisms with their respective genotypes, and allelic frequencies in PCa group (n = 132), BPH (n = 41) and population control (n = 169). All genotypic frequencies were in Hardy–Weinberg equilibrium, but no differences in genotypes and alleles were observed between the three groups. Parameters as age and PSA levels were not different comparing PCa and BPH groups. We observed strong linkage disequilibrium between TaqI, BsmI and ApaI polymorphisms located in exon 8 and exon 9 of VDR gene (Table 3).
Table 2.
Genotypic and allelic frequencies for VDR gene polymorphisms and clinical parameters of patients with prostate cancer, benign prostatic hyperplasia and healthy volunteers
| SNP | PCa (N = 132) | BPH (N = 41) | Healthy volunteers (N = 169) | |||||
|---|---|---|---|---|---|---|---|---|
| N (%) | Age* | PSA* | N (%) | Age* | PSA* | N (%) | Age* | |
| FokI | ||||||||
| CC | 54.00 (40.91) | 65.08 (8.79) | 14.08 (18.35) | 18.00 (43.90) | 66.25 (10.67) | 18.30 (46.56) | 79.00 (46.75) | 23.00 (4.57) |
| CT | 62.00 (46.97) | 67.51 (8.41) | 19.21 (32.75) | 19.00 (46.34) | 70.00 (10.59) | 12.75 (16.73) | 76.00 (44.97) | 22.98 (4.31) |
| TT | 16.00 (12.12) | 65.53 (11.29) | 50.68 (123.35) | 4.00 (9.76) | 72.33 (5.77) | 8.27 (2.22) | 14.00 (8.38) | 22.86 (6.18) |
| Alleles C/T | 170/94 (64.39/35.61) | 55/27 (67.07/32.93) | 234/104 (69.23/30.77) | |||||
| §PHWE | 0.78 | 0.75 | 0.47 | |||||
| Pχ2 | PCa x BPH = 0.90 | PCa x Healthy = 0.42 | BPH x Healthy = 0.93 | |||||
| BsmI | ||||||||
| AA | 14.00 (10.61) | 64.31 (9.66) | 30.11 (57.48) | 5.00 (12.20) | 71.00 (8.21) | 3.53 (2.40) | 28.00 (16.57) | 23.37 (4.50) |
| AG | 63.00 (47.73) | 67.69 (8.24) | 17.09 (22.57) | 25.00 (60.98) | 66.80 (11.25) | 10.41 (14.68) | 70.00 (41.42) | 22.93 (4.78) |
| GG | 55.00 (41.67) | 65.31 (9.44) | 21.49 (61.54) | 11.00 (26.86) | 71.45 (8.71) | 26.14 (55.23) | 71.00 (42.01) | 22.91 (4.15) |
| Alleles A/G | 91/173 (34.47/65.53) | 35/47 (42.68/57.32) | 126/212 (37.28/62.72) | |||||
| §PHWE | 0.52 | 0.11 | 0.14 | |||||
| Pχ2 | PCa x BPH = 0.23 | PCa x Healthy = 0.28 | BPH x Healthy = 0.08 | |||||
| ApaI | ||||||||
| TT | 49.00 (37.12) | 65.74 (9.22) | 19.98 (36.07) | 18.00 (43.90) | 66.56 (10.78) | 9.72 (17.38) | 51.00 (30.18) | 22.36 (4.94) |
| TG | 59.00 (44.70) | 65.44 (9.29) | 25.48 (63.84) | 16.00 (39.02) | 69.57 (10.50) | 8.45 (6.10) | 89.00 (52.66) | 22.91 (4.33) |
| GG | 24.00 (18.18) | 69.30 (7.18) | 10.66 (12.26) | 7.00 (17.07) | 71.14 (9.23) | 37.11 (68.48) | 29.00 (17.16) | 24.77 (3.94) |
| Alleles T/G | 157/107 (59.47/40.53) | 52/30 (63.41/36.59) | 191/147 (56.51/43,49) | |||||
| §PHWE | 0.40 | 0.31 | 0.35 | |||||
| Pχ2 | PCa x BPH = 0.73 | PCa x Healthy = 0.35 | BPH x Healthy = 0.21 | |||||
| TaqI | ||||||||
| TT | 60.00 (45.45) | 66.09 (8.94) | 20.77 (60.47) | 13.00 (31.71) | 70.75 (8.66) | 23.79 (53.24) | 71.00 (42.01) | 23.33 (4.01) |
| TC | 62.00 (46.97) | 66.93 (8.92) | 17.59 (21.26) | 23.00 (56.10) | 66.38 (11.44) | 10.69 (15.05) | 75.00 (44.38) | 22.90 (5.00) |
| CC | 10.00 (7.58) | 63.25 (9.78) | 37.62 (72.71) | 5.00 (12.20) | 73.50 (6.24) | 6.03 (2.65) | 23.00 (13.61) | 22.40 (4.47) |
| Alleles T/C | 182/82 (68.94/31.06) | 49/33 (59.76/40.24) | 217/121 (64.20/35.80) | |||||
| §PHWE | 0.27 | 0.29 | 0.65 | |||||
| Pχ2 | PCa x BPH = 0.26 | PCa x Healthy = 0.25 | BPH x Healthy = 0.39 | |||||
Pχ2: P value of Chi square test
SNP single nucleotide polymorphism
* Mean (±SD)
§PHWE: Hardy–Weinberg equilibrium
Table 3.
Contingency table for analysis of linkage disequilibrium between ApaI, BsmI and TaqI polymorphisms
| TaqI versus BsmIa | AA | AG | GG |
|---|---|---|---|
| TT | 1 (0.29) | 19 (5.56) | 124 (36.26) |
| TC | 11 (3.22) | 137 (40.06) | 12 (3.51) |
| CC | 35 (10.23) | 2 (0.58) | 1 (0.29) |
| ApaI versus TaqIb | TT | TC | CC |
|---|---|---|---|
| TT | 21 (6.14) | 60 (17.54) | 37 (10.82) |
| GT | 65 (19.01) | 99 (28.95) | 0 |
| GG | 58 (16.96) | 1 (0.29) | 1 (0.29) |
| BsmI versus ApaIc | TT | GT | GG |
|---|---|---|---|
| AA | 46 (13.45) | 1 (0.29) | 0 |
| AG | 55 (16.08) | 102 (29.82) | 1 (0.29) |
| GG | 17 (4.97) | 61 (17.84) | 59 (17.25) |
a TaqI versus BsmI: χ2 = 426.57, P < 0.0001*
b ApaI versus TaqI: χ2 = 158.77, P < 0.0001*
c BsmI versus ApaI: χ2 = 188.96, P < 0.0001*
* Significant data
Combined genotypes analysis demonstrated a statistically significant correlation between genotypes and the parameters of diagnosis. The estimated OR for BPH occurrence compared to healthy volunteers for AG/TT genotype (BsmI/ApaI) was 3.90 (95 % CI 1.19–12.84, P = 0.04). The OR for AG/TT (BsmI/TaqI) comparing PCa group and healthy volunteers was 3.30 (95 % CI 1.09–10.00, P = 0.05). GG/TT (BsmI/ApaI) genotypes were responsible for providing higher chances to healthy volunteers to develop PCa (OR 3.54, 95 % CI 1.08–11.57, P = 0.007).
Although not significant, a 2.37-fold chance for the occurrence of hyperplasia was observed in heterozygous individuals AG/TC (BsmI/TaqI) compared to healthy volunteers (95 % CI 1.05–5.38, P = 0.06) (Table 4). Genotype-level associations between diagnosis, clinical parameters and the four vitamin D variants considering Coefficient C are displayed in Table 5. TT genotype (TaqI) was more frequent in patients with lower PSA levels (30.21 %; P = 0.09), and GG genotypes (ApaI) seems to be associated to older men in the BPH group (P = 0.07).
Table 4.
Combined genotypic frequency for ApaI, TaqI and BsmI polymorphisms in VDR gene
| Genotypes | PCa X BPH | PCa X healthy volunteers | BPH X healthy volunteers | |||
|---|---|---|---|---|---|---|
| OR (95 % CI) | P | OR (95 % CI) | P | OR (95 % CI) | P | |
| Bsm I - Apa I | ||||||
| AATT | 1.00 | Reference | 1.00 | Reference | 1.00 | Reference |
| AGTT | 0.66 (0.19–2.24) | 0.71 | 2.57 (1.06–6.26) | 0.06 | 3.90 (1.19–12.84) | 0.04* |
| GGGT | 1.88 (0.43–8.22) | 0.64 | 1.13 (0.49–2.61) | 0.95 | 0.60 (0.15–2.45) | 0.72 |
| AGGT | 1.13 (0.34–3.79) | 0.91 | 1.41 (0.65–3.04) | 0.49 | 1.25 (0.40–3.91) | 0.93 |
| GGGG | 1.17 (0.31–4.42) | 0.92 | 1.53 (0.66–3.57) | 0.44 | 1.30 (0.37–4.60) | 0.93 |
| GGTT | ND | ND | 3.54 (1.08–11.57) | 0.07 | ND | ND |
| Apa I - Taq I | ||||||
| TTTT | 1.00 | Reference | 1.00 | Reference | 1.00 | Reference |
| TTTC | 0.69 (0.16–2.97) | 0.89 | 0.85 (0.28–2.56) | 0.99 | 1.22 (0.26–5.68) | 0.90 |
| GTTT | 1.71 (0.33–8.93) | 0.85 | 0.44 (0.15–1.30) | 0.22 | 0.26 (0.05–1.42) | 0.26 |
| GTTC | 0.77 (0.18–3.25) | 1.00 | 0.41 (0.14–1.16) | 0.15 | 0.53 (0.12–2.35) | 0.67 |
| TTCC | 0.68 (0.12–3.83) | 1.00 | 0.28 (0.08–0.92) | 0.07 | 0.41 (0.07–2.27) | 0.57 |
| GGTT | 1.09 (0.23–5.19) | 0.77 | 0.55 (0.18–1.63) | 0.41 | 0.50 (0.10–2.51) | 0.69 |
| Bsm I - Taq I | ||||||
| GGTT | 1.00 | Reference | 1.00 | Reference | 1.00 | Reference |
| AGTC | 0.45 (0.20–1.06) | 0.10 | 1.07 (0.64–1.82) | 0.89 | 2.37 (1.05–5.38) | 0.06 |
| AATC | ND | ND | 1.15 (0.33–3.97) | 0.92 | ND | ND |
| AACC | 0.47 (0.12–1.83) | 0.47 | 0.56 (0.24–1.33) | 0.27 | 1.20 (0.34–4.21) | 0.96 |
| AGTT | 1.25 (0.24–6.48) | 0.89 | 3.30 (1.09–10.00) | 0.05* | 2.64 (0.45–15.49) | 0.58 |
| GGTC | ND | ND | 1.93 (0.58–6.53) | 0.44 | ND | ND |
| AGCC | ND | ND | 1.38 (0.08–22.54) | 0.62 | ND | ND |
| HAPLOTYPES | ||||||
| Bsm I - Apa I | ||||||
| AT | 1.00 | Reference | 1.00 | Reference | 1.00 | Reference |
| GT | 1.28 (0.71–2.32) | 0.51 | 1.08 (0.72–1.62) | 0.78 | 0.85 (0.48–1.51) | 0.67 |
| GG | 1.43 (0.76–2.66) | 0.34 | 0.92 (0.61–1.38) | 0.75 | 0.64 (0.35–1.18) | 0.20 |
| AG | 1.29 (0.59–2.78) | 0.66 | 0.95 (0.57–1.58) | 0.94 | 0.74 (0.35–1.56) | 0.54 |
| Apa I - Taq I | ||||||
| TT | 1.00 | Reference | 1.00 | Reference | 1.00 | Reference |
| TC | 0.82 (0.45–1.49) | 0.61 | 0.89 (0.59–1.33) | 0.63 | 1.09 (0.61–1.95) | 0.90 |
| GT | 1.16 (0.62–2.15) | 0.77 | 0.84 (0.57–1.24) | 0.43 | 0.73 (0.40–1.33) | 0.38 |
| GC | 0.80 (0.38–1.71) | 0.71 | 0.75 (0.45 to1.25) | 0.33 | 0.94 (0.45–1.94) | 0.99 |
| Bsm I - Taq I | ||||||
| AT | 1.00 | Reference | 1.00 | Reference | 1.00 | Reference |
| GC | 0.94 (0.49–1.81) | 0.98 | 0.93 (0.57–1.50) | 0.85 | 1.0 (0.52–1.88) | 0.90 |
| GT | 1.30 (0.72–2.34) | 0.48 | 0.94 (0.62–1.41) | 0.83 | 0.72 (0.40–1.29) | 0.34 |
| AC | 0.93 (0.49–1.77) | 0.96 | 0.78 (0.50–1.24) | 0.35 | 0.84 (0.45–1.56) | 0.69 |
ND no data, CI confidence interval
* Significant data
Table 5.
Coefficient C for VDR polymorphisms and clinical data
| SNPs | PCa | BPH | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Age < 58 years old | Age ≥ 58 years old | P | PSA < 10 ng/mL | PSA ≥ 10 ng/mL | P | Age < 58 years old | Age ≥ 58 years old | P | |
| TaqI | |||||||||
| TT | 2 (1.82) | 6 (5.45) | <0.0001* | 29 (30.21) | 15 (15.63) | 0.09 | 0 | 4 (10.81) | 0.0004* |
| TC | 38 (34.55) | 8 (7.27) | 22 (22.92) | 23 (23.96) | 15 (40.54) | 6 (16.22) | |||
| CC | 12 (10.91) | 44 (40.00) | 2 (2.08) | 5 (5.21) | 1 (2.70) | 11 (29.73) | |||
| BsmI | |||||||||
| AA | 3 (2.50) | 10 (8.33) | 0.35 | 2 (2.08) | 9 (9.38) | 0.02* | 0 | 4 (10.81) | 0.27 |
| AG | 7 (5.83) | 48 (40.00) | 23 (23.96) | 20 (20.83) | 6 (16.22) | 16 (43.24) | |||
| GG | 12 (10.00) | 40 (33.33) | 28 (29.17) | 14 (14.58) | 1 (2.70) | 10 (27.03) | |||
| ApaI | |||||||||
| GG | 1 (0.83) | 22 (18.33) | 0.71 | 18 (18.75) | 19 (19.79) | 0.30 | 1 (2.70) | 6 (16.22) | 0.07 |
| GT | 14 (11.67) | 40 (33.33) | 21 (21.88) | 18 (18.75) | 2 (5.41) | 12 (32.43) | |||
| TT | 7 (5.83) | 36 (30.00) | 14 (14.58) | 6 (6.25) | 4 (10.81) | 12 (32.43) | |||
SNP single nucleotide polymorphism
* Significant data
In a significant manner, CC polymorphism (TaqI) was associated to men over 58 years of age in PCa (P < 0.0001) and BPH (P = 0.0004) groups. In addition, GG genotype (BsmI) was associated to lower PSA levels (PSA < 10 ng/mL) in PCa group (P = 0.02), which is considered a prognostic factor.
Additional analyses were performed to clarify the effect of genotypes and haplotypes in PCa group and their correlation with clinical pathological features (Table 6 and Additional file 1 for complete data). All polymorphism were analyzed and those which were not significant were omitted. The data indicated that patients containing the GG genotype (BsmI) have a prevalence 9.00 times higher to present PSA levels <10 ng/mL (95 % CI 1.71–47.39, P = 0.01). Moreover, G allele (BsmI) significantly increased the chances of lower PSA levels in the recessive (OR 6.75, 95 % CI 1.37–33.18, P = 0.02), and dominant models (OR 2.32, 95 % CI 1.01–5.35, P = 0.07). It is also observed that the T allele of the polymorphism TaqI in recessive model was associated to PSA < 10 ng/mL in PCa group (P = 0.08), but it was not significant.
Table 6.
VDR polymorphims sub grouping in PCa cases according to clinical parameters
| Polymorphisms | PSA N (%) | Gleason N (%) | TNM N (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotypes | <10 ng/mL | ≥10 ng/mL | Odds (95 % CI) | P | <7 | ≥7 | Odds (95 % CI) | P | T1-T2 | T3 | Odds (95 % CI) | P |
| FokI | ||||||||||||
| CC | 21 (21.88) | 16 (16.67) | 1.00 | 26 (24.53) | 18 (16.98) | 1.00 | 16 (22.22) | 16 (22.22) | 1.00 | |||
| CT | 25 (26.04) | 24 (25.00) | 0.79 (0.34–1.87) | 0.76 | 27 (25.47) | 21 (19.81) | 0.89 (0.39–2.04) | 0.95 | 24 (33.33) | 8 (11.11) | 3.00 (1.04–8.65) | 0.07 |
| TT | 7 (7.29) | 3 (3.13) | 1.78 (0.40–7.97) | 0.69 | 7 (6.60) | 7 (6.60) | 0.69 (0.21–2.32) | 0.77 | 5 (6.94) | 3 (4.17) | 1.67 (0.34–8.18) | 0.81 |
| BsmI | ||||||||||||
| AA | 2 (2.08) | 9 (9.38) | 1.00 | 8 (7.55) | 4 (3.77) | 1.00 | 8 (11.11) | 2 (2.78) | 1.00 | |||
| AG | 23 (23.96) | 20 (20.83) | 5.18 (1.00–26.82) | 0.08 | 25 (23.58) | 21 (19.81) | 0.60 (0.16–2.26) | 0.66 | 16 (22.22) | 11 (15.28) | 0.36 (0.07–2.05) | 0.43 |
| GG | 28 (29.17) | 14 (14.58) | 9.00 (1.71–47.39) | 0.01* | 27 (25.47) | 21 (19.81) | 0.64 (0.17–2.43) | 0.74 | 21 (29.17) | 14 (19.44) | 0.38 (0.07–2.03) | 0.43 |
| Genotypes in pairs | ||||||||||||
| BsmI - ApaI | ||||||||||||
| AATT | 2 (2.11) | 9 (9.47) | 1.00 | 8 (8.25) | 4 (4.12) | 1.00 | 8 (12.70) | 2 (3.17) | 1.00 | |||
| AGTT | 10 (10.53) | 7 (7.37) | 6.43 (1.05–39.33) | 0.08 | 9 (9.28) | 6 (6.19) | 0.75 (0.15–3.65) | 0.97 | 7 (11.11) | 4 (6.35) | 0.44 (ND) | 0.73 |
| GGGT | 9 (9.47) | 5 (5.26) | 8.10 (1.23–53.20) | 0.06 | 4 (4.12) | 6 (6.19) | 0.33 (0.06–1.91) | 0.41 | 4 (6.35) | 4 (6.35) | 0.25 (ND) | 0.40 |
| AGGT | 12 (12.63) | 13 (13.68) | 4.15 (0.74–23.23) | 0.19 | 15 (15.46) | 15 (15.46) | 0.50 (0.12–2.02) | 0.52 | 9 (14.29) | 6 (9.52) | 0.38 (0.06–2.41) | 0.54 |
| GGGG | 13 (13.68) | 6 (6.32) | 9.75 (1.59–59.70) | 0.02* | 12 (12.37) | 7 (7.22) | 0.86 (0.19–3.92) | 0.85 | 7 (11.11) | 4 (6.35) | 0.44 (ND) | 0.73 |
| GGTT | 6 (6.32) | 3 (3.16) | 9.00 (ND) | 0.08 | 4 (4.12) | 6 (6.19) | 0.33 (0.06–1.91) | 0.41 | 4 (6.35) | 4 (6.35) | 0.25 (ND) | 0.40 |
| Dominant model | ||||||||||||
| BsmI | ||||||||||||
| AA + AG | 25 (26.04) | 29 (30.21) | 1.00 | 33 (31.43) | 25 (23.81) | 1.00 | 24 (33.33) | 13 (18.06) | 1.00 | |||
| GG | 28 (29.17) | 14 (14.58) | 2.32 (1.01–5.35) | 0.07 | 26 (24.76) | 21 (20.00) | 0.94 (0.43–2.04) | 0.97 | 21 (29.17) | 14 (19.44) | 0.81 (0.31–2.11) | 0.86 |
| Recessive model | ||||||||||||
| FokI | ||||||||||||
| CC | 21 (21.88) | 16 (16.67) | 1.00 | 25 (23.81) | 18 (17.14) | 1.00 | 16 (22.22) | 16 (22.22) | 1.00 | |||
| CT + TT | 32 (33.33) | 27 (28.13) | 0.90 (0.40–2.07) | 0.98 | 34 (32.38) | 28 (26.67) | 0.87 (0.40–1.92) | 0.89 | 29 (40.28) | 11 (15.28) | 2.64 (0.99–7.03) | 0.09 |
| TaqI | ||||||||||||
| TT | 29 (30.21) | 15 (15.63) | 1.00 | 27 (25.71) | 22 (20.95) | 1.00 | 21 (29.17) | 17 (23.61) | 1.00 | |||
| TC + CC | 24 (25.00) | 28 (29.17) | 0.44 (0.19–1.02) | 0.08 | 32 (30.48) | 24 (22.86) | 1.09 (0.50–2.35) | 0.99 | 24 (33.33) | 10 (13.89) | 1.94 (0.73–5.16) | 0.27 |
| BsmI | ||||||||||||
| AA | 2 (2.08) | 9 (9.38) | 1.00 | 8 (7.62) | 4 (3.81) | 1.00 | 8 (11.11) | 2 (2.78) | 1.00 | |||
| AG + GG | 51 (53.13) | 34 (35.42) | 6.75 (1.37–33.18) | 0.02* | 51 (48.57) | 42 (40.00) | 0.61 (0.17–2.16) | 0.64 | 37 (51.39) | 25 (34.72) | 0.37 (0.07–1.89) | 0.38 |
ND no data, CI confidence interval
* Significant data
Individuals displaying GG/GG (BsmI/ApaI) are 9.75 times more likely to present low levels of PSA (95 % CI 1.59–59.70, P = 0.02). The combination of BsmI and ApaI variants showed that AG/TT (OR 6.43, 95 % CI 1.05–39.33, P = 0.08), and GG/GT (OR 8.10, 95 % CI 1.23–53.20, P = 0.06) genotypes provide an increased chance of presenting PSA < 10 ng/mL in PCa patients (Table 6).
Evaluating pathological data, CT (FokI) individuals showed a 3.00-fold increased risk for confined PCa tumors (T1–T2) compared to disease progression (T3) (95 % CI 1.04–8.65, P = 0.07). Moreover, evaluating the recessive model, the T allele from the same SNP presented a 2.64-fold decreased risk to T3 tumors development (95 % CI 0.99–7.03 P = 0.09).
Discussion
While VDR regulates numerous genes across the genome, much remains to be learned about pathways and cellular interactions. VDR functions have a broad impact, contributing to cancer development once it regulates cell proliferation and cell cycle control. A better understanding of the genetic factors related to this receptor may facilitate the development of improved strategies for the diagnostic and prognostic of prostate cancer (Saccone et al. 2015). In the analyses presented herein, we did not find associations between prostate cancer risk and FokI, BsmI, ApaI and TaqI polymorphisms, when considered individually.
A functional activity of the FokI polymorphism and its involvement with the incidence of prostate cancer has already been reported (John et al. 2005). Although the SNP FokI seems to be functional and the 424 amino acids (aa) VDR variant is somewhat more active than the 427 aa in terms of its transactivation capacity as a transcription factor, no association between this mutation, prostate cancer and BPH has been reported to date (Hayes et al. 2005; Zeng et al. 2014).
VDR gene polymorphisms may affect the binding of 1,25-(OH)2D3 to its receptor and thereby compromise the anti-proliferative effects of vitamin D. Various polymorphisms in 3′ cluster of the gene have been identified and it is not known whether these represent functional genetic differences or just mark the disease risk alleles (Whitfield et al. 2001). Although genetic variants in the VDR itself do not appear to be linked to 25(OH)D levels, they exert an influence in VDR expression and function, especially considering mRNA stability. Through the 3′ untranslated region, the SNPs BsmI, ApaI and TaqI are silent polymorphisms.
Age is a well-established risk factor that contributes to the etiology of PCa. The chance of PCa occurrence rises after 50-years of age, and about 6 cases in 10 are diagnosed in men aged 65 or older (Prostate Cancer 2015). It has been reported that the onset of disease at a young age has correlated with more aggressive tumor types and subsequent mortality, contributing to poorer prognosis (Bratt et al. 1998). In patients with PCa and BPH, we found an association between TaqI polymorphism (CC genotype) and increased age (>58-year-old) suggesting that the detection of this polymorphism could help to determine prognosis.
The BsmI polymorphism in the VDR gene has been described as a possible genetic marker for different clinical conditions such as type 1 diabetes, obesity and some types of cancers (Cavalcante et al. 2015). A previous study investigating the BsmI polymorphism in multiple sclerosis showed that the G allele contributed to providing a protective effect, while the A allele showed a positive association with the pathology (Narooie-Nejad et al. 2015). Moreover, functional data on VDR level in peripheral blood mononuclear cells of healthy subjects demonstrated that homozygosity for BsmI ‘A’ allele and TaqI ‘C’ allele is associated with lower levels of VDR protein (Saccone et al. 2015; Selvaraj 2009). In our work, G allele was correlated to lower levels of PSA in PCa patients, which also represents a protective effect.
Vitamin D also impacts prostate cancer, regulating androgen-responsive and androgen-metabolizing genes. Androgens act through their receptor to regulate prostate growth and play an important role in the development and progression of PCa (Krishnan et al. 2003). A crosstalk between VDR and the Androgen Receptor (AR) has already been suggested, since LNCaP cells have intensively responded to dihydrotestosterone in the presence of 1,25-(OH)2D3. Furthermore, 1,25-(OH)2D3 and dihydrotestosterone exhibit synergistic interaction to regulate the LNCaP cell proliferation and PSA secretion in a heterologous up-regulation of AR by 1,25-(OH)2D3 (Zhao et al. 1997). Considering BsmI polymorphisms, our results demonstrating the association between G allele and lower PSA levels highly support the idea that VDR and AR share the same coregulators in a crosstalk between both receptors (Williams et al. 2004). The overexpression of AR in PC-3 cell line and the activation of AR in LNCaP cells can suppress VDR transactivation corroborating with these two-way molecular interaction, which may be coordinated by ARA70 coregulator (Ting et al. 2005).
The PSA expression is rigidly controlled by androgens via the AR (Young et al. 1991). Partin et al. (1993) demonstrated that patients with pre-operative serum PSA concentrations higher than 10.0 ng/mL are at a statistically increased risk of PCa recurrence. Additionally, PSA levels higher than 2.0 ng/mL during the year before the diagnosis increased the risk of mortality, despite undergoing radical prostatectomy (D’Amico et al. 2004).
It has been shown that the A allele is protective for men with locally advanced disease, and is correlated with a poorer prognosis among men with organ-confined disease (Williams et al. 2004). In our study the G allele may be associated with higher levels of VDR protein, lower levels of PSA and contributes to organ-confined disease, decreasing the possibility of PCa recurrence. Based on these findings, we hypothesize that VDR polymorphisms associated to PSA levels may be useful as a prognostic factor.
Strong linkage disequilibrium between the BsmI, ApaI, and TaqI polymorphisms have been reported and are linked to the risk of prostate cancer (Mikhak et al. 2007). In fact, polymorphisms tend to be inherited, and their various possible combinations may have a significant association with the disease phenotype (Jingwi et al. 2015). In our study, this linkage disequilibrium was confirmed (P < 0.0001). Haplotypes G/T (SNPs ApaI/TaqI), A/T and G/G (SNPs BsmI/ApaI), T/G and C/A (SNPs TaqI/BsmI) were the most frequent in our population, and the four polymorphisms were found in the Hardy–Weinberg equilibrium.
Our data demonstrated that AG/TT genotype (BsmI/ApaI) conferred higher risk of hyperplasic disease development. This data is supported by a previous study in which TaqI and BsmI variants were associated with the susceptibility to the development of benign prostatic hyperplasia within an Indian population (Manchanda et al. 2010). Furthermore, studies have shown that the BsmI and TaqI variants can play a significant role in the development of prostate cancer (Taylor et al. 1996; Huang et al. 2004). In our study, the AG/TT genotype (BsmI/TaqI) was responsible for an increased chance for PCa occurrence compared to controls.
These genetic polymorphisms associated with PCa have been extensively studied generating contradictory results (Jingwi et al. 2015; Xu et al. 2014). Here we demonstrated the association between VDR polymorphism and critical clinical parameters such as PSA levels and age.
Conclusion
This is the first study that describes an association between VDR polymorphisms and clinical data. We found an association between TaqI polymorphism (CC genotype) and increased age (>58-year-old) in patients with PCa and BPH. Besides, BsmI G allele was correlated to lower levels of PSA in PCa patients.
Future studies with a large population cohort focused on genotyping additional polymorphisms to capture more of the variations in the VDR gene, and haplotype analysis to elucidate the role of the VDR gene as a prostate cancer risk factor may benefit the expansion of significant data. Our work demonstrated a correlation between age and PSA in PCa, opening new perspectives on using polymorphic markers clinically.
Acknowledgements
The authors would like to thank the financial support by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and Fundação de Amparo à Pesquisa do estado de Minas Gerais (FAPEMIG), and the Urology Division of the University Hospital of Uberlandia for providing the biological samples.
Authors’ contributions
SBRN, TGA, LRG, GRA: Experimental design and manuscript writing. AFN, FMO, KM: Statistical analysis and manuscript writing. SBRN: Genetic studies and draft the manuscript. TGA: Project coordinator and senior author. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. We also confirm that this work was approved by the University Research Ethics Committee under the number 005/2001.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional file
10.1186/s40064-016-2009-8 ApaI, FokI, TaqI and BsmI polymorphims sub grouping in PCa cases according toclinical parameters
Contributor Information
Sarah Braga Rodrigues Nunes, Email: sarahhbraga@hotmail.com.
Fabrícia de Matos Oliveira, Email: fabriciamatos@famat.ufu.br.
Adriana Freitas Neves, Email: neves.af@gmail.com.
Galber Rodrigues Araujo, Email: galber.araujo@gmail.com.
Karina Marangoni, Email: marangonik@yahoo.com.br.
Luiz Ricardo Goulart, Email: lrgoulart@ufu.br.
Thaise Gonçalves Araújo, Phone: + 55 34 3822-3714/29, Email: thaisegaraujo@gmail.com.
References
- Barve A, Jin W, Cheng K. Prostate cancer relevant antigens and enzymes for targeted drug delivery. J Control Release. 2014;187:118–132. doi: 10.1016/j.jconrel.2014.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratt O, Kristoffersson U, Olsson H, Lundgren R. Clinical course of early onset prostate cancer with special reference to family history as a prognostic factor. Eur Urol. 1998;34(1):19–24. doi: 10.1159/000019672. [DOI] [PubMed] [Google Scholar]
- Cavalcante IG, Silva AS, Costa MJ, Persuhn DC, Issa CI, Freire TL, et al. Effect of vitamin D3 supplementation and influence of BsmI polymorphism of the VDR gene of the inflammatory profile and oxidative stress in elderly women with vitamin D insufficiency: Vitamin D3 megadose reduces inflammatory markers. Exp Gerontol. 2015;66:10–16. doi: 10.1016/j.exger.2015.03.011. [DOI] [PubMed] [Google Scholar]
- Cuzick J, Thorat MA, Andriole G, Brawley OW, Brown PH, Culig Z, et al. Prevention and early detection of prostate cancer. Lancet Oncol. 2014;15(11):e484–e492. doi: 10.1016/S1470-2045(14)70211-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amico AV, Chen M-H, Roehl KA, Catalona WJ. Preoperative PSA velocity and the risk of death from prostate cancer after radical prostatectomy. N Engl J Med. 2004;351(2):125–135. doi: 10.1056/NEJMoa032975. [DOI] [PubMed] [Google Scholar]
- Gandini S, Gnagnarella P, Serrano D, Pasquali E, Raimondi S. Vitamin D receptor polymorphisms and cancer. Adv Exp Med Biol. 2014;810:69–105. doi: 10.1007/978-1-4939-0437-2_5. [DOI] [PubMed] [Google Scholar]
- Gommersall LM, Khanim FL, Peehl DM, Doherty AH, Campbell MJ. Epigenetic repression of transcription by the Vitamin D3 receptor in prostate cancer cells. J Steroid Biochem Mol Biol. 2004;89–90(1–5):251–256. doi: 10.1016/j.jsbmb.2004.03.080. [DOI] [PubMed] [Google Scholar]
- Hayes VM, Severi G, Padilla EJ, Eggleton SA, Southey MC, Sutherland RL, et al. Genetic variants in the vitamin D receptor gene and prostate cancer risk. Cancer Epidemiol Biomarkers Prev. 2005;14(4):997–999. doi: 10.1158/1055-9965.EPI-04-0660. [DOI] [PubMed] [Google Scholar]
- Huang S-P, Chou Y-H, Wayne Chang W-S, Wu M-T, Chen Y-Y, Yu C-C, et al. Association between vitamin D receptor polymorphisms and prostate cancer risk in a Taiwanese population. Cancer Lett. 2004;207(1):69–77. doi: 10.1016/j.canlet.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Jingwi EY, Abbas M, Ricks-Santi L, Winchester D, Beyene D, Day A, et al. Vitamin D receptor genetic polymorphisms are associated with PSA Level, gleason score and prostate cancer risk in African-American Men. Anticancer Res. 2015;35(3):1549–1558. [PMC free article] [PubMed] [Google Scholar]
- John EM, Schwartz GG, Koo J, Van Den Berg D, Ingles SA. Sun exposure, vitamin D receptor gene polymorphisms, and risk of advanced prostate cancer. Cancer Res. 2005;65(12):5470–5479. doi: 10.1158/0008-5472.CAN-04-3134. [DOI] [PubMed] [Google Scholar]
- Krishnan AV, Peehl DM, Feldman D. Inhibition of prostate cancer growth by vitamin D: regulation of target gene expression. J Cell Biochem. 2003;88(2):363–371. doi: 10.1002/jcb.10334. [DOI] [PubMed] [Google Scholar]
- Li L, Shang F, Zhang W, Zhang C, Li J, Wang C, et al. Role of vitamin D receptor gene polymorphisms in pancreatic cancer: a case–control study in China. Tumor Biol. 2015 doi: 10.1007/s13277-015-3119-6. [DOI] [PubMed] [Google Scholar]
- Manchanda PK, Konwar R, Nayak VL, Singh V, Bid HK. Association of genetic variants of the vitamin D receptor (VDR) gene (Fok-I, Taq-I and Bsm-I) with susceptibility of benign prostatic hyperplasia in a North Indian population. Asian Pac J Cancer Prev. 2010;11(4):1005–1008. [PubMed] [Google Scholar]
- Mehta RG, Peng X, Alimirah F, Murillo G, Mehta R. Vitamin D and breast cancer: emerging concepts. Cancer Lett. 2013;334(1):95–100. doi: 10.1016/j.canlet.2012.10.034. [DOI] [PubMed] [Google Scholar]
- Mikhak B, Hunter DJ, Spiegelman D, Platz EA, Hollis BW, Giovannucci E. Vitamin D receptor (VDR) gene polymorphisms and haplotypes, interactions with plasma 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D, and prostate cancer risk. Prostate. 2007;67(9):911–923. doi: 10.1002/pros.20570. [DOI] [PubMed] [Google Scholar]
- Narooie-Nejad M, Moossavi M, Torkamanzehi A, Moghtaderi A, Salimi S. Vitamin D receptor gene polymorphism and the risk of multiple sclerosis in South Eastern of Iran. J Mol Neurosci. 2015 doi: 10.1007/s12031-015-0513-x. [DOI] [PubMed] [Google Scholar]
- Partin AW, Pound CR, Clemens JQ, Epstein JI, Walsh PC. Serum PSA after anatomic radical prostatectomy. The Johns Hopkins experience after 10 years. Urol Clin North Am. 1993;20(4):713–725. [PubMed] [Google Scholar]
- Prostate Cancer [database on the Internet]. American Cancer Society. 2015. http://www.cancer.org/cancer/prostatecancer/. Accessed 6 June 2015
- Saccone D, Asani F, Bornman L. Regulation of the vitamin D receptor gene by environment, genetics and epigenetics. Gene. 2015;561(2):171–180. doi: 10.1016/j.gene.2015.02.024. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Selvaraj P, Anand SP, Harishankar M, Alagarasu K. Plasma 1,25 dihydroxy vitamin D3 level and expression of vitamin d receptor and cathelicidin in pulmonary tuberculosis. J Clin Immunol. 2009;29(4):470–478. doi: 10.1007/s10875-009-9277-9. [DOI] [PubMed] [Google Scholar]
- Taylor JA, Hirvonen A, Watson M, Pittman G, Mohler JL, Bell DA. Association of prostate cancer with vitamin D receptor gene polymorphism. Cancer Res. 1996;56(18):4108–4110. [PubMed] [Google Scholar]
- Ting HJ, Bao BY, Hsu CL, Lee YF. Androgen-receptor coregulators mediate the suppressive effect of androgen signals on vitamin D receptor activity. Endocrine. 2005;26(1):1–9. doi: 10.1385/ENDO:26:1:001. [DOI] [PubMed] [Google Scholar]
- Whitfield GK, Remus LS, Jurutka PW, Zitzer H, Oza AK, Dang HT, et al. Functionally relevant polymorphisms in the human nuclear vitamin D receptor gene. Mol Cell Endocrinol. 2001;177(1–2):145–159. doi: 10.1016/S0303-7207(01)00406-3. [DOI] [PubMed] [Google Scholar]
- Williams H, Powell IJ, Land SJ, Sakr WA, Hughes MR, Patel NP, et al. Vitamin D receptor gene polymorphisms and disease free survival after radical prostatectomy. Prostate. 2004;61(3):267–275. doi: 10.1002/pros.20103. [DOI] [PubMed] [Google Scholar]
- Wyatt AW, Mo F, Wang Y, Collins CC. The diverse heterogeneity of molecular alterations in prostate cancer identified through next-generation sequencing. Asian J Androl. 2013;15(3):301–308. doi: 10.1038/aja.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, He B, Pan Y, Deng Q, Sun H, Li R, et al. Systematic review and meta-analysis on vitamin D receptor polymorphisms and cancer risk. Tumour Biol. 2014;35(5):4153–4169. doi: 10.1007/s13277-013-1544-y. [DOI] [PubMed] [Google Scholar]
- Yang B, Liu S, Yang X, Wang Y, Zhao X, Zheng D, et al. Current evidence on the four polymorphisms of VDR and breast cancer risk in Caucasian women. Meta Gene. 2014;2:41–49. doi: 10.1016/j.mgene.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CY, Montgomery BT, Andrews PE, Qui SD, Bilhartz DL, Tindall DJ. Hormonal regulation of prostate-specific antigen messenger RNA in human prostatic adenocarcinoma cell line LNCaP. Cancer Res. 1991;51(14):3748–3752. [PubMed] [Google Scholar]
- Zeng X-T, Yao Q-S, Weng H, Li S, Huang J-Y, Wang X-H. Meta-analysis of vitamin D receptor gene polymorphisms and benign prostatic hyperplasia risk. Mol Biol Rep. 2014;41(10):6713–6717. doi: 10.1007/s11033-014-3554-2. [DOI] [PubMed] [Google Scholar]
- Zhao XY, Ly LH, Peehl DM, Feldman D. 1alpha,25-dihydroxyvitamin D3 actions in LNCaP human prostate cancer cells are androgen-dependent. Endocrinology. 1997;138(8):3290–3298. doi: 10.1210/endo.138.8.5328. [DOI] [PubMed] [Google Scholar]