

Abstract
We screened a collection of synthetic compounds consisting of natural-product-like substructural motifs to identify a spirocyclic chromane as a novel antiplasmodial pharmacophore using an unbiased cell-based assay. The most active spirocyclic compound UCF 201 exhibits a 50% effective concentration (EC50) of 350 nM against the chloroquine-resistant Dd2 strain and a selectivity over 50 using human liver HepG2 cells. Our analyses of physicochemical properties of UCF 201 showed that it is in compliance with Lipinski's parameters and has an acceptable physicochemical profile. We have performed a limited structure-activity-relationship study with commercially available chromanes preserving the spirocyclic motif. Our evaluation of stage specificities of UCF 201 indicated that the compound is early-acting in blocking parasite development at ring, trophozoite and schizont stages of development as well as merozoite invasion. SPC is an attractive lead candidate scaffold because of its ability to act on all stages of parasite's aexual life cycle unlike current antimalarials.
Keywords: Antimalarials, Antiplasmodials, Natural-product-like compounds, Spirocyclic chromane, Novel mechanism of action
Graphical abstract

Highlights
-
•
SPC is a novel antiplasmodial scaffold.
-
•
SPC has an EC50 of 350 nM against chloroquine-resistant parasite and is selective.
-
•
SPC inhibits all stages of parasite growth in erythrocytes.
1. Introduction
There are more than 200 million global clinical cases of malaria resulting in over 600,000 deaths each year (Murray et al., 2012, World Health Organization et al., 2014). In addition to contributing significantly towards overall childhood mortality in the poorest nations, the disease is estimated to cause considerable reduction in economic growth in countries that bear a heavy malaria burden (Gallup and Sachs, 2001). The situation is dire because widespread prevalence of drug resistant parasites is making most of the available drugs for treatment to rapidly lose their effectiveness (Greenwood, 1995, Rieckmann, 2006). Most of the drugs that are currently being used for malaria treatment were developed more than 30 years ago, and many are derivatives of older drugs. Furthermore, only a handful of antimalarials primarily belonging to three main chemical classes (4- and 8-aminoquinolines, antifolates. and artemisinin-based agents) are being used for therapy (Grimberg and Mehlotra, 2011). While artemisinin-based combination treatments (ACTs) have played an effective role in controlling the disease in many malaria endemic areas, the appearance of resistant parasites to artemisinin derivatives in wide area of Southeast Asia encompassing south Vietnam to central Myanmar underscores the fragility of malaria treatment measures (Cui, 2011, Miotto et al., 2013, Ashley et al., 2014). Therefore, there is a pressing need for novel therapeutic options to treat resistant malaria. Historically, natural products were the most important source for the majority of therapeutics, including antimalarials (Li and Vederas, 2009). Quinine and artemisinin and their synthetic derivatives are prime examples in the malaria field (Woodward and Doering, 1944, Klayman, 1985). Current trends in lead discovery are biased towards high-throughput screening (HTS) of synthetic compound collections instead of the testing of natural products (NPs). The complexity of natural product structures are often accompanied by synthetic challenges and supply problems, whereas compound collections of commercial sources or “in-house” origin are easily accessible. Nevertheless, because NPs are known to occupy biologically important chemical space, they continue to be valuable for the discovery of new biologically active compounds. The synthesis of NP-inspired compound libraries is a promising alternative covering chemical space similar to NPs while being synthetic tractable (Cordier et al., 2008, Rishton, 2008, Vasilevich et al., 2012). Critical evaluation of known drugs and NPs has been used to identify NP-based substructural motifs, termed as “BioCores” (Kombarov et al., 2010), which have been incorporated into the scaffolds of a BioDesign synthetic library to increase drug-likeness of compounds. In an effort to identify novel antimalarial scaffolds we have screened a subset of the anti-infective BioDesign collection of the vendor Asinex that incorporates “BioCore” features and structures (e.g., enriched oxygen and saturated rings), which were filtered against peroxide bridges that are present in many current antimalarials. Herein, we report that spirocyclic chromane compounds demonstrate potent antiplasmodial activities with excellent selectivity. Furthermore, we demonstrate that this chemotype exhibits a cellular mechanism of action distinct from current antimalarials.
2. Materials and methods
2.1. Plasmodium falciparum culture and EC50 determination
Plasmodium falciparum Dd2 and 3D7 strains were maintained at 37 °C in 5% CO2 and 95% air using a modified Trager and Jensen (Trager and Jensen, 1976) method in RPMI media with l-glutamine (Invitrogen) and supplemented with 25 mM HEPES, 26 mM NaHCO3, 2% dextrose, 15 mg/L hypoxanthine (Sigma–Aldrich), 25 mg/L gentamicin (Life Technologies), and 0.5% Albumax II (Life Technologies). Different dilutions of the compound in RPMI 1640 (Life Technologies) from a stock of 10 mM in dimethyl sulfoxide (DMSO) were added to the culture at a 1% parasitemia and 2% hematocrit in 96-well black plates (Santa Cruz Biotechnology). Maximum DMSO concentration in the culture never exceeded 0.125%. Chloroquine at 1 μM was used as a positive control to determine the baseline value. Following 72 h incubation at 37 °C, the ability of the compounds to inhibit growth of the parasite was determined by a SYBR green I-based DNA quantification assay (Bennett et al., 2004, Smilkstein et al., 2004, Johnson et al., 2007). EC50 was calculated (n = 3) from a dose response curve that was generated from a concentration range of 0–20 μM using GraphPad Prism v5.0.
2.2. Cytotoxicity determination
Compounds were evaluated for cytotoxicity using HepG2 human hepatoma cells. A 384-well clear bottom plate (Santa Cruz Biotechnology) was seeded with 2500 cells/well and incubated for 24 h. Serial dilutions (0–40 μM concentration range) of the compound were added to the plate followed by incubation for an additional 48 h. Viability of cells were assessed by MTS [(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) cell proliferation assay (CellTiter 96® Aqueous non-radioactive cell proliferation assay, Promega) (Cory et al., 1991).
2.3. Physicochemical parameters
The aqueous solubility at pH 7.4 was determined by UV–visible absorption based method (Avdeef, 2001). The permeability was assessed by the in vitro double-sink parallel artificial membrane permeability assay (Kansy et al., 1998) that is a model for the passive transport from the gastrointestinal tract into the blood stream. The microsomal stability (Janiszewski et al., 2001) was determined in the presence of mouse liver microsomes with or without NADPH.
2.4. Determination of cellular mechanism of action
P. falciparum Dd2 cultures were tightly synchronized using a combination of magnetic separation of schizonts (Ribaut et al., 2008) followed by sorbitol treatment at the early ring stage (Lambros and Vanderberg, 1979). Inhibitory effects of UCF 201 on parasite development, merozoite egress and invasion were analyzed by treating the synchronized culture (1.5% hematocrit and 5% parasitemia) at four different time-points post-merozoite invasion (6, 18, 30, and 42 h post invasion or hpi) at 5×EC50 final concentration for a duration of 36 h (or 24 h for treatments at the 42 hpi) under regular culture conditions. Giemsa-stained thin smears were prepared at 12 h intervals for microscopic evaluations of the intraerythrocytic maturation of parasite.
2.5. Synthesis of spirocyclic chromane UCF 201
In order to meet the amount of compound needed for in vivo efficacy studies, hit compound UCF 201 has been prepared “in-house” (Fig. 1). Compound 1 was synthesized via an acetylation of α-naphthol using hot glacial acetic acid and zinc chloride (Vyawahare et al., 2010). It was then converted to compound 2 by refluxing with tert-butyl 4-oxopiperidine-1-carboxylate and pyrrolidine in methanol, which was then deprotected in dichloromethane using trifluoroacetic acid to afford compound 3. Compound 3 was then reacted with epoxide 4, which was obtained from the sulfur-ylide epoxidation of 3,4-difluorobenzaldehyde in ethanol, to yield compound 5 which was subsequently converted to UCF 201 using sodium borohydride in absolute ethanol.
Fig. 1.

Synthetic Scheme of UCF 201. Starting from commercially available α-naphthol, UCF 201 was synthesized in 6 steps in good yields.
tert-butyl 4-oxo-3,4-dihydrospiro[benzo[h]chromene-2,4'-piperidine]-1'-carboxylate (2): A solution of intermediate 1 (Vyawahare et al., 2010) (500 mg, 2.7 mmol), tert-butyl 4-oxopiperidine-1-carboxylate (536 mg, 2.7 mmol) and pyrrolidine (517 mg, 7.2 mmol) in absolute methanol (5 mL) was refluxed for 8 h under N2. The methanol was removed under reduced pressure and the residue was purified by flash chromatography (hexanes:EtOAc = 7:1) to afford 2 (908 mg, 92%) as a yellow solid. Rf (hexanes:EtOAc = 5:1) = 0.37. 1H NMR (400 MHz, CDCl3) δ: 8.40–8.24 (m, 1H), 7.84 (d, J = 8.7 Hz, 1H), 7.82–7.76 (m, 1H), 7.65–7.59 (m, 1H), 7.58–7.50 (m, 1H), 7.40 (d, J = 8.6 Hz, 1H), 3.97 (d, J = 13.5 Hz, 2H), 3.42–3.19 (m, 2H), 2.82 (s, 2H), 2.29–2.14 (m, 2H), 1.70 (s, 2H), 1.47 (s, 9H) ppm. 13C NMR (101 MHz, CDCl3) δ: 191.19, 157.06, 154.78, 137.89, 129.76, 128.14, 126.51, 125.36, 123.24, 121.55, 121.00, 115.20, 79.99, 79.16, 47.80, 39.55, 34.29, 28.55 ppm.
spiro[benzo[h]chromene-2,4'-piperidin]-4(3H)-one (3): A stirred solution of 2 (1.5 g, 4.0 mmol) in DCM (40 mL) was cooled to 0 C TFA (13 mL) was then added drop wise and the reaction mixture was allowed to warm to room temperature. After 2 h, the reaction was diluted with water (40 mL) and extracted with DCM (40 mL×2). The aqueous layer was basified to pH 9.0 with 10% NaOH and then extracted with DCM (40 mL×3). The organic layer was combined, dried with sodium sulfate and concentrated under reduced pressure to afford 3 (1.05 g, 97%) as a brown solid). Rf (DCM:MeOH = 10:1) = 0.3. 1H NMR (600 MHz, CDCl3) δ: 8.37 (d, J = 8.4 Hz, 1H), 7.82 (d, J = 8.7 Hz, 1H), 7.77 (d, J = 8.1 Hz, 1H), 7.62–7.58 (m, 1H), 7.55–7.50 (m, 1H), 7.37 (d, J = 8.6 Hz, 1H), 3.21–3.12 (m, 2H), 2.98–2.90 (m, 2H), 2.80 (d, J = 1.5 Hz, 2H), 2.22–2.13 (m, 2H), 2.08 (s, 2H), 1.77–1.67 (m, 2H) ppm. 13C NMR (151 MHz, CDCl3) δ: 191.66, 157.29, 137.85, 129.63, 128.04, 126.32, 125.49, 123.44, 121.54, 120.67, 115.14, 79.62, 48.17, 42.13, 35.36 ppm. HRMS (ESI) calcd for C17H18NO2 [M+H]+: 268.1332, found: 268.1325.
2-(3,4-difluorophenyl)oxirane (4): Potassium hydroxide pellets 3.92 mg, 7.0 mmol) was added to a solution of 3,4-difluorobenzaldehyde (500 mg, 3.5 mmol) and trimethylsulfonium iodide (Me3SI) (1.1 g, 5.4 mmol) in acetonitrile (8 mL) and water (20 μL). The mixture was stirred at 80 °C for 4 h and then cooled; the acetonitrile was removed by rotatory evaporator. The residue was treated with a solution of water (20 mL) and sodium hypochlorite or bleach (5 mL) followed by extraction with diethyl ether (15 mL×3). The organic layer was combined, dried with sodium sulfate and the solvent was removed under reduced pressure. The crude was used without further purification.
1'-(2-(3,4-difluorophenyl)-2-hydroxyethyl)spiro[benzo[h]chromene-2,4'-piperidin]-4(3H)-one (5): A solution of 3 (500 mg, 1.8 mmol) and 4 (292 mg, 1.8 mmol) in ethanol (30 mL) was heated to reflux for 18 h. The ethanol was removed under reduced pressure and the residue was purified by flash chromatography (hexanes:EtOAc = 2:1) to afford 5 (498 mg, 63%) as a yellow solid. Rf = 0.69 in EtOAc. 1H NMR (600 MHz, CDCl3) δ: 8.34 (d, J = 8.3 Hz, 1H), 7.85 (d, J = 8.6 Hz, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.65–7.60 (m, 1H), 7.55 (t, J = 7.6 Hz, 1H), 7.40 (d, J = 8.7 Hz, 1H), 7.25–7.21 (m, 1H), 7.15–7.09 (m, 1H), 7.08 (d, J = 3.4 Hz, 1H), 4.73–4.69 (m, 1H), 2.97 (t, J = 11.4 Hz, 1H), 2.93–2.86 (m, 1H), 2.83 (d, J = 16.1 Hz, 2H), 2.72–2.59 (m, 3H), 2.49–2.43 (m, 1H), 2.31–2.22 (m, 2H), 1.92–1.81 (m, 2H) ppm. 13C NMR (151 MHz, CDCl3) δ: 191.39, 157.16, 150.6 (dd, J = 249.0, 16.2 Hz), 149.8 (dd, J = 248.2, 12.7 Hz), 139.30 (dd, J = 5.3, 3.5 Hz), 137.93, 129.72, 128.17, 126.43, 125.47, 123.32, 121.75 (dd, J = 6.3, 3.5 Hz) 121.63, 120.91, 117.22 (d, J = 17.2 Hz), 115.26, 114.95 (d, J = 18.0 Hz), 78.79, 68.07, 66.19, 50.63, 47.86, 47.73, 34.78, 34.60 ppm. HRMS (ESI) calcd for C25H24F2NO3 [M+H]+: 424.1719, found: 424.1705.
1'-(2-(3,4-difluorophenyl)-2-hydroxyethyl)-3,4-dihydrospiro[benzo[h]chro-mene-2,4'-piperidin]-4-ol (UCF 201): Sodium borohydride (27 mg, 0.7 mmol) was added to a solution of 5 (100 mg, 0.23 mmol) in absolute ethanol (2 mL). The reaction mixture was heated at reflux overnight and then cooled. The ethanol was removed under reduced pressure, the residue was diluted with water (2 mL) and extracted with ethyl acetate (5 mL×3). The combined organic layer was dried with sodium sulfate, the solvent was evaporated and the residue was purified by flash chromatography (DCM:MeOH = 30:1) to afford final compound 1 (88 mg, 88%) as a white solid. Rf (DCM:MeOH = 10:1) = 0.37. 1H NMR (600 MHz, CDCl3) δ: 8.30–8.18 (m, 1H), 7.83–7.70 (m, 1H), 7.57–7.44 (m, 3H), 7.46–7.34 (m, 1H), 7.26–7.20 (m, 1H), 7.15–7.08 (m, 1H), 7.08–7.02 (m, 1H), 4.93 (t, J = 6.7 Hz, 1H), 4.78–4.59 (m, 1H), 2.99–2.73 (m, 2H), 2.74–2.50 (m, 3H), 2.49–2.35 (m, 1H), 2.22–2.05 (m, 2H), 2.02–1.88 (m, 2H), 1.88–1.62 (m, 2H) ppm. 13C NMR (151 MHz, CDCl3) δ: 150.53 (dd, J = 247.1, 13.6 Hz), 149.73 (dd, J = 248.6, 13.5 Hz), 147.46, 139.43 (dd, J = 4.9, 3.5 Hz), 134.37, 127.68, 126.64, 125.60, 125.49, 125.47, 122.12, 121.75 (dd, J = 6.4, 3.3 Hz), 120.22, 117.97, 117.14 (d, J = 17.2 Hz), 114.90 (d, J = 17.8 Hz), 73.90, 73.88, 67.95, 66.23, 63.33, 50.89, 50.75, 47.92, 47.78, 41.99, 36.10, 35.84, 34.47, 34.19 ppm. HRMS (ESI) calcd for C25H26F2NO3 [M+H]+: 426.1875, found: 426.1861.
2.6. Cytological profiling of inhibition mechanisms
Effects of UCF 201 on merozoite egress and invasion were analyzed using a recently reported high content image-based assay for malaria parasitemia detection and stage classification in a 384-well format (Moon et al., 2013). Sorbitol synchronized cultures (P. falciparum HB3 infected cultures) at 1.5% hematocrit and 5% parasitemia were treated at 42 h post-merozoite invasion with UCF 201 or reference compounds (N-acetylglucosamine (GlcNAc), E-64, or artemisinin) each at 10 μM final concentration for a duration 24 h under a culture condition of 37 C in gassed chambers (5% CO2, 1% O2 and 94% N2). The cultures were then diluted in a staining solution comprising wheat germ agglutinin-Alexa Fluor 488 conjugate and Mitotracker Red CMXRos each at 1 nM final concentrations, and maintained for 20 min at 37 C to allow for complete incorporation of the dyes. The cultures were further diluted to a final hematocrit of 0.02% in a fixing solution comprising 4% paraformaldehyde and 5 μg/ml DAPI and left to stand at room temperature for 10 min. Following a brief vortex at 1700 rpm for 45 s, and a short spin at 1000 rpm for 1 min, five image fields were captured using the Operetta 2.0 automated imaging system (Perkin Elmer) from each assay well (384-well glass plate, Matrical) with a 40X high numerical aperture objective. Operetta automated high content imaging system is equipped with a 50 μM pinwheel array and images were captured at three wavelengths (405 nM, DAPI; 488 nM, Alexa fluor; and 635 nM, Mitoracker deep red).
2.7. Flow cytometric profiling of inhibition
Synchronized P. falciparum Dd2 cultures were treated (5×EC50) with UCF 201 at 6 hpi, samples were collected at 18, 30, 42, and 54 hpi time intervals, fixed in a solution containing 0.04% glutaraldehyde in PBS, permeabilized with 0.25% Triton X-100, treated with RNAse (50 μg/ml) and stained with 10.24 μM YOYO-1 (Bouillon et al., 2013). Flow cytometry acquisition was performed in BD Accuri C6 using FL-1 channel (488 nm Laser with 533/30 filter).
2.8. In vivo efficacy of UCF 201
Female pathogen-free Balb/c mice (8 weeks old, ∼25 g, 5 animals per group) were infected with 1 × 106 P. berghei ANKA parasitized RBC (from a donor mouse) by intraperitoneal injection. UCF 201 was formulated in 0.5% hydroxyethylcellulose-0.1% Tween 80 for oral delivery (0.2 ml/dose). The control group had vehicle only. Parasitemia was monitored by microscopic evaluations of Giemsa-stained thin smears of daily superficial temporal vein bleed samplings. This animal study protocol was approved by the Institutional Animal Care and Use Committee at the University of Central Florida.
3. Results and discussion
To identify novel compounds that interact with cellular targets different from those of current antimalarials, we screened a 594 compound collection containing NP-like structural motifs (e.g. alkaloids, stereogenic carbons, sp3-rich frameworks, basic nitrogens and others) lacking the presence of a peroxide bridge (which is typical for the artemisinin class of antimalarials) from a natural BioDesign scaffold library. To the best of our knowledge, no report of screening of such a compound selection for antimalarial activities exists. We used an unbiased cell-based screen utilizing SYBR green I-based DNA quantification assay (Bennett et al., 2004, Smilkstein et al., 2004, Johnson et al., 2007) to identify antiplasmodial activities. For our primary screen, we used the chloroquine-resistant Dd2 strain, as our main goal was to overcome the problem of drug-resistant malaria. For selecting initial hits, we used a stringent criteria of >75% inhibition of the parasite growth at 1 μM. We identified 98 (16%) initial hits from 22 different scaffolds based on this criterion. The half maximal effective concentration (EC50) values of hits prioritized by this criteria were determined against both the chloroquine-resiatnt (Dd2) and chloroquine-sensitive (3D7) strains. UCF 201, which contains two unique structural elements consisting of a chromane ring and a spirocyclic carbon center belongs to the compound class of spirocyclic chromanes, exhibited antiplasmodial potency with an EC50 value of 350 nM (Table 1) was among the most active compounds. Furthermore, this chemotype showed similar or better EC50 values with the chloroquine resistant Dd2 strain compared to the chloroquine sensitive 3D7 line, indicating that it is not influenced by the efflux mechanism of PfCRT of chloroquine-resistant strains (Ecker et al., 2012). The cytotoxicity of UCF 201 was determined in human HepG2 hepatoma cells using the MTS cell proliferation assay. The EC50 value of UCF 201 in HepG2 cells was 19 μM suggesting an excellent selectivity of over 50-fold (Table 1).
Table 1.
Activity of the Spirocyclic Chromane Chemotype. Z’ factors of these assays were >0.8. For the antimalarial screening different dilutions of compounds were added to an asynchronous culture at a 1% parasitemia and 2% hematocrit and incubated at 37 °C for 72 h. Each assay was done in triplicate and repeated at least 3 times (n = 3) and the standard deviation of values are shown. For the cytotoxicity assessment the incubation time with compounds was 48 h.
| UCF201 | Spirocyclic chromane (SPC) |  |
Dd2 EC50 = 0.35 μM ± 0.02 3D7 EC50 = 0.39 μM ± 0.03 HepG2 EC50 = 19.0 μM ± 0.7 |
||||
|---|---|---|---|---|---|---|---|
| ID | Structure | Dd2 EC50 (μM) | HepG2 EC50 (μM) | ID | Structure | Dd2 EC50 (μM) | HepG2 EC50 (μM) |
| 6 |  |
0.41 ± 0.02 | 13.4 ± 0.3 | 14 |  |
0.85 ± 0.10 | 19.3 ± 1.3 |
| 7 |  |
0.45 ± 0.07 | 17.4 ± 0.6 | 15 |  |
0.62 ± 0.08 | 19.1 ± 0.7 |
| 8 |  |
0.51 ± 0.07 | 19.4 ± 1.0 | 16 |  |
0.67 ± 0.07 | 16.3 ± 0.7 |
| 9 |  |
0.56 ± 0.08 | 13.3 ± 0.8 | 17 |  |
0.75 ± 0.09 | 13.3 ± 1.1 |
| 10 |  |
0.56 ± 0.09 | 17.3 ± 0.8 | 18 |  |
0.87 ± 0.07 | 19.1 ± 1.1 |
| 11 |  |
0.61 ± 0.06 | 15.3 ± 0.9 | 19 | 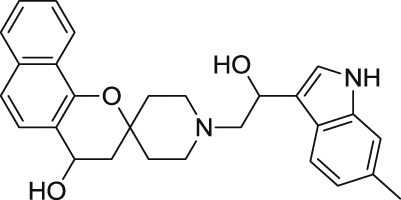 |
1.04 ± 0.05 | 16.4 ± 1.1 |
| 12 |  |
0.61 ± 0.07 | 18.4 ± 1.0 | 20 | 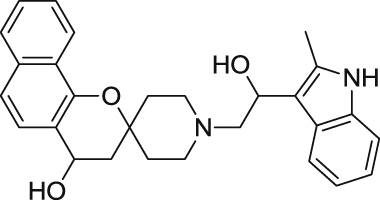 |
1.07 ± 0.06 | 18.9 ± 0.9 |
| 13 |  |
0.62 ± 0.08 | 15.4 ± 2.0 | 21 |  |
1.18 ± 0.10 | 15.3 ± 0.8 |
UCF 201 is in compliance with Lipinski's parameters. The data as shown in Table 2 suggest that the properties of this compound are in line with the preferred parameters in respect of distribution coefficient (clogP), molecular weight, number of hydrogen bond donors and acceptors, as well as polar surface area. We next determined the in vitro physiochemical profiles of the compound. The aqueous solubility of UCF 201 was moderate at pH 7.4 with a value of 24.3 μg/ml (57.1 μM), and even better at lower pH. UCF 201 also exhibited excellent permeability (logPe of <−2.9) compared to reference compounds as assessed by the in vitro double-sink parallel artificial membrane permeability assay (Kansy et al., 1998) that is a model for the passive transport from the gastrointestinal tract into the blood stream. The microsomal stability (Janiszewski et al., 2001) of UCF 201 in the presence of mouse liver microsomes is low (16% remaining at 60 min). Measurements from the aforementioned assays provided initial evidence that the spirocyclic compound UCF 201 displayed acceptable physiochemical profiles.
Table 2.
Physicochemical properties of spirocyclic chromane UCF 201.
| Property | UCF 201 |
|---|---|
| clogP | 3.85 |
| Molecular Weight (g/mol) | 425.5 |
| Number of H Bond Donor | 2 |
| Number of N & O Atoms | 4 |
| Polar Surface Area (A2) | 52.9 |
| Aqueous Solubility pH 7.4/6.2/5.0(μg/mL) | 24.3/>42/>42 |
| Permeability pH 7.4/6.2/5.0 (−logPe) | <2.9/3.2/3.6 |
| Mouse Microsome Stability (% remaining at 60 min) | 15.6 |
Guidelines:
Aqueous solubility: <10 μg/ml-low; 10–60 μg/ml-moderate; >60-high Reference permeability (−logPe) at pH 7.4/6.2/5.0: Verapamil-HCl -2.7/2.9/3.7; metoprolol- 3.6/4.4/>5.4; rantidine- >5.9/5.1/5.2. Verapamil-HCl is considered highly permeable, metoprolol is moderately permeable; and rantidine is poorly permeable. Reference microsomal stability (% remaining at 60 min): Verapamil- 68.8%; Testosterone- 26.1%
In order to perform an expedient SAR evaluation on various skeleton motifs of UCF 201, we chose related analogues from Asinex's compound collections. We tested spirocyclic analogues (6–21) with an emphasis on the substitutions of the piperidine nitrogen and the results are summarized in Table 1. An important observation from this limited SAR reveals that simple phenyl derivatives are much more tolerated than large aromatics such as indoles and naphthalenes in terms of potency. Within the selected phenyl groups, it seems that electron density plays no significant role for antimalarial activities as the EC50 values of the electron-poor UCF 201 and the electron-rich analogue 7 are only marginally different from their reference counterpart 6. Mono-fluoro analogue 13 shows an inferior potency profile to electron-rich analogues; however, additional fluorine atom boosts the potency by almost two folds, which indicates that the lipophilicity, rather than the electron density, may have a significant impact on the EC50. Our study also suggests that the benzo[h]chromane of UCF 201 may be replaced by a simple chromane scaffold without sacrificing submicromolar antimalarial activity. Because the role of naphthalene on piperidine nitrogen could not be determined by the current SAR study, a thorough screening with phenyl-derived chromanes is necessary to better understand such structural features.
To further define the stage-specificities and speeds of action of UCF 201, we investigated its effects on (i) schizont development, and (ii) merozoite egress and invasion processes. In this assay, we treated synchronized cultures with UCF 201 at different stages of intraerythrocytic developmental cycle and followed its effect on parasite development by quantifying culture parasitemias, and proportions of each parasite stage in culture (early rings, mid-late rings, trophozoites, and schizonts) microscopically. As can be seen from Fig. 2, while the control culture matures through the developmental cycle, UCF 201 blocks maturation. UCF 201 was early acting in inhibiting intraerythrocytic developmental cycle when added at either the early (Fig. 2A 6 h post invasion) or late (Fig. 2B, 18 h post invasion) ring stages. UCF 201 also exhibited inhibitory activity against schizont development from the mid-trophozoite stage (30 hpi) (Fig. 2C). In addition, when added at the late schizont stage (42-hpi), UCF 201 displayed notable reductions in culture parasitemias with increased accumulation of free merozoites not schizonts in the culture (Fig. 3A and B). Artemisinin was used as a reference antimalarial compound acting on asexual stages and E-64, a cysteine protease inhibitor, blocking egress (Blackman, 2008). As expected artemisinin exposure at 42 hpi did not block merozoite egress or invasion, whereas E-64 treatment blocked egress as evidenced by presence of intracellular schizont. Detection of extracellular merozoites (Fig. 3B) suggests an inhibitory effect of UCF 201 on the merozoite invasion process in a manner similar to that of the reference invasion inhibitor N-acetylglucosamine (Howard and Miller, 1981). Additionally, we conducted a flow cytometric analysis of the synchronized parasite culture following exposure to UCF 201. The 6 hpi culture at the ring stage was treated with UCF 201 at 5×EC50 and aliquots were withdrawn at 12 h intervals for flow cytrometric analysis following staining with YOYO-1 dye. As shown in Fig. 3C, the peaks on the right in control culture at 6 hpi represent singly, double or triply-infected RBC based on DNA content. Following maturation of the parasite the peaks merge as DNA content increases. At 42 hpi peaks begin to reappear as reinvasion occurs. At 54 hpi parasites are at the ring stage of the next growth cycle, peak height increases, and individual peaks reappear. In contrast, exposure to UCF 201 and artemisinin at the ring stage of maturation is blocked. Collectively, the above data suggest that UCF 201 may act by inhibiting parasite target(s) that are common to all major intra-erythrocytic forms of P. falciparum.
Fig. 2.

Activity Profile of UCF 201 on P. falciparum Asexual Stages. The effect of UCF 201 on parasite life cycle stage progression upon treatment at (A) 6 h, (B) 18 h, or (C) 30 h post-invasion time-points at 5×EC50 for 36 h. For the (D) 42 h post-invasion time-point the effect was followed for 24 h after compound addition. Giemsa-stained thin smears were prepared at 12 h intervals for microscopic evaluations. Microscopic images represent parasite stages after evaluating 1000 infected RBC.
Fig. 3.

(A) Effect of UCF 201 on Parasitemia When Treated at the Late Schizont/Segmenter Stage. Synchronized culture at 42 h post invasion was treated with 5×EC50 of the compound. Giemsa stained thin smears were prepared every 12 h for microscopic evaluations of parasitemia to quantify parasitemia. (B) Confocal plate micrograph of parasite phenotype following 24 h exposure at 42 hpi. Synchronized cultures were exposed to UCF 201 or reference compounds E-64, GlcNac, or artemisinin at 10 μM concentration. (C) Effect of UCF 201 on P. falciparum growth by flow cytometric analysis of YOYO-1 labeled cells. The synchronized culture was exposed to UCF 201 at 6 hpi with 5×EC50 of UCF 201. Plots represent cell count in y-axis versus FL1 channel representing DNA content.
To overcome the problems of drug-resistance in malaria, a major goal is to identify the next-generation of lead compounds that are acting early on parasite's developmental cycle with novel mechanism of action. The majority of the current antimalarial drugs target the digestive vacuole, mitochondrial electron transport chain, or apicoplast processes of the parasite (Baggish and Hill, 2002, Famin and Ginsburg, 2002, Krishna et al., 2004, Dahl and Rosenthal, 2007). Recent analysis of 10 widely used antimalarials show that none of the drugs were active in blocking merozoite invasion, whereas only artemisinin and artesunate demonstrated significant ring stage activity (Wilson et al., 2013). A major significance of our work is that the spirocyclic chromane scaffold inhibits early cellular processes of the parasite invasion as well as all development stages of the asexual development of the parasite. Activity of UCF 201 against the ring stage is noteworthy given that latent ring stage has been shown to promote in vitro resistance to dihydroartesunate (Hoshen et al., 2000, Teuscher et al., 2010). Inhibitory effect of UCF 201 against all stages of asexual life cycle of the parasite (Fig. 2A and B) makes it particularly useful for the treatment of acute infection where rapid clearance is needed. Because UCF 201 acts on all intraerythrocytic developmental forms including rings and merozoites, we can predict that the spirocyclic chromane chemotype action is likely to be distinct from known antimalarial compounds.
Our evaluation of in vivo antimalarial efficacy of UCF 201 using the murine malaria model infected with P. berghei ANKA strain demonstrated that the compound prolonged survivability >1.7 times (12 versus 7 days) compared to the control and delayed the increase in parasitemia (Fig. 4). The P. berghei mouse model is quite demanding; requires total clearance of parasites otherwise fatal parasitaemia recrudesce (Nallan et al., 2005). However, these results are the proof-of-concept of the utility of spirocyclic chromanes for malaria therapy. Given that this novel antiplasmodial scaffold exhibits excellent selectivity, acceptable physicochemical properties and broad activity on asexual developmental stages, we believe it will be a good candidate for hit to lead optimization.
Fig. 4.

Efficacy of UCF 201 in the Mouse Malaria Model. (A) Effect of UCF 201 on the survivability of P. berghei ANKA infected Balb/c mice. (B) Effect of UCF 201 on increase in parasitemia in the P. berghei ANKA infected mice. The experiments were conducted according to the approved protocol of UCF IACUC committee.
Acknowledgments
This study is supported by grants from NIH R03 AI117298 (DC) and R01 GM097118 (RM and DEK) and the Burnett School of Biomedical Sciences Director's Impact award.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ijpddr.2016.02.004.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- Ashley E.A., Dhorda M., Fairhurst R.M., Amaratunga C., Lim P., Suon S., Sreng S., Anderson J.M., Mao S., Sam B., Sopha C., Chuor C.M., Nguon C., Sovannaroth S., Pukrittayakamee S., Jittamala P., Chotivanich K., Chutasmit K., Suchatsoonthorn C., Runcharoen R., Hien T.T., Thuy-Nhien N.T., Thanh N.V., Phu N.H., Htut Y., Han K.T., Aye K.H., Mokuolu O.A., Olaosebikan R.R., Folaranmi O.O., Mayxay M., Khanthavong M., Hongvanthong B., Newton P.N., Onyamboko M.A., Fanello C.I., Tshefu A.K., Mishra N., Valecha N., Phyo A.P., Nosten F., Yi P., Tripura R., Borrmann S., Bashraheil M., Peshu J., Faiz M.A., Ghose A., Hossain M.A., Samad R., Rahman M.R., Hasan M.M., Islam A., Miotto O., Amato R., MacInnis B., Stalker J., Kwiatkowski D.P., Bozdech Z., Jeeyapant A., Cheah P.Y., Sakulthaew T., Chalk J., Intharabut B., Silamut K., Lee S.J., Vihokhern B., Kunasol C., Imwong M., Tarning J., Taylor W.J., Yeung S., Woodrow C.J., Flegg J.A., Das D., Smith J., Venkatesan M., Plowe C.V., Stepniewska K., Guerin P.J., Dondorp A.M., Day N.P., White N.J., Tracking Resistance to Artemisinin, C Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2014;371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdeef A. Physicochemical profiling (solubility, permeability and charge state) Curr. Top. Med. Chem. 2001;1:277–351. doi: 10.2174/1568026013395100. [DOI] [PubMed] [Google Scholar]
- Baggish A.L., Hill D.R. Antiparasitic agent atovaquone. Antimicrob. Agents Chemother. 2002;46:1163–1173. doi: 10.1128/AAC.46.5.1163-1173.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett T.N., Paguio M., Gligorijevic B., Seudieu C., Kosar A.D., Davidson E., Roepe P.D. Novel, rapid, and inexpensive cell-based quantification of antimalarial drug efficacy. Antimicrob. Agents Chemother. 2004;48:1807–1810. doi: 10.1128/AAC.48.5.1807-1810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman M.J. Malarial proteases and host cell egress: an 'emerging' cascade. Cell Microbiol. 2008;10:1925–1934. doi: 10.1111/j.1462-5822.2008.01176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillon A., Gorgette O., Mercereau-Puijalon O., Barale J.C. Screening and evaluation of inhibitors of Plasmodium falciparum merozoite egress and invasion using cytometry. Methods Mol. Biol. 2013;923:523–534. doi: 10.1007/978-1-62703-026-7_36. [DOI] [PubMed] [Google Scholar]
- Cordier C., Morton D., Murrison S., Nelson A., O'Leary-Steele C. Natural products as an inspiration in the diversity-oriented synthesis of bioactive compound libraries. Nat. Prod. Rep. 2008;25:719–737. doi: 10.1039/b706296f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory A.H., Owen T.C., Barltrop J.A., Cory J.G. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Comm. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- Cui W. WHO urges the phasing out of artemisinin based monotherapy for malaria to reduce resistance. Bmj. 2011;342:d2793. doi: 10.1136/bmj.d2793. [DOI] [PubMed] [Google Scholar]
- Dahl E.L., Rosenthal P.J. Multiple antibiotics exert delayed effects against the Plasmodium falciparum apicoplast. Antimicrob. Agents Chemother. 2007;51:3485–3490. doi: 10.1128/AAC.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecker A., Lehane A.M., Clain J., Fidock D.A. PfCRT and its role in antimalarial drug resistance. Trends Parasitol. 2012;28:504–514. doi: 10.1016/j.pt.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famin O., Ginsburg H. Differential effects of 4-aminoquinoline-containing antimalarial drugs on hemoglobin digestion in Plasmodium falciparum-infected erythrocytes. Biochem. Pharmacol. 2002;63:393–398. doi: 10.1016/s0006-2952(01)00878-4. [DOI] [PubMed] [Google Scholar]
- Gallup J.L., Sachs J.D. The economic burden of malaria. Am. J. Trop. Med. Hyg. 2001;64:85–96. doi: 10.4269/ajtmh.2001.64.85. [DOI] [PubMed] [Google Scholar]
- Greenwood D. Conflicts of interest: the genesis of synthetic antimalarial agents in peace and war. J. Antimicrob. Chemother. 1995;36:857–872. doi: 10.1093/jac/36.5.857. [DOI] [PubMed] [Google Scholar]
- Grimberg B.T., Mehlotra R.K. Expanding the antimalarial drug arsenal-now, but how? Pharm. (Basel) 2011;4:681–712. doi: 10.3390/ph4050681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshen M.B., Na-Bangchang K., Stein W.D., Ginsburg H. Mathematical modelling of the chemotherapy of Plasmodium falciparum malaria with artesunate: postulation of 'dormancy', a partial cytostatic effect of the drug, and its implication for treatment regimens. Parasitology. 2000;121(Pt 3):237–246. doi: 10.1017/s0031182099006332. [DOI] [PubMed] [Google Scholar]
- Howard R.J., Miller L.H. Invasion of erythrocytes by malaria merozoites: evidence for specific receptors involved in attachment and entry. Ciba Found. Symp. 1981;80:202–219. doi: 10.1002/9780470720639.ch13. [DOI] [PubMed] [Google Scholar]
- Janiszewski J.S., Rogers K.J., Whalen K.M., Cole M.J., Liston T.E., Duchoslav E., Fouda H.G. A high-capacity LC/MS system for the bioanalysis of samples generated from plate-based metabolic screening. Anal. Chem. 2001;73:1495–1501. doi: 10.1021/ac0013251. [DOI] [PubMed] [Google Scholar]
- Johnson J.D., Dennull R.A., Gerena L., Lopez-Sanchez M., Roncal N.E., Waters N.C. Assessment and continued validation of the malaria SYBR green I-based fluorescence assay for use in malaria drug screening. Antimicrob. Agents Chemother. 2007;51:1926–1933. doi: 10.1128/AAC.01607-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansy M., Senner F., Gubernator K. Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem. 1998;41:1007–1010. doi: 10.1021/jm970530e. [DOI] [PubMed] [Google Scholar]
- Klayman D.L. Qinghaosu (artemisinin): an antimalarial drug from China. Science. 1985;228:1049–1055. doi: 10.1126/science.3887571. [DOI] [PubMed] [Google Scholar]
- Kombarov R., Altieri A., Genis D., Kirpichenok M., Kochubey V., Rakitina N., Titarenko Z. BioCores: identification of a drug/natural product-based privileged structural motif for small-molecule lead discovery. Mol. Divers. 2010;14:193–200. doi: 10.1007/s11030-009-9157-5. [DOI] [PubMed] [Google Scholar]
- Krishna S., Uhlemann A.C., Haynes R.K. Artemisinins: mechanisms of action and potential for resistance. Drug Resist. Updat. Rev. Comment. Antimicrob. anticancer Chemother. 2004;7:233–244. doi: 10.1016/j.drup.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Lambros C., Vanderberg J.P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979;65:418–420. [PubMed] [Google Scholar]
- Li J.W., Vederas J.C. Drug discovery and natural products: end of an era or an endless frontier? Science. 2009;325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- Miotto O., Almagro-Garcia J., Manske M., Macinnis B., Campino S., Rockett K.A., Amaratunga C., Lim P., Suon S., Sreng S., Anderson J.M., Duong S., Nguon C., Chuor C.M., Saunders D., Se Y., Lon C., Fukuda M.M., Amenga-Etego L., Hodgson A.V., Asoala V., Imwong M., Takala-Harrison S., Nosten F., Su X.Z., Ringwald P., Ariey F., Dolecek C., Hien T.T., Boni M.F., Thai C.Q., Amambua-Ngwa A., Conway D.J., Djimde A.A., Doumbo O.K., Zongo I., Ouedraogo J.B., Alcock D., Drury E., Auburn S., Koch O., Sanders M., Hubbart C., Maslen G., Ruano-Rubio V., Jyothi D., Miles A., O'Brien J., Gamble C., Oyola S.O., Rayner J.C., Newbold C.I., Berriman M., Spencer C.C., McVean G., Day N.P., White N.J., Bethell D., Dondorp A.M., Plowe C.V., Fairhurst R.M., Kwiatkowski D.P. Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nat. Genet. 2013;45:648–655. doi: 10.1038/ng.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon S., Lee S., Kim H., Freitas-Junior L.H., Kang M., Ayong L., Hansen M.A. An image analysis algorithm for malaria parasite stage classification and viability quantification. PloS one. 2013;8:e61812. doi: 10.1371/journal.pone.0061812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray C.J., Rosenfeld L.C., Lim S.S., Andrews K.G., Foreman K.J., Haring D., Fullman N., Naghavi M., Lozano R., Lopez A.D. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet. 2012;379:413–431. doi: 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- Nallan L., Bauer K.D., Bendale P., Rivas K., Yokoyama K., Horney C.P., Pendyala P.R., Floyd D., Lombardo L.J., Williams D.K., Hamilton A., Sebti S., Windsor W.T., Weber P.C., Buckner F.S., Chakrabarti D., Gelb M.H., Van Voorhis W.C. Protein farnesyltransferase inhibitors exhibit potent antimalarial activity. J. Med. Chem. 2005;48:3704–3713. doi: 10.1021/jm0491039. [DOI] [PubMed] [Google Scholar]
- Ribaut C., Berry A., Chevalley S., Reybier K., Morlais I., Parzy D., Nepveu F., Benoit-Vical F., Valentin A. Concentration and purification by magnetic separation of the erythrocytic stages of all human Plasmodium species. Malar. J. 2008;7:45. doi: 10.1186/1475-2875-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieckmann K.H. The chequered history of malaria control: are new and better tools the ultimate answer? Ann. Trop. Med. Parasitol. 2006;100:647–662. doi: 10.1179/136485906X112185. [DOI] [PubMed] [Google Scholar]
- Rishton G.M. Molecular diversity in the context of leadlikeness: compound properties that enable effective biochemical screening. Curr. Opin. Chem. Biol. 2008;12:340–351. doi: 10.1016/j.cbpa.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Smilkstein M., Sriwilaijaroen N., Kelly J.X., Wilairat P., Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuscher F., Gatton M.L., Chen N., Peters J., Kyle D.E., Cheng Q. Artemisinin-induced dormancy in plasmodium falciparum: duration, recovery rates, and implications in treatment failure. J. Infect. Dis. 2010;202:1362–1368. doi: 10.1086/656476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager W., Jensen J.B. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Vasilevich N.I., Kombarov R.V., Genis D.V., Kirpichenok M.A. Lessons from natural products chemistry can offer novel approaches for synthetic chemistry in drug discovery. J. Med. Chem. 2012;55:7003–7009. doi: 10.1021/jm300344v. [DOI] [PubMed] [Google Scholar]
- Vyawahare D., Ghodke M., Nikalje A.P. Green synthesis and pharmacological screening of novel 1,5-benzothiazepines. Int. J. Pharm. Pharm. Sci. 2010;2:27–29. [Google Scholar]
- Wilson D.W., Langer C., Goodman C.D., McFadden G.I., Beeson J.G. Defining the timing of action of antimalarial drugs against Plasmodium falciparum. Antimicrob. Agents Chemother. 2013;57:1455–1467. doi: 10.1128/AAC.01881-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward R.B., Doering W.E. The total synthesis of quinine. J. Am. Chem. Soc. 1944;14:926–928. [Google Scholar]
- World Health Organization, UNICEF, World Health Organization Global Malaria Programme . World Health Organization; Geneva, Switzerland: 2014. World Malaria Report. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.