

Abstract
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) targeting mitochondria are major causative factors in disease pathogenesis. The mitochondrial permeability transition pore (PTP) is a mega-channel modulated by calcium and ROS/RNS modifications and it has been described to play a crucial role in many pathophysiological events since prolonged channel opening causes cell death. The recent identification that dimers of ATP synthase form the PTP and the fact that posttranslational modifications caused by ROS/RNS also affect cellular bioenergetics through the modulation of ATP synthase catalysis reveal a dual function of these modifications in the cells. Here, we describe mitochondria as a major site of production and as a target of ROS/RNS and discuss the pathophysiological conditions in which oxidative and nitrosative modifications modulate the catalytic and pore-forming activities of ATP synthase.
1. Introduction
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) play important physiological functions but can also cause extensive cellular damage, in a balance that is determined by their relative rates of formation and removal. Usually, these species are removed rapidly before they cause cell dysfunction and death. Oxidative/nitrosative stress generated by an imbalance between formation of ROS/RNS and antioxidant defense capacity can affect major cellular components, including lipids, proteins, carbohydrates, and DNA. Mitochondria are recognized as a critical site in the cell for the formation of ROS/RNS and as their target.
Mitochondrial processes are highly compartmentalized because of the existence of two limiting membranes allowing the selective localization of proteins, nucleotides, and coenzymes in the intermembrane and matrix spaces. The outer mitochondrial membrane (OMM) is the interface between mitochondria and the cell components and its permeabilization is essential to allow the release of mitochondrial proteins involved in apoptosis such as cytochrome c [1]. The inner mitochondrial membrane (IMM), whose permeability to solutes is controlled by highly specific transporters and tightly regulated channels, is the site of coupling between substrate oxidation and ATP synthesis in the process of oxidative phosphorylation. Mitochondria operate a sequence of energy conversion processes through which the exergonic flow of electrons along the respiratory complexes supports the endergonic pumping of protons from the matrix to the intermembrane space. The resulting proton motive force drives the rotation of the FO sector of ATP synthase leading to the synthesis of ATP in the F1 sector, but the electron flow through the respiratory chain also generates ROS/RNS. In addition, the mitochondrial permeability transition pore (PTP), a large-conductance channel, is also located at the level of the IMM and prolonged opening of this channel leads to mitochondrial swelling, rupture of the OMM, and cell death [2]. PTP opening is dependent on the presence of matrix calcium, but the threshold calcium load which is required is modulated by inducers of the pore such as oxidants [3]. Although the existence of the PTP was established as early as the 1970s [4–6], its molecular nature has been the subject of controversy as many potential components were ruled out by the use of targeted gene deletion in mice [2]. The only candidate remaining is cyclophilin D (CyPD), which was found to act not as a structural component of the pore but as a modulator whose binding to the PTP decreases the threshold calcium concentration necessary to induce permeability transition [7–10]. CyPD was shown to interact with the lateral stalk of the ATP synthase in mammals [11], a finding which was the basis for the characterization of the molecular structure of the PTP as formed by ATP synthase itself [12–16]. Genetic ablation of the Ppif gene (which encodes for CyPD) in the mouse or its displacement from the PTP by the treatment with cyclosporin A (CsA), a known inhibitor of the PTP, has been also used to demonstrate the important role of PTP in the pathophysiological mechanism of several diseases such as neurodegenerative diseases, muscular dystrophies, ischemia/reperfusion (I/R), and diabetes [2, 17, 18]. Besides the PTP, mitochondrial function and bioenergetics (including the modulation of the catalytic activity of ATP synthase) are also affected in most of these pathophysiological conditions and ROS/RNS are presumably involved as causative factors.
While numerous mechanisms of oxidant-induced injury have been identified, the impact of oxidants on the mitochondrial proteome has been investigated only recently. Oxidative or nitrosative stress may not only alter levels of mitochondrial proteins, but also induce posttranslational modifications of proteins. These modifications involve reversible changes at the level of cysteine, tyrosine, methionine, histidine, and tryptophan residues and irreversible protein carbonylation [19]. Thiol groups can be S-nitrosylated by nitric oxide (NO) or reversibly oxidized by ROS to form disulfide bonds or sulfenic acid; the latter can be further oxidized to sulfinic and sulfonic acids [20]. Sulfenic acid can also interact with glutathione to become glutathionylated. Tyrosine residues instead are target for peroxynitrite (ONOO−) which leads to irreversible formation of 3-nitrotyrosine. All these modifications lead to changes in protein structure and/or activity, thereby affecting their roles in cell function. In this review we discuss the role of mitochondria as a source and target of ROS/RNS during the switch between cell death and survival. We will focus on the effects of these reactive species on critical ATP synthase amino acid residues as part of the mechanism affecting ATP synthesis and/or permeability transition.
2. Mitochondria Are Sources of ROS/RNS
A number of sites responsible for ROS/RNS formation within the cell have been identified and shown to play a role in different pathologies. NADPH oxidases, xanthine oxidase, uncoupled nitric oxide synthase (NOS), and mitochondria are all relevant sources of ROS that in certain pathological conditions contribute to oxidative damage of cells and tissues. Indeed, these processes have been identified as disease-relevant enzymatic sources of ROS and their inhibition has been shown to afford protection in a number of diseases both in experimental models and in patients [21, 22].
2.1. Mitochondrial Sources of ROS
It is well accepted that mitochondria are a major source of ROS in the cell. These organelles contain several enzymes that catalyze ROS formation either as the obligatory product or as the result of an occasional, possibly undesired, reaction [23]. The best example of “accidental” ROS formation is represented by the mitochondrial respiratory chain. A small fraction of the electrons (about 0.1%) flowing through the respiratory chain is diverted causing the partial reduction of O2 into superoxide [24]. This process occurs at the level of the first three complexes where flavins or quinones are able to act as single electron donors. The electron detour at these sites is favored when flow is hampered downstream as a result of either protein alterations in respiratory complexes or their inhibition. Other mitochondrial enzymes, such as flavin containing glycerol-3-phosphate-, proline-, and dihydroorotate-dehydrogenase, and the electron transferring flavoprotein/ETF:Q oxidoreductase system of fatty acid β-oxidation have also been described as potential ROS producers [25]. Nevertheless, all these enzymes and respiratory complexes normally catalyze reactions required for energy metabolism, cell function, and viability maintenance, making it difficult to envision their inhibition as a potential therapeutic tool.
Mitochondria also contain other enzymes that may generate hydrogen peroxide (H2O2) as a direct and obligatory product. One such example is p66Shc, cytosolic adaptor protein that upon stress translocates to mitochondria where it catalyzes electron transfer from cytochrome c to oxygen, a process that can result in the formation of ROS [37]. Genetic deletion of p66Shc protects against I/R injury in mice [38, 39], obesity [40], and diabetic complications such as cardiomyopathy [41], nephropathy [42], delayed wound healing [43], pancreatic cell death [44], and endothelial dysfunction [45]. On the other hand, the observation that mice lacking p66Shc actually live shorter when exposed to natural conditions (low temperatures and food competition) [46] may suggest that p66Shc dependent ROS also serve a physiological role. NADPH oxidase 4 (Nox4), an enzyme belonging to the NADPH oxidase family, is another ROS generating enzyme that has been reported to localize in the plasma membrane and apparently also in the mitochondria, focal adhesions, nucleus, and endoplasmic reticulum [47]. This enzyme associates with p22phox for its activation; it is constitutively active, and, unlike other members of the Nox family, generates H2O2 rather than superoxide [47, 48]. However, the relevance of Nox4 for mitochondrial ROS generation remains controversial. A recent study has shown that Nox4 can (in principle) interact with complex I subunits, but under physiological conditions Nox4 protein or ROS formation could not be detected in kidney or heart mitochondria [49]. Other studies have shown that cardiac-specific targeting of Nox4 can be both protective and harmful in different models of cardiac pressure overload [50, 51]. Moreover, while certain studies reported Nox4 to be deleterious, contributing to mitochondrial dysfunction and several pathologies such as ischemic stroke [52], diabetic cardiomyopathy [53], vascular inflammation, and remodeling [54], others concluded that Nox4 might be vascular-protective rather than vascular-damaging [55]. These apparently contradictory findings reflect the need for further investigation in order to address the pathophysiological role and regulation of Nox4. Another major source of H2O2 in the mitochondria is monoamine oxidase (MAO). Activation of this enzyme, localized at the level of the OMM, leads to H2O2 formation and has been shown to contribute to neuronal disorders, such as Parkinson's (PD) or Alzheimer's disease (AD), most likely due to formation of ROS responsible for oxidative damage to neurons [56]. In recent years, the contribution of MAO to oxidative stress that ultimately leads to mitochondrial dysfunction and cell damage has been demonstrated also in the cardiovascular field where MAO inhibition has been shown to be protective in I/R injury, pressure overload, vascular damage, and diabetes [23, 57–62].
2.2. Mitochondrial Sources of RNS
In addition to ROS, cells are also capable of generating RNS. NOS exist in three isoforms (endothelial (eNOS), neuronal (nNOS), and inducible (iNOS)) which catalyze the conversion of L-arginine into citrulline and NO. This reaction also requires flavin adenine dinucleotide, flavin mononucleotide, tetrahydrobiopterin (BH4), heme, and calmodulin. These cofactors are important (e.g., when BH4 levels are limited) because NOS may become uncoupled and generate superoxide instead of NO [63, 64]. Another possibility is that, in conditions of high oxidative stress, NO and superoxide interact to generate ONOO−, a very reactive species capable of nitrating tyrosine residues, thus amplifying oxidative damage.
It is generally accepted that NO has protective effects on mitochondria although it remains unclear whether mitochondria actually possess a mitochondrial NOS [65–69]. Nevertheless, in addition to being generated by NOS, NO can be formed from dietary inorganic nitrite (NO2 −) and nitrate (NO3 −) [70]. Several in vitro studies have demonstrated that mitochondria are able to metabolize NO2 − into NO, since cytochrome c and respiratory chain complexes III and IV possess nitrite reductase activities that can be stimulated under hypoxic or acidic conditions [71–73]. Either way, the bioactivity of newly synthesized NO is rapidly terminated by its oxidation into NO2 − and NO3 −, thus completing the NO cycle. The protective effects exerted by NO on mitochondria are due to S-nitrosylation of critical cysteine residues. One such example is represented by the mechanism through which MitoSNO, a mitochondria selective S-nitrosylating agent, affords protection in cardiac I/R injury in vivo [74]. Administration of MitoSNO within the first minutes of reperfusion temporarily inhibits complex I and thus blocks the reverse electron flow that leads to generation of ROS, oxidative damage, and tissue necrosis [74, 75]. The inhibition of complex I is due to S-nitrosylation of a critical cysteine residue (Cys39) within the ND3 subunit, underlining the importance of (i) the reversibility of cysteine modification and (ii) mitochondria as both source and target of ROS/RNS. This latter notion is further supported by the observations that mice lacking Mn superoxide dismutase (SOD), enzyme responsible for conversion of superoxide into H2O2, develop ROS toxicity and dilated cardiomyopathy [76] and that targeting catalase expression in mitochondria affords beneficial effects in a number of pathologies [77–80].
3. The Role of ATP Synthase in Mitochondrial ROS Formation
Recent evidence suggests that ATP synthase activity can modulate ROS formation in mitochondria. Although not a direct source of ROS, ATP synthase is able to regulate energy metabolism and modulate pathways leading to ROS formation, cell death, and survival. A recent study by Ni et al. [81] very elegantly showed that α subunit is a target for calpain-1 in diabetic hearts, leading to its proteolytic degradation, reduction in ATP synthase activity, increase in mitochondrial superoxide formation, and diabetic cardiomyopathy in mice. Indeed, genetic inhibition of calpain-1 or upregulation of α subunit increased ATP synthase activity and reduced mitochondrial ROS generation and all the downstream changes occurring in diabetic hearts. It is likely that, besides insufficient ATP production that can directly contribute to myocardial dysfunction, disruption in ATP synthase activity leads to the accumulation of the electrons in the upstream complexes of the respiratory chain, promoting superoxide generation through complexes I and III [81].
Whereas inhibition of ATP synthase in normal cells leads to mitochondrial oxidative stress and cell death [81–83], most cancer cells are highly glycolytic and have adopted several molecular strategies to reduce oxidative phosphorylation including ATP synthase activity inhibition [84, 85], thus modulating the level of ROS in mitochondria. This does not only occur through downregulation of ATP synthase, but also through overexpression of its inhibitor protein (IF1) [86]. IF1 is the endogenous inhibitor of ATP synthase that reversibly binds to the enzyme. Its binding is promoted by low matrix pH and membrane potential, thereby limiting ATP hydrolysis and energy dissipation [87, 88]. In fact, its role has been extensively studied in myocardial ischemia and ischemic preconditioning (IPC), since IF1 seems to mediate ATP synthase inhibition and thus sparing of ATP during myocardial preconditioning [89–92]. Moreover, IF1 seems to regulate the oligomeric state of ATP synthase, increasing the density of cristae and formation of ATP synthase dimers [93–96]. Mitochondria from different types of carcinoma show a remarkable increase in IF1 expression [86] that has been linked to the inhibition of ATP synthase, glycolytic switch in energy metabolism, and production of ROS [97–99]. A critical role of the phosphorylation status of IF1 has been recently suggested in the modulation of ATP synthesis and respiration in tumors and heart [100]. ROS generated when ATP synthesis is inhibited are used as nuclear signals to initiate transcription of genes necessary to support tumor development [86, 97]. Indeed, IF1 triggers ROS-induced activation of transcription factors (such as NFkB and HIF1α) in cancer cells causing enhanced proliferation, invasion, and survival [97]. These findings highlight how cancer cells, as opposed to normal cells, rely on a subtle mechanism of redox equilibrium: on the one hand, ROS enhance proliferation and favor genomic instability by damaging DNA, while on the other hand, excess ROS is harmful as it could promote PTP opening and cell death [101]. Moreover, it is tempting to speculate that PTP might be differently modulated by IF1 and CyPD in cancer cells, as observed for CyPD in osteosarcoma and prostatic cell models [102]. This suggests that this different PTP modulation might render cancer cells more resistant to cell death. Nevertheless, the ROS signaling pathways triggered by IF1 overexpression in many other cancer types still remain to be elucidated.
4. Mitochondrial ROS/RNS Induce Posttranslational Modification of ATP Synthase Residues
ROS/RNS induced posttranslational modifications are known to modulate the catalytic activity of ATP synthase in several pathophysiological conditions. Matrix calcium levels and oxidative/nitrosative stress play a crucial role also in the modulation of PTP. Recent finding that ATP synthase dimers form the PTP [12] makes the identification of the sites involved in the two different functions of ATP synthase more feasible (Figure 1). The most frequently modified sites of ATP synthase possibly involved in mitochondrial pathophysiology are listed in Table 1.
Figure 1.

Schematic representation of ATP synthase and PTP. Dimers of ATP synthase that form the PTP are shown from a lateral view. F1 catalytic part of ATP synthase is from bovine crystal structure (PDB 2WSS, modified by PyMOL 1.3 software) and is composed of α, β, and γ subunits in red, yellow, and blue, respectively, as indicated in the left part of the dimer. FO and lateral stalk subunits are also indicated in the left part in pink, light-blue, and green regions. On the right part of the dimer arrows indicate the critical cysteine residues modified by ROS/RNS in pathophysiological conditions. Cysteines are highlighted in cyan.
Table 1.
Residues of ATP synthase modified by ROS/RNS in pathophysiology.
| Residue | Condition | Modification | Reference(s) |
|---|---|---|---|
| α-Cys201 | DHF | Oxidation, S-S bonds | [26] |
|
| |||
| α-Cys251 | DHF | Oxidation, S-S bonds | [26] |
| Rat brain and liver | S-glutathionylation | [27] | |
| Mouse heart overexpressing iNOS | S-glutathionylation | [28] | |
| Preconditioning and I/R | S-nitrosylation, | [29, 30] | |
| Aging-mouse skeletal muscles | Carbonylation, | [31] | |
| Rat brain mitochondria | Tyr-phosphorylation | [32] | |
|
| |||
| α subunit | PD, HtrA2 KO mouse | Truncation | [33] |
| γ-Cys78 | DHF | Oxidation, S-S bonds | [26] |
|
| |||
| OSCP-Cys118 | Bovine heart mitochondria | NEM incorporation | [34] |
| β-Tyr345 | Rat liver | S-nitrosylation | [35] |
| β-Tyr368 | Rat liver | S-nitrosylation | [35] |
| d-Trp | Human heart mitochondria | Oxidation | [36] |
Residues refer to bovine ATP synthase subunits without mitochondrial targeting sequence.
DHF: dyssynchronous heart failure; iNOS: inducible nitric oxide synthase; I/R: ischemia/reperfusion; PD: Parkinson's disease; KO: knockout; OSCP: oligomycin-sensitivity conferring protein; and NEM: N-ethylmaleimide.
4.1. Matrix Residues
Critical thiols facing the matrix are involved in the modulation of ATP synthase catalysis. H2O2 inactivates F1-ATPase activity from bovine heart through formation of iron-protein adducts [103]. ROS- and RNS-mediated modifications responsible for ATPase inactivation are mostly localized at the level of cysteine residues in the α and γ subunits [104]. A special feature of higher plant CF1-ATPase is a regulatory domain in the γ subunit consisting of three methionines and a cysteine that were identified by mass spectrometry to be oxidized by ROS. Reduction of the disulfide bonds of γ subunit elevates ATP hydrolysis and synthesis [105].
Although in mammalian cells the chloroplast redox-sensitive γ subunit region is not present, mitochondrial ATP synthase F1 domain might be a major site for oxidative/nitrosative posttranslational modifications. Garcia et al. [27] have reported that α subunit is S-glutathionylated under oxidative stress in rat brain or liver mitochondria leading to decreased ATPase activity. The same inhibitory effect due to α subunit S-glutathionylation was observed in transgenic mouse hearts overexpressing iNOS [28]. Sun et al. [29], on the other hand, reported inhibition of ATPase activity which is protective in preconditioned mouse hearts during myocardial ischemia due to α subunit S-nitrosylation and an additive cardioprotective effect of this posttranslational modification which causes desensitization of the PTP [30]. An age-associated decline of ATPase activity due to carbonylation of α subunit was also observed in mouse skeletal muscle [31]. An indirect effect of ROS on α subunit was also found following H2O2 treatment of mitochondria that caused tyrosine-phosphorylation of α (but not β) chain [32]. This is likely due to the inactivation of phosphatases mediated by H2O2 [106].
Two disulfide bonds were identified between cysteines of ATP synthase α and γ subunits and between those of α subunits in heart failure [26] and both S-glutathionylation and S-nitrosylation of α-cysteine 294 (corresponding to bovine Cys251, Figures 1 and 2) can prevent the formation of these disulfide bridges. Other selective targets of ROS in the matrix are tryptophan residues of d subunit of ATP synthase in mammals [36] or tryptophan 503 of α subunit in Podospora anserina [107]. In bovine heart mitochondria treated with ONOO−, 3-nitrotyrosine modified residues were identified at the level of β and d subunits of ATP synthase [108, 109]. Although it is not yet known which of the tyrosine residues present in this subunit were modified, one might speculate that Tyr345 and Tyr368 in the β subunit are involved, since they have been suggested to be a major target for nitrosative stress in rat liver under in vivo conditions [35] (Figure 3). Carbonylation at the level of β subunit was observed in Escherichia coli after treatment with H2O2 [110]. Whether sites targeted by ROS/RNS that modulate ATP synthase are also involved in PTP formation/modulation has not been clarified yet.
Figure 2.

Lateral view of a section of the catalytic core of ATP synthase (PDB 2WSS, modified by PyMOL 1.3 software) composed of α, β, and γ subunits in red, yellow, and blue, respectively. Critical cysteine residues subjected to posttranslational modifications are highlighted in cyan. Distances between α-Cys251 and α-Cys201 or α-Cys251 and γ-Cys78 are indicated by black dashed lines and are 12 Å or 61.5 Å, respectively.
Figure 3.
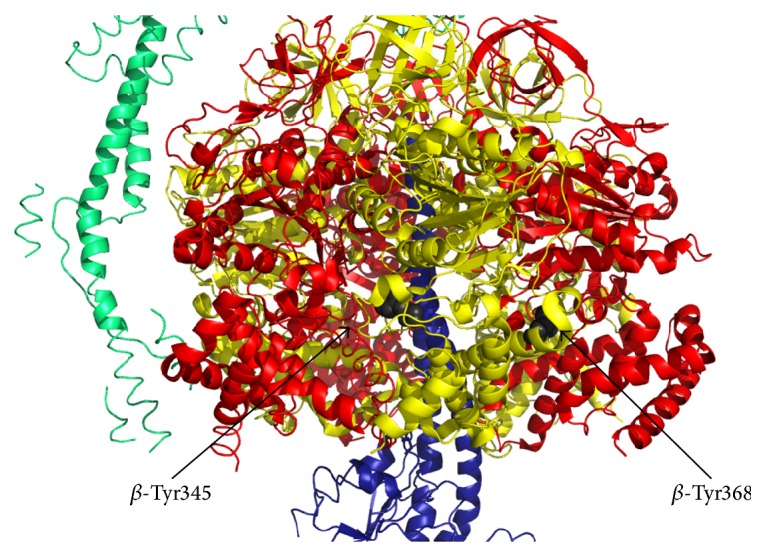
Lateral view of a section of the catalytic core of ATP synthase (PDB 2WSS, modified by PyMOL 1.3 software) composed of α, β, and γ subunits in red, yellow, and blue, respectively. Critical β-Tyr345 and β-Tyr368 residues that might be modified by RNS are highlighted in gray.
The first indication of the presence of ROS-sensitive modulatory thiols on the matrix-facing side of the PTP was revealed in isolated mitochondria through the use of chemical thiol-oxidants, such as diamide, arsenite anion (AsO), copper-o-phenanthroline, phenylarsine oxide (PhAsO), and β-hydroxybutyrate, or thiol-modifiers, such as N-ethylmaleimide (NEM) or monobromobimane (MBM) [111–114]. In these studies the authors proposed that the PTP can be modulated by oxidation-reduction effectors at two sites that can be distinguished experimentally. One site (which was called the “P site”) appears to be modulated through the oxidation-reduction state of pyridine nucleotides even when glutathione is fully reduced and can be blocked by NEM, but not by MBM. The other site (which was called the “S site”) coincides with the oxidation/reduction-sensitive dithiol(s) [111], and it can be activated by reaction with AsO or PhAsO. The S site can be blocked by both NEM and MBM. Irrespective of the precise mechanism by which glutathione and pyridine nucleotides affect the PTP, oxidative stress causes an increased probability of pore opening when the concentrations of reduced glutathione and pyridine nucleotides decrease.
Some of the above-mentioned oxidants and thiol-modifiers of the PTP sites are also known to be reagents effective on ATP synthase [115] and since the PTP is formed by ATP synthase dimers, an accurate analysis of the amino acid residues of ATP synthase might clarify the molecular localization for the S and P sensitive sites of the PTP. Moreover, the characterization of these residues could be useful to discriminate between the two effects of ROS/RNS on ATP synthase: the modulation of its catalytic activity and the induction of the PTP.
Upon treatment of beef heart mitochondria with radioactive NEM and dithiobis(nitrobenzoate), these radiolabeled compounds were incorporated into the oligomycin-sensitivity conferring protein (OSCP) subunit, which faces the matrix side, through its only cysteine (OSCP-Cys118 in Figure 1) without any detectable effect on the ATPase activity [34, 116]. Intriguingly, the OSCP subunit has been identified as the binding site for CyPD to ATP synthase [12], an interaction favored by phosphate [11]. In mouse liver mitochondria high phosphate (5 mM) protects PTP from the oxidative stress, something that is not observed in CyPD knockout (KO) mitochondria [8], suggesting that the binding of CyPD on OSCP, which is favored by phosphate, might affect PTP thiols availability. Taken together, these results suggest that the OSCP subunit in mammals might play a crucial modulatory role for PTP opening and ROS sensitivity. This hypothesis might explain the previous and never fully understood finding that overexpression of CyPD in HEK293 cells protects from oxidative stress and apoptosis [117].
4.2. Intermembrane Space/IMM Residues
Many possible sites of oxidation are present in the FO sector and in the noncatalytic subunits of ATP synthase in the IMM and facing the intermembrane space (Figure 1). These conserved cysteines are involved in dimer/oligomer formation.
Preferential interactions in yeast dimers occur through subunits e [118, 119], g [120], 6 [121], and 4 [122] and also through subunit h and the yeast-specific subunit i [123], most of which harbor cysteine residues. Among the mammalian corresponding subunits e, g, a, b, and F6 (Figure 1) only b presents a conserved cysteine residue. However, in both yeast [121, 124] and mammals [95, 125, 126], the stabilizing contribution of the different subunits seems to be additive. In yeast mutants lacking e and/or g subunits, the PTP was desensitized to opening [14]. These data support the hypothesis that dimerization of ATP synthase is necessary for PTP formation. Moreover, the effect of ROS is conserved from human to yeast and Drosophila [127], since dimers extracted from blue native gels and tested for their channel activity generated currents in the presence of oxidants in all these species [12, 14, 15]. In yeast, both e and g subunits harbor a cysteine residue (eCys28 and gCys75) and form e/g interactions in the dimerization interface through GXXXG motifs [117]. They also participate in the putative oligomerization interface through e/e and g/g interactions [124], as established in a yeast gCys75Ser/Leu109Cys mutant, which formed eCys28-eCys28 and gLeu109Cys-gLeu109Cys cross-links [118, 120]. A similar arrangement of e and g subunits in the IMM of bovine heart has been hypothesized based on cross-linking experiments [128]. The treatment of yeast mitochondria lacking e and/or g subunits with copper(II) chloride (CuCl2), which mimics the effect of oxidants, stabilized preexisting dimers by formation of disulfide bridges between adjacent monomers [14]. This suggests the involvement of species-specific cysteine residues located in subunits other than e and g that might stabilize dimers, as it was observed in subunit 6 in yeast [124] or factor B [129] and subunit b in bovine heart, whose modification also affects enzyme function [130–132]. Subunit b spans the membrane without contacting the c8-ring in the enzyme monomer, suggesting that two subunits b are in close proximity in the dimer [133].
It still remains to be defined whether these or other ATP synthase residues might coincide with cysteines of the PTP facing the intermembrane space showed to be sensitive to membrane-impermeant photooxidants in isolated mitochondria [134]. Moreover, their involvement in the modulation of the PTP in mitochondrial pathophysiology in humans remains to be addressed.
5. PTP Modulators as Targets for ROS/RNS
S-nitrosylation of CyPD cysteine 203 seems to be important for the modulation of the PTP during I/R injury [135–137], since its modification inhibits PTP opening. Moreover, the substitution of this residue with a serine desensitizes PTP in MEF cells exposed to oxidative stress similarly to CyPD ablation [137]. Indeed, cysteine 203 has been identified as a major cysteine residue susceptible to oxidation in human cells, leading to conformational changes of CyPD and induction of PTP [138]. Another possible effect of ROS on the modulation of the PTP is the direct phosphorylation of CyPD by mitochondrial glycogen synthase kinase 3 (mGSK3) [102, 139]. This kinase has been involved in the regulation of PTP in pathological conditions, such as cardiac I/R injury or hypoxia [140–144], in tumor cells [102, 139], and in cells lacking mtDNA [145]. ROS-dependent activation of mGSK3 enhances cell death in neurons, probably following PTP induction [146, 147], and this suggests that mGSK3 coordinates diverse signaling pathways to connect PTP opening with stress and survival signals by modulating ROS and/or calcium threshold of PTP opening.
Another recently identified interactor of CyPD that might be involved in the modulatory effect of ROS on the PTP is the IMM protein spastic paraplegia 7 (SPG7), but the mechanisms of action through which it could sensitize the PTP to opening in the presence of oxidative stress need to be clarified [148].
6. The Dual Role of ATP Synthase in Cardiac Disease
The cardiac muscle heavily depends on mitochondrial bioenergetics and metabolism for its function. This becomes particularly evident during I/R, a scenario in which low levels of oxygen lead to the inhibition of electron flow through the respiratory chain and thus an impairment of energy conservation and oxidative metabolism. The resulting loss in proton motive force prevents ADP phosphorylation to ATP at the level of ATP synthase, which rather works in “reverse mode” coupling ATP hydrolysis to proton pumping. The net result is that mitochondria no longer produce ATP and become very powerful in hydrolyzing glycolytic ATP [149]. IMM permeabilization due to the opening of the PTP leads to depolarization and worsening of ATP depletion and precipitates cell death [150]. Indeed, the crucial role of the pore opening in mediating I/R induced damage was extensively corroborated by the protective effects of CsA [151] and lack of injury in CyPD KO mice [7, 9]. The opening of the PTP was thought for years to occur due to mitochondrial calcium overload, but this hypothesis was questioned recently by the observation that MCU KO mice are not protected from I/R injury [152]. Nevertheless, recent reports showed that cardiac-specific ablation of MCU affords protection from I/R injury [153, 154]. Considering that MCU KO MEFs are equally susceptible to oxidative damage as their wild type counterparts [152], it is tempting to speculate that PTP opening might be induced by oxidative stress independently of mitochondrial calcium overload, either by directly targeting critical cysteine residues of ATP synthase to induce conformational changes and dimer formation or through oxidative changes of CyPD. Other amino acid residues might also be involved in the oxidative modulation of the enzyme activity and/or conformation, such as tyrosine nitration in the α subunit occurring in cardiac I/R injury [155] or tryptophan oxidation described at the level of subunits d and a [36]. The functional consequences resulting from these modifications are yet to be elucidated.
It is well accepted that low levels of ROS are key signals mediating physiological or cardioprotective responses, whereas high levels of ROS contribute to oxidative stress and cell or tissue damage. This dual effect has been named mitohormesis [156, 157]. One such example of beneficial effects of ROS is represented by IPC. IPC consists of brief periods of ischemia followed by brief periods of reperfusion, which prepare the heart and render cardiomyocytes more resistant to sustained ischemic insults. Although IPC involves complex mechanisms that are not completely understood, it has been established that ROS/RNS mediate signal transduction in the early phase of IPC through posttranslational modifications of redox-sensitive proteins [158]. Again, mitochondrial proteins come into play as both the source and the target of these species. More specifically, IPC resulted in the S-nitrosylation of subunit α [30]. S-nitrosylation has been suggested to be protective, shielding the cysteine residues from oxidation and thus protecting the proteins from irreversible oxidation occurring at the start of reperfusion. It has also been reported that S-nitrosoglutathione (GSNO) treatment, also protective against I/R injury, leads to S-nitrosylation of the α subunit and this results in decreased ATP hydrolytic activity [29]. Thus, in addition to shielding the cysteine residues from irreversible oxidative damage, S-nitrosylation might also afford protection by decreasing ATP synthase activity and therefore reduce the consumption of ATP following myocardial ischemia. S-nitrosylation of ATP synthase subunit α occurs in both IPC and postconditioning and is dependent on NOS activity [30, 159]. Moreover, cysteine 19 of the subunit ε was also found to be S-nitrosylated following postconditioning but the functional significance of this finding remains to be elucidated [159].
Oxidation and S-nitrosylation have also been shown to occur at the level of ATP synthase in a canine model of dyssynchronous heart failure (DHF) [26]. Indeed, DHF was correlated with disulfide bond formation in the ATP synthase complex occurring between either cysteine 294 of the neighboring α subunits or cysteine 294 of the α subunit and cysteine 103 of the γ subunit. This cross-linking was negatively correlated with ATP hydrolytic activity. Furthermore, cysteine 294 of the ATP synthase α subunit was also S-glutathionylated in DHF, another modification that correlated with a reduction in ATP synthase activity [104]. Interestingly, during chronic resynchronization therapy (CRT, the only clinically effective therapy for DHF) the disulfide bond at cysteine 294 was found to be reversed and partially replaced by S-nitrosylation, resulting in the recovery of ATPase activity. Indeed, the same group has shown that cysteine 294 is critical for ATP synthase function in vitro and that it may play a major role in redox regulation of ATP production acting as a redox-sensor [160]. Nevertheless, the S-nitrosylation of the α subunit and improvement in ATPase activity observed in this study are in contrast with the previously mentioned results from Sun et al. [29], showing that S-nitrosylation of ATP synthase α subunit in IPC or after GSNO treatment leads to a reduction of its hydrolytic activity. One cannot exclude that different cysteine residues were modified in the two aforementioned studies, a fact that could explain this discrepancy. Another possible explanation is that cross-link reversal and recovery of ATPase activity observed in CRT occur at a higher level compared to the extent of S-nitrosylation, suggesting that the majority of cysteine residues are actually free in CRT hearts [26]. On the other hand, the distances between the two cysteines 294 of neighboring α subunits or between α cysteine 294 and γ cysteine 103 are more than 5 Å (corresponding to α-Cys251 and γ-Cys78, respectively, in Figures 1 and 2), making it unlikely that these disulfide bonds actually occur in the assembled complex. Rather, these bonds could form in the misfolded/aggregated enzyme, suggesting that the reversal of cysteine cross-links observed in CRT may promote the correct assembly of the complex, thus resulting in improved ATPase activity. Of note, CuCl2 treatment of isolated ATP synthase complex from rat mitochondria in vitro showed that disulfide bonds can form also between α and OSCP subunits as well as between the two γ subunits and γ and OSCP [26]. The effect of these disulfide crossbridges on ATP synthase activity was not addressed, and whether these modifications could also occur in vivo still needs to be assessed. Further efforts should also be put in addressing the effect of these oxidative posttranslational modifications on PTP formation.
7. Neurodegenerative Diseases
Mitochondria have a central role in aging-related neurodegenerative diseases. Oxidative stress generated by mitochondria has been inversely correlated with longevity in model organisms [161, 162] and defects in mitochondrial bioenergetics have been implicated in a number of neurodegenerative diseases [163].
In vitro, ATP synthase is susceptible to ROS [103, 164, 165] and to oxidative/nitrosative stress associated with disorders of the central nervous system [166, 167] and aging [35, 168]. The fact that ROS/RNS modifications can alter the mitochondrial oxidative phosphorylation efficiency may explain the mitochondrial involvement in neurological diseases.
Moreover, the involvement of PTP in neurodegenerative diseases has been demonstrated by the use of CsA both in vitro and in vivo and the ablation in mouse models of its target, CyPD [17, 18]. CyPD plays an important dual function on the modulation of ATP synthase; on the one hand, it sensitizes PTP to matrix calcium, while, on the other hand, it inhibits both ATP hydrolysis and synthesis by 30% [11]. In cellular mechanisms of neurodegenerative diseases, when mitochondria are hydrolyzing ATP, CyPD binding and inhibition of ATP synthase can be an advantage avoiding ATP consumption and cell death, or a disadvantage in cases in which PTP is sensitized to calcium and the displacement of CyPD would promote cell survival. This is only one of the complex relationships between ATP synthase and PTP in neurodegeneration, and in this paragraph we attempt to summarize possible mechanisms linking ATP synthase/PTP and ROS/RNS modifications.
7.1. Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a progressive degeneration of motor neurons. In familial ALS, mutations have been found in Cu,Zn-SOD that are suggested to increase generation of ROS [169, 170], RNS, and nitrosylation [171]. Furthermore, Beal et al. [172] detected increased levels of nitrotyrosine staining in motor neurons of both sporadic ALS and familial ALS, suggesting that ONOO− mediated oxidative damage may play a role in both forms of the disease. Of note, as already mentioned, tyrosine modifications at the level of the β subunit of ATP synthase have been described to be modulatory of the catalytic ATPase activity in the presence of ROS/RNS [108, 109], even if their direct involvement in ALS remains to be addressed.
In light of the susceptibility of the mitochondrial respiratory chain to nitrosative stress, it is not surprising that mitochondrial function is impaired in ALS [173]. Spinal cord mitochondria in ALS mouse model display decreased calcium retention capacity long before the onset of motor weakness and neuronal death [174], and this was corrected by ablation of the Ppif gene which encodes CyPD [175]. In these mice, an improved mitochondrial ATP synthesis was matched by PTP inhibition and significant suppression of motor neuron death throughout disease, although survival was not improved confirming the role of ATP synthesis and permeability transition in ALS neuron cell death [176, 177]. Increased ATP synthesis in this mouse ALS model lacking CyPD could also be explained by our observation that ablation of CyPD increases the catalysis of ATP synthase [11].
7.2. Alzheimer's Disease
AD, the most common form of dementia in aged people, is characterized by deposition of amyloid plaques formed by the amyloid β peptide [178]. Amyloid β can be imported in mitochondria [179]. Several studies have observed activity changes in key mitochondrial enzymes in AD. While the exact mechanism for this loss of activity is unclear, evidence suggests that ROS/RNS production is increased in AD. Nitrotyrosine residues have been detected in postmortem Alzheimer's tissue but not in age-matched controls [180], indicating the presence of ONOO−. The induction of iNOS in cultured rat astrocytes causes NO-mediated neuronal death in a coculture system [181].
Amyloid β is also reported to stimulate glial NO production [182–184] and it has been shown to inhibit both purified complex IV [185] and complex IV in isolated brain mitochondria [186]. Additionally, in neuronal cultures amyloid β caused a loss of activity of all the mitochondrial complexes and a loss of mitochondrial integrity, due to PTP opening [187]. It has been shown that amyloid β oligomers alter calcium homeostasis [188].
Upon import in mitochondria, amyloid β interacts with CyPD and enhances PTP induction, since neurons from CyPD KO mice are protected from cell death induced by amyloid β dependent PTP opening [189]. Interestingly, a novel association with AD risk has been recently identified in the ATP synthase ATP5H locus, which encodes subunit d of the lateral stalk [190]. Intriguingly, in a proteomic analysis of human heart mitochondria in normal condition, a tryptophan residue at the level of d subunit was identified as a “hot spot” for oxidation [36], the latter being another important pathophysiological factor in AD. Moreover, in the filamentous fungus Podospora anserina, a model with a clear defined mitochondrial etiology of aging, another tryptophan residue on the α subunit of ATP synthase (Trp503) has been described to be crucial for the selective targeting of oxidative damage [107]. Although this residue is not conserved in unicellular fungi and higher eukaryotes, the example of P. anserina is instructive because the authors proposed that oxidized cysteine and methionine can be efficiently reduced by repair mechanisms, whereas tryptophan oxidation products are irreversible and have the potential to form markers detected by the mitochondrial quality control system.
7.3. Multiple Sclerosis
In multiple sclerosis (MS), the myelin sheath of neurons in the central nervous system is destroyed leading to axonal degeneration [191]. This is associated with mitochondrial calcium overload and bioenergetic dysfunction [192]. CyPD KO mice with autoimmune encephalomyelitis display a marked protection from axonal degeneration and a milder clinical picture despite a normal inflammatory response, thus suggesting that PTP might be involved in disease pathogenesis [193], even if other effects on bioenergetics caused by CyPD ablation cannot be excluded. A further indication of the important role played by the PTP in MS and its activation by ROS is provided by the observation that axonal damage of mice undergoing experimental autoimmune encephalomyelitis is reduced by genetic ablation of p66Shc [194]. Importantly, a proteomic study of experimental autoimmune encephalomyelitis identified the ATP synthase dimer-forming subunit e differentially expressed [195]. Moreover, mutations at the level of the genes encoding subunits a and A6L were observed in MS patients [196].
A large body of evidence exists implicating increased generation of RNS in MS. The observation that the concentration of NO2 − plus NO3 − in cerebrospinal fluid is elevated by 70% in MS patients supports this hypothesis [197]. Furthermore, increased iNOS activity and iNOS mRNA have been demonstrated in astrocytes associated with demyelinating lesions in postmortem MS brain [198] and in experimental models of demyelination [199, 200]. Nitrotyrosine residues indicating the presence of ONOO− have also been detected in MS brain [201]. Whether NO-mediated mitochondrial damage is the cause of the disease remains to be established; however, the first direct evidence of altered mitochondrial function in MS came from a study by Lu et al. [202]. These authors described a decrease of respiratory complex I activity and a compensatory increase in complex IV in MS. Major targets for nitrosative stress in rat liver under in vivo conditions are tyrosine 345 and tyrosine 368 in the β subunit of the mitochondrial ATP synthase that is suggested as an early marker of nitrosative stress and aging [35]. A comparison between α and β subunits allowed the speculation that the latter is more accessible for RNS modification and that the catalytic conformation of this subunit also affects tyrosine residues exposure to nitration (higher exposure to RNS modifications in the absence of bound nucleotide). Moreover, Ding et al. [203] demonstrated in ND4 mice (an in vivo model for MS) an impaired transport of ATP5b mRNA (which generates subunit β) to mitochondria. This caused decreased ATP synthesis in MS mice due to lower levels of this subunit and not to impaired ATP synthase assembly, as suggested by the authors. However, the possibility that tyrosine modifications in this subunit might affect enzyme activity cannot be excluded.
7.4. Parkinson's Disease
PD is caused by death of dopaminergic neurons in the substantia nigra pars compacta. A sensitization to PTP opening has been proposed as a major cause of neurodegeneration in several models of the disease characterized by altered homeostasis of intracellular calcium [204–206], including the forms caused by complex I inhibition [207, 208] and by inactivation of the Ser/Thr kinase PINK1 [209], in which changes in calcium storage capacity [210, 211], impairment of respiratory complex I [212], and altered mitophagy [213] are observed. Moreover, the PTP can be induced in dopaminergic neurons because of the inability to buffer increased intracellular ROS levels [214]. In a PD mouse model induced by parkin deficiency, state 3 and state 4 respiration rates were both affected indicating a more likely direct modulation of the respiratory chain compared to an effect on ATP synthase [215]. On the other hand, a mouse model lacking chaperone protein HtrA2 and showing a parkinsonian phenotype displays mitochondrial uncoupling at the level of ATP synthase and a truncated α subunit that might be involved in the neurodegeneration observed in these mice [33].
Postsynaptic density protein 95 (PSD-95) binds to neuronal nNOS and the neuroprotective effects of small-molecule inhibitors of this interaction were tested in an in vitro PD model. The observed protective effects were associated with suppressed mitochondrial dysfunction, as evidenced by decreased ROS generation, preserved ATP synthesis, and PTP inhibition [216]. Moreover, Darios et al. [217] demonstrated that in PC12 cells the overexpression of parkin protects from ceramide-induced swelling suggesting that parkin may act directly to prevent PTP mediated cell death, but the exact mechanism remains to be addressed.
8. Conclusions
Posttranslational modifications of ATP synthase due to ROS/RNS generation might play a dual role by promoting cell death or survival depending on their relative effects on mitochondrial ATP synthase catalysis and PTP. We have illustrated how ATP synthase is the target for oxidative/nitrosative modifications that affect its activity and might promote formation of the PTP. Identification of specific residues involved in the latter event is still lacking and will help to elucidate the mechanisms that mediate the role of ATP synthase in modulating cell survival or death. Finally, we discussed how changes in the ATP synthase activity regulate mitochondrial ROS formation and thus may represent an attractive strategy for the treatment of pathologies such as cancer.
Acknowledgments
The authors thank Paolo Bernardi, Fabio Di Lisa, and Andrea Rasola for the critical reading of the paper and helpful discussions. The authors also thank Manuela Antoniel, University of Padova, for the generous help in the use of PyMOL 1.3 software. Valentina Giorgio's research activities are supported by the Telethon Grant GGP14187.
Abbreviations
- AD:
Alzheimer's disease
- ALS:
Amyotrophic lateral sclerosis
- AsO:
Arsenite anion
- BH4:
Tetrahydrobiopterin
- CRT:
Chronic resynchronization therapy
- CsA:
Cyclosporin A
- CuCl2:
Copper(II) chloride
- CyPD:
Cyclophilin D
- DHF:
Dyssynchronous heart failure
- eNOS:
Endothelial nitric oxide synthase
- GSNO:
S-Nitrosoglutathione
- H2O2:
Hydrogen peroxide
- IF1:
Inhibitor protein
- IMM:
Inner mitochondrial membrane
- iNOS:
Inducible nitric oxide synthase
- IPC:
Ischemic preconditioning
- I/R:
Ischemia/reperfusion
- KO:
Knockout
- MAO:
Monoamine oxidase
- MBM:
Monobromobimane
- mGSK3:
Mitochondrial glycogen synthase kinase 3
- PTP:
Mitochondrial permeability transition pore
- MS:
Multiple sclerosis
- NEM:
N-Ethylmaleimide
- nNOS:
Neuronal nitric oxide synthase
- NO:
Nitric oxide
- NO2−:
Nitrite
- NO3−:
Nitrate
- Nox4:
NADPH oxidase 4
- OMM:
Outer mitochondrial membrane
- ONOO−:
Peroxynitrite
- OSCP:
Oligomycin-sensitivity conferring protein
- PD:
Parkinson's disease
- PhAsO:
Phenylarsine oxide
- RNS:
Reactive nitrogen species
- ROS:
Reactive oxygen species
- SOD:
Superoxide dismutase
- SPG7:
Spastic paraplegia 7.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Gustafsson Å. B., Gottlieb R. A. Bcl-2 family members and apoptosis, taken to heart. The American Journal of Physiology—Cell Physiology. 2007;292(1):C45–C51. doi: 10.1152/ajpcell.00229.2006. [DOI] [PubMed] [Google Scholar]
- 2.Bernardi P., Rasola A., Forte M., Lippe G. The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiological Reviews. 2015;95(4):1111–1155. doi: 10.1152/physrev.00001.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le-Quoc K., Le-Quoc D. Crucial role of sulfhydryl groups in the mitochondrial inner membrane structure. The Journal of Biological Chemistry. 1985;260(12):7422–7428. [PubMed] [Google Scholar]
- 4.Haworth R. A., Hunter D. R. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Archives of Biochemistry and Biophysics. 1979;195(2):460–467. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- 5.Hunter D. R., Haworth R. A. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Archives of Biochemistry and Biophysics. 1979;195(2):453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- 6.Hunter D. R., Haworth R. A. The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Archives of Biochemistry and Biophysics. 1979;195(2):468–477. doi: 10.1016/0003-9861(79)90373-4. [DOI] [PubMed] [Google Scholar]
- 7.Baines C. P., Kaiser R. A., Purcell N. H., et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434(7033):658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 8.Basso E., Fante L., Fowlkes J., Petronilli V., Forte M. A., Bernardi P. Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. The Journal of Biological Chemistry. 2005;280(19):18558–18561. doi: 10.1074/jbc.c500089200. [DOI] [PubMed] [Google Scholar]
- 9.Nakagawa T., Shimizu S., Watanabe T., et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434(7033):652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 10.Schinzel A. C., Takeuchi O., Huang Z., et al. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(34):12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giorgio V., Bisetto E., Soriano M. E., et al. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. The Journal of Biological Chemistry. 2009;284(49):33982–33988. doi: 10.1074/jbc.m109.020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giorgio V., von Stockum S., Antoniel M., et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(15):5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonora M., Bononi A., De Marchi E., et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle. 2013;12(4):674–683. doi: 10.4161/cc.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carraro M., Giorgio V., Sileikyte J., et al. Channel formation by yeast F-ATP synthase and the role of dimerization in the mitochondrial permeability transition. The Journal of Biological Chemistry. 2014;289(23):15980–15985. doi: 10.1074/jbcc114.559633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Stockum S., Giorgio V., Trevisan E., et al. F-ATPase of Drosophila melanogaster forms 53-picosiemen (53-pS) channels responsible for mitochondrial Ca2+-induced Ca2+ release. The Journal of Biological Chemistry. 2015;290(8):4537–4544. doi: 10.1074/jbc.c114.629766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alavian K. N., Dworetzky S. I., Bonanni L., et al. The mitochondrial complex V—associated large-conductance inner membrane current is regulated by cyclosporine and dexpramipexole. Molecular Pharmacology. 2015;87(1):1–8. doi: 10.1124/mol.114.095661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giorgio V., Soriano M. E., Basso E., et al. Cyclophilin D in mitochondrial pathophysiology. Biochimica et Biophysica Acta (BBA)—Bioenergetics. 2010;1797(6-7):1113–1118. doi: 10.1016/j.bbabio.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elrod J. W., Molkentin J. D. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circulation Journal. 2013;77(5):1111–1122. doi: 10.1253/circj.CJ-13-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butterfield D. A., Dalle-Donne I. Redox proteomics: from protein modifications to cellular dysfunction and disease. Mass Spectrometry Reviews. 2014;33(1):1–6. doi: 10.1002/mas.21404. [DOI] [PubMed] [Google Scholar]
- 20.Steinberg S. F. Oxidative stress and sarcomeric proteins. Circulation Research. 2013;112(2):393–405. doi: 10.1161/CIRCRESAHA.111.300496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Casas A. I., Dao V. T., Daiber A., et al. Reactive oxygen-related diseases: therapeutic targets and emerging clinical indications. Antioxidants & Redox Signaling. 2015;23(14):1171–1185. doi: 10.1089/ars.2015.6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dao V. T., Casas A. I., Maghzal G. J., et al. Pharmacology and clinical drug candidates in redox medicine. Antioxidants & Redox Signaling. 2015;23(14):1113–1129. doi: 10.1089/ars.2015.6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaludercic N., Mialet-Perez J., Paolocci N., Parini A., Di Lisa F. Monoamine oxidases as sources of oxidants in the heart. Journal of Molecular and Cellular Cardiology. 2014;73:34–42. doi: 10.1016/j.yjmcc.2013.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy M. P. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brand M. D. The sites and topology of mitochondrial superoxide production. Experimental Gerontology. 2010;45(7-8):466–472. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang S.-B., Foster D. B., Rucker J., O'Rourke B., Kass D. A., Van Eyk J. E. Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circulation Research. 2011;109(7):750–757. doi: 10.1161/circresaha.111.246124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia J., Han D., Sancheti H., Yap L.-P., Kaplowitz N., Cadenas E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. Journal of Biological Chemistry. 2010;285(51):39646–39654. doi: 10.1074/jbc.m110.164160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.West M. B., Hill B. G., Xuan Y.-T., Bhatnagar A. Protein glutathiolation by nitric oxide: an intracellular mechanism regulating redox protein modification. The FASEB Journal. 2006;20(10):1715–1717. doi: 10.1096/fj.06-5843fje. [DOI] [PubMed] [Google Scholar]
- 29.Sun J., Morgan M., Shen R.-F., Steenbergen C., Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circulation Research. 2007;101(11):1155–1163. doi: 10.1161/circresaha.107.155879. [DOI] [PubMed] [Google Scholar]
- 30.Sun J., Nguyen T., Aponte A. M., et al. Ischaemic preconditioning preferentially increases protein S-nitrosylation in subsarcolemmal mitochondria. Cardiovascular Research. 2015;106(2):227–236. doi: 10.1093/cvr/cvv044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das N., Jana C. K. Age-associated oxidative modifications of mitochondrial α-subunit of F1 ATP synthase from mouse skeletal muscles. Free Radical Research. 2015;49(8):954–961. doi: 10.3109/10715762.2015.1017477. [DOI] [PubMed] [Google Scholar]
- 32.Augereau O., Claverol S., Boudes N., et al. Identification of tyrosine-phosphorylated proteins of the mitochondrial oxidative phosphorylation machinery. Cellular and Molecular Life Sciences. 2005;62(13):1478–1488. doi: 10.1007/s00018-005-5005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plun-Favreau H., Burchell V. S., Holmström K. M., et al. HtrA2 deficiency causes mitochondrial uncoupling through the F(1)F(0)-ATP synthase and consequent ATP depletion. Cell Death and Disease. 2012;3, article e335 doi: 10.1038/cddis.2012.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dupuis A., Lunardi J., Issartel J.-P., Vignais P. V. Interactions between the oligomycin sensitivity conferring protein (OSCP) and beef heart mitochondrial F1ATPase. 2. Identification of the interacting F1 subunits by cross-linking. Biochemistry. 1985;24(3):734–739. doi: 10.1021/bi00324a030. [DOI] [PubMed] [Google Scholar]
- 35.Haynes V., Traaseth N. J., Elfering S., Fujisawa Y., Giulivi C. Nitration of specific tyrosines in FoF1 ATP synthase and activity loss in aging. American Journal of Physiology—Endocrinology and Metabolism. 2010;298(5):E978–E987. doi: 10.1152/ajpendo.00739.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor S. W., Fahy E., Murray J., Capaldi R. A., Ghosh S. S. Oxidative post-translational modification of tryptophan residues in cardiac mitochondrial proteins. Journal of Biological Chemistry. 2003;278(22):19587–19590. doi: 10.1074/jbc.c300135200. [DOI] [PubMed] [Google Scholar]
- 37.Giorgio M., Migliaccio E., Orsini F., et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122(2):221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 38.Carpi A., Menabò R., Kaludercic N., Pelicci P., Di Lisa F., Giorgio M. The cardioprotective effects elicited by p66Shc ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochimica et Biophysica Acta—Bioenergetics. 2009;1787(7):774–780. doi: 10.1016/j.bbabio.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 39.Spescha R. D., Shi Y., Wegener S., et al. Deletion of the ageing gene p66Shc reduces early stroke size following ischaemia/reperfusion brain injury. European Heart Journal. 2013;34(2):96–103. doi: 10.1093/eurheartj/ehs331. [DOI] [PubMed] [Google Scholar]
- 40.Ciciliot S., Albiero M., Menegazzo L., et al. p66Shc deletion or deficiency protects from obesity but not metabolic dysfunction in mice and humans. Diabetologia. 2015;58(10):2352–2360. doi: 10.1007/s00125-015-3667-8. [DOI] [PubMed] [Google Scholar]
- 41.Rota M., LeCapitaine N., Hosoda T., et al. Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. Circulation Research. 2006;99(1):42–52. doi: 10.1161/01.RES.0000231289.63468.08. [DOI] [PubMed] [Google Scholar]
- 42.Menini S., Amadio L., Oddi G., et al. Deletion of p66Shc longevity gene protects against experimental diabetic glomerulopathy by preventing diabetes-induced oxidative stress. Diabetes. 2006;55(6):1642–1650. doi: 10.2337/db05-1477. [DOI] [PubMed] [Google Scholar]
- 43.Fadini G. P., Albiero M., Menegazzo L., et al. The redox enzyme p66Shc contributes to diabetes and ischemia-induced delay in cutaneous wound healing. Diabetes. 2010;59(9):2306–2314. doi: 10.2337/db09-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Natalicchio A., Tortosa F., Labarbuta R., et al. The p66(Shc) redox adaptor protein is induced by saturated fatty acids and mediates lipotoxicity-induced apoptosis in pancreatic beta cells. Diabetologia. 2015;58(6):1260–1271. doi: 10.1007/s00125-015-3563-2. [DOI] [PubMed] [Google Scholar]
- 45.Camici G. G., Schiavoni M., Francia P., et al. Genetic deletion of p66Shc adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(12):5217–5222. doi: 10.1073/pnas.0609656104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giorgio M., Berry A., Berniakovich I., et al. The p66Shc knocked out mice are short lived under natural condition. Aging Cell. 2012;11(1):162–168. doi: 10.1111/j.1474-9726.2011.00770.x. [DOI] [PubMed] [Google Scholar]
- 47.Touyz R. M., Montezano A. C. Vascular Nox4: a multifarious NADPH oxidase. Circulation Research. 2012;110(9):1159–1161. doi: 10.1161/circresaha.112.269068. [DOI] [PubMed] [Google Scholar]
- 48.Martyn K. D., Frederick L. M., von Loehneysen K., Dinauer M. C., Knaus U. G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cellular Signalling. 2006;18(1):69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 49.Hirschhäuser C., Bornbaum J., Reis A., et al. NOX4 in mitochondria: yeast two-hybrid-based interaction with complex I without relevance for basal reactive oxygen species? Antioxidants & Redox Signaling. 2015;23(14):1106–1112. doi: 10.1089/ars.2014.6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang M., Brewer A. C., Schröder K., et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(42):18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuroda J., Ago T., Matsushima S., Zhai P., Schneider M. D., Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(35):15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kleinschnitz C., Grund H., Wingler K., et al. Post-stroke inhibition of induced NADPH Oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biology. 2010;8(9) doi: 10.1371/journal.pbio.1000479.e1000479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maalouf R. M., Eid A. A., Gorin Y. C., et al. Nox4-derived reactive oxygen species mediate cardiomyocyte injury in early type 1 diabetes. The American Journal of Physiology—Cell Physiology. 2012;302(3):C597–C604. doi: 10.1152/ajpcell.00331.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sorescu D., Weiss D., Lassègue B., et al. Superoxide production and expression of Nox family proteins in human atherosclerosis. Circulation. 2002;105(12):1429–1435. doi: 10.1161/01.CIR.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 55.Schröder K., Zhang M., Benkhoff S., et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circulation Research. 2012;110(9):1217–1225. doi: 10.1161/circresaha.112.267054. [DOI] [PubMed] [Google Scholar]
- 56.Youdim M. B. H., Edmondson D., Tipton K. F. The therapeutic potential of monoamine oxidase inhibitors. Nature Reviews Neuroscience. 2006;7(4):295–309. doi: 10.1038/nrn1883. [DOI] [PubMed] [Google Scholar]
- 57.Bianchi P., Kunduzova O., Masini E., et al. Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation. 2005;112(21):3297–3305. doi: 10.1161/CIRCULATIONAHA.104.528133. [DOI] [PubMed] [Google Scholar]
- 58.Pchejetski D., Kunduzova O., Dayon A., et al. Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circulation Research. 2007;100(1):41–49. doi: 10.1161/01.res.0000253900.66640.34. [DOI] [PubMed] [Google Scholar]
- 59.Kaludercic N., Carpi A., Nagayama T., et al. Monoamine oxidase B prompts mitochondrial and cardiac dysfunction in pressure overloaded hearts. Antioxidants and Redox Signaling. 2014;20(2):267–280. doi: 10.1089/ars.2012.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaludercic N., Takimoto E., Nagayama T., et al. Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circulation Research. 2010;106(1):193–202. doi: 10.1161/circresaha.109.198366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sturza A., Leisegang M. S., Babelova A., et al. Monoamine oxidases are mediators of endothelial dysfunction in the mouse aorta. Hypertension. 2013;62(1):140–146. doi: 10.1161/HYPERTENSIONAHA.113.01314. [DOI] [PubMed] [Google Scholar]
- 62.Umbarkar P., Singh S., Arkat S., Bodhankar S. L., Lohidasan S., Sitasawad S. L. Monoamine oxidase-A is an important source of oxidative stress and promotes cardiac dysfunction, apoptosis, and fibrosis in diabetic cardiomyopathy. Free Radical Biology and Medicine. 2015;87:263–273. doi: 10.1016/j.freeradbiomed.2015.06.025. [DOI] [PubMed] [Google Scholar]
- 63.Takimoto E., Champion H. C., Li M., et al. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. The Journal of Clinical Investigation. 2005;115(5):1221–1231. doi: 10.1172/jci200521968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takimoto E., Kass D. A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007;49(2):241–248. doi: 10.1161/01.HYP.0000254415.31362.a7. [DOI] [PubMed] [Google Scholar]
- 65.Brookes P. S. Mitochondrial nitric oxide synthase. Mitochondrion. 2004;3(4):187–204. doi: 10.1016/j.mito.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 66.Venkatakrishnan P., Nakayasu E. S., Almeida I. C., Miller R. T. Absence of nitric-oxide synthase in sequentially purified rat liver mitochondria. The Journal of Biological Chemistry. 2009;284(30):19843–19855. doi: 10.1074/jbc.m109.003301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Venkatakrishnan P., Nakayasu E. S., Almeida I. C., Miller R. T. Arginase activity in mitochondria—an interfering factor in nitric oxide synthase activity assays. Biochemical and Biophysical Research Communications. 2010;394(3):448–452. doi: 10.1016/j.bbrc.2009.10.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Elfering S. L., Sarkela T. M., Giulivi C. Biochemistry of mitochondrial nitric-oxide synthase. The Journal of Biological Chemistry. 2002;277(41):38079–38086. doi: 10.1074/jbc.m205256200. [DOI] [PubMed] [Google Scholar]
- 69.Forner F., Kumar C., Luber C. A., Fromme T., Klingenspor M., Mann M. Proteome differences between brown and white fat mitochondria reveal specialized metabolic functions. Cell Metabolism. 2009;10(4):324–335. doi: 10.1016/j.cmet.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 70.Figueira T. R., Barros M. H., Camargo A. A., et al. Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxidants and Redox Signaling. 2013;18(16):2029–2074. doi: 10.1089/ars.2012.4729. [DOI] [PubMed] [Google Scholar]
- 71.Kozlov A. V., Staniek K., Nohl H. Nitrite reductase activity is a novel function of mammalian mitochondria. FEBS Letters. 1999;454(1-2):127–130. doi: 10.1016/S0014-5793(99)00788-7. [DOI] [PubMed] [Google Scholar]
- 72.Nohl H., Staniek K., Kozlov A. V. The existence and significance of a mitochondrial nitrite reductase. Redox Report. 2005;10(6):281–286. doi: 10.1179/135100005X83707. [DOI] [PubMed] [Google Scholar]
- 73.Basu S., Azarova N. A., Font M. D., et al. Nitrite reductase activity of cytochrome c. The Journal of Biological Chemistry. 2008;283(47):32590–32597. doi: 10.1074/jbc.m806934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chouchani E. T., Methner C., Nadtochiy S. M., et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nature Medicine. 2013;19(6):753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chouchani E. T., Pell V. R., Gaude E., et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515(7527):431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li Y., Huang T.-T., Carlson E. J., et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genetics. 1995;11(4):376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 77.Dai D.-F., Chen T., Wanagat J., et al. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell. 2010;9(4):536–544. doi: 10.1111/j.1474-9726.2010.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dai D.-F., Santana L. F., Vermulst M., et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119(21):2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee H.-Y., Choi C. S., Birkenfeld A. L., et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metabolism. 2010;12(6):668–674. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schriner S. E., Linford N. J., Martin G. M., et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308(5730):1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 81.Ni R., Zheng D., Xiong S., et al. Mitochondrial calpain-1 disrupts ATP synthase and induces superoxide generation in type-1 diabetic hearts: a novel mechanism contributing to diabetic cardiomyopathy. Diabetes. 2016;65(1):255–268. doi: 10.2337/db15-0963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Johnson K. M., Chen X., Boitano A., Swenson L., Opipari A. W., Jr., Glick G. D. Identification and validation of the mitochondrial F1F0-ATPase as the molecular target of the immunomodulatory benzodiazepine Bz-423. Chemistry and Biology. 2005;12(4):485–496. doi: 10.1016/j.chembiol.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 83.Santamaría G., Martínez-Diez M., Fabregat I., Cuezva J. M. Efficient execution of cell death in non-glycolytic cells requires the generation of ROS controlled by the activity of mitochondrial H+-ATP synthase. Carcinogenesis. 2006;27(5):925–935. doi: 10.1093/carcin/bgi315. [DOI] [PubMed] [Google Scholar]
- 84.Gaude E., Frezza C. Defects in mitochondrial metabolism and cancer. Cancer & Metabolism. 2014;2, article 10 doi: 10.1186/2049-3002-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martínez-Reyes I., Cuezva J. M. The H(+)-ATP synthase: a gate to ROS-mediated cell death or cell survival. Biochimica et Biophysica Acta—Bioenergetics. 2014;1837(7):1099–1112. doi: 10.1016/j.bbabio.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 86.Sánchez-Aragó M., Formentini L., Martínez-Reyes I., et al. Expression, regulation and clinical relevance of the ATPase inhibitory factor 1 in human cancers. Oncogenesis. 2013;2, article e46 doi: 10.1038/oncsis.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cabezon E., Butler P. J. G., Runswick M. J., Walker J. E. Modulation of the oligomerization state of the bovine F1-ATPase inhibitor protein, IF1, by pH. The Journal of Biological Chemistry. 2000;275(33):25460–25464. doi: 10.1074/jbc.m003859200. [DOI] [PubMed] [Google Scholar]
- 88.Gledhill J. R., Montgomery M. G., Leslie A. G. W., Walker J. E. How the regulatory protein, IF1, inhibits F1-ATPase from bovine mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(40):15671–15676. doi: 10.1073/pnas.0707326104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Di Pancrazio F., Mavelli I., Isola M., et al. In vitro and in vivo studies of F0F1ATP synthase regulation by inhibitor protein IF1 in goat heart. Biochimica et Biophysica Acta. 2004;1659(1):52–62. doi: 10.1016/j.bbabio.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 90.Ylitalo K., Ala-Rämi A., Vuorinen K., et al. Reversible ischemic inhibition of F1F0-ATPase in rat and human myocardium. Biochimica et Biophysica Acta—Bioenergetics. 2001;1504(2-3):329–339. doi: 10.1016/s0005-2728(00)00261-9. [DOI] [PubMed] [Google Scholar]
- 91.Green D. W., Grover G. J. The IF1 inhibitor protein of the mitochondrial F1F0-ATPase. Biochimica et Biophysica Acta (BBA)—Bioenergetics. 2000;1458(2-3):343–355. doi: 10.1016/s0005-2728(00)00085-2. [DOI] [PubMed] [Google Scholar]
- 92.Rouslin W., Broge C. W. Mechanisms of ATP conservation during ischemia in slow and fast heart rate hearts. The American Journal of Physiology—Cell Physiology. 1993;264(1):C209–C216. doi: 10.1152/ajpcell.1993.264.1.C209. [DOI] [PubMed] [Google Scholar]
- 93.Campanella M., Casswell E., Chong S., et al. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1 . Cell Metabolism. 2008;8(1):13–25. doi: 10.1016/j.cmet.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 94.García J. J., Morales-Ríos E., Cortés-Hernández P., Rodríguez-Zavala J. S. The inhibitor protein (IF1) promotes dimerization of the mitochondrial F1F0-ATP synthase. Biochemistry. 2006;45(42):12695–12703. doi: 10.1021/bi060339j. [DOI] [PubMed] [Google Scholar]
- 95.Bisetto E., Comelli M., Salzano A. M., et al. Proteomic analysis of F1F0-ATP synthase super-assembly in mitochondria of cardiomyoblasts undergoing differentiation to the cardiac lineage. Biochimica et Biophysica Acta. 2013;1827(7):807–816. doi: 10.1016/j.bbabio.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 96.Cabezón E., Arechaga I., Jonathan P., Butler G., Walker J. E. Dimerization of bovine F1-ATPase by binding the inhibitor protein, IF1. Journal of Biological Chemistry. 2000;275(37):28353–28355. doi: 10.1074/jbc.c000427200. [DOI] [PubMed] [Google Scholar]
- 97.Formentini L., Sánchez-Aragó M., Sánchez-Cenizo L., Cuezva J. M. C. The mitochondrial ATPase inhibitory factor 1 triggers a ROS-mediated retrograde prosurvival and proliferative response. Molecular Cell. 2012;45(6):731–742. doi: 10.1016/j.molcel.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 98.Sánchez-Aragó M., Formentini L., Cuezva J. M. Mitochondria-mediated energy adaption in cancer: the H+-ATP synthase-geared switch of metabolism in human tumors. Antioxidants & Redox Signaling. 2013;19(3):285–298. doi: 10.1089/ars.2012.4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sánchez-Cenizo L., Formentini L., Aldea M., et al. Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a warburg phenotype. Journal of Biological Chemistry. 2010;285(33):25308–25313. doi: 10.1074/jbc.m110.146480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Garcia-Bermudez J., Sanchez-Arago M., Soldevilla B., Del Arco A., Nuevo-Tapioles C., Cuezva J. M. PKA phosphorylates the ATPase inhibitory factor 1 and inactivates its capacity to bind and inhibit the mitochondrial H+-ATP synthase. Cell Reports. 2015;12(12):2143–2155. doi: 10.1016/j.celrep.2015.08.052. [DOI] [PubMed] [Google Scholar]
- 101.Gorrini C., Harris I. S., Mak T. W. Modulation of oxidative stress as an anticancer strategy. Nature Reviews Drug Discovery. 2013;12(12):931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 102.Rasola A., Sciacovelli M., Chiara F., Pantic B., Brusilow W. S., Bernardi P. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(2):726–731. doi: 10.1073/pnas.0912742107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lippe G., Comelli M., Mazzilis D., Sala F. D., Mavelli I. The inactivation of mitochondrial F1 ATPase by H2O2 is mediated by iron ions not tightly bound in the protein. Biochemical and Biophysical Research Communications. 1991;181(2):764–770. doi: 10.1016/0006-291x(91)91256-c. [DOI] [PubMed] [Google Scholar]
- 104.Wang S.-B., Murray C. I., Chung H. S., Van Eyk J. E. Redox regulation of mitochondrial ATP synthase. Trends in Cardiovascular Medicine. 2013;23(1):14–18. doi: 10.1016/j.tcm.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Buchert F., Schober Y., Römpp A., Richter M. L., Forreiter C. Reactive oxygen species affect ATP hydrolysis by targeting a highly conserved amino acid cluster in the thylakoid ATP synthase γ subunit. Biochimica et Biophysica Acta. 2012;1817(11):2038–2048. doi: 10.1016/j.bbabio.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 106.Kaludercic N., Deshwal S., Di Lisa F. Reactive oxygen species and redox compartmentalization. Frontiers in Physiology. 2014;5, article 285 doi: 10.3389/fphys.2014.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rexroth S., Poetsch A., Rögner M., et al. Reactive oxygen species target specific tryptophan site in the mitochondrial ATP synthase. Biochimica et Biophysica Acta. 2012;1817(2):381–387. doi: 10.1016/j.bbabio.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 108.Murray J., Taylor S. W., Zhang B., Ghosh S. S., Capaldi R. A. Oxidative damage to mitochondrial complex I due to peroxynitrite: identification of reactive tyrosines by mass spectrometry. Journal of Biological Chemistry. 2003;278(39):37223–37230. doi: 10.1074/jbc.m305694200. [DOI] [PubMed] [Google Scholar]
- 109.Nesci S., Ventrella V., Trombetti F., Pirini M., Pagliarani A. Preferential nitrite inhibition of the mitochondrial FF-ATPase activities when activated by Ca in replacement of the natural cofactor Mg. Biochimica et Biophysica Acta. 2015;1860(2):345–353. doi: 10.1016/j.bbagen.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 110.Tamarit J., Cabiscol E., Ros J. Identification of the major oxidatively damaged proteins in Escherichia coli cells exposed to oxidative stress. Journal of Biological Chemistry. 1998;273(5):3027–3032. doi: 10.1074/jbc.273.5.3027. [DOI] [PubMed] [Google Scholar]
- 111.Petronilli V., Costantini P., Scorrano L., Colonna R., Passamonti S., Bernardi P. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. Journal of Biological Chemistry. 1994;269(24):16638–16642. [PubMed] [Google Scholar]
- 112.Costantini P., Chernyak B. V., Petronilli V., Bernardi P. Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. Journal of Biological Chemistry. 1996;271(12):6746–6751. doi: 10.1074/jbc.271.12.6746. [DOI] [PubMed] [Google Scholar]
- 113.Costantini P., Chernyak B. V., Petronilli V., Bernardi P. Selective inhibition of the mitochondrial permeability transition pore at the oxidation-reduction sensitive dithiol by monobromobimane. FEBS Letters. 1995;362(2):239–242. doi: 10.1016/0014-5793(95)00256-9. [DOI] [PubMed] [Google Scholar]
- 114.Costantini P., Colonna R., Bernardi P. Induction of the mitochondrial permeability transition by N-ethylmaleimide depends on secondary oxidation of critical thiol groups. Potentiation by copper-ortho-phenanthroline without dimerization of the adenine nucleotide translocase. Biochimica et Biophysica Acta. 1998;1365(3):385–392. doi: 10.1016/s0005-2728(98)00090-5. [DOI] [PubMed] [Google Scholar]
- 115.Hong S., Pedersen P. L. ATP synthase and the actions of inhibitors utilized to study its roles in human health, disease, and other scientific areas. Microbiology and Molecular Biology Reviews. 2008;72(4):590–641. doi: 10.1128/mmbr.00016-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dupuis A., Issartel J.-P., Lunardi J., Satre M., Vignais P. V. Interactions between the oligomycin sensitivity conferring protein (OSCP) and beef heart mitochondrial F1-ATPase. 1. Study of the binding parameters with a chemically radiolabeled OSCP. Biochemistry. 1985;24(3):728–733. doi: 10.1021/bi00324a029. [DOI] [PubMed] [Google Scholar]
- 117.Lin D.-T., Lechleiter J. D. Mitochondrial targeted cyclophilin D protects cells from cell death by peptidyl prolyl isomerization. The Journal of Biological Chemistry. 2002;277(34):31134–31141. doi: 10.1074/jbc.m112035200. [DOI] [PubMed] [Google Scholar]
- 118.Arselin G., Giraud M.-F., Dautant A., et al. The GxxxG motif of the transmembrane domain of subunit e is involved in the dimerization/oligomerization of the yeast ATP synthase complex in the mitochondrial membrane. European Journal of Biochemistry. 2003;270(8):1875–1884. doi: 10.1046/j.1432-1033.2003.03557.x. [DOI] [PubMed] [Google Scholar]
- 119.Everard-Gigot V., Dunn C. D., Dolan B. M., Brunner S., Jensen R. E., Stuart R. A. Functional analysis of subunit e of the F1Fo-ATP synthase of the yeast Saccharomyces cerevisiae: importance of the N-terminal membrane anchor region. Eukaryotic Cell. 2005;4(2):346–355. doi: 10.1128/ec.4.2.346-355.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bustos D. M., Velours J. The modification of the conserved GXXXG motif of the membrane-spanning segment of subunit g destabilizes the supramolecular species of yeast ATP synthase. The Journal of Biological Chemistry. 2005;280(32):29004–29010. doi: 10.1074/jbc.m502140200. [DOI] [PubMed] [Google Scholar]
- 121.Wittig I., Velours J., Stuart R., Schägger H. Characterization of domain interfaces in monomeric and dimeric ATP synthase. Molecular and Cellular Proteomics. 2008;7(5):995–1004. doi: 10.1074/mcp.M700465-MCP200. [DOI] [PubMed] [Google Scholar]
- 122.Spannagel C., Vaillier J., Arselin G., Graves P.-V., Grandier-Vazeille X., Velours J. Evidence of a subunit 4 (subunit b) dimer in favor of the proximity of ATP synthase complexes in yeast inner mitochondrial membrane. Biochimica et Biophysica Acta—Biomembranes. 1998;1414(1-2):260–264. doi: 10.1016/s0005-2736(98)00174-6. [DOI] [PubMed] [Google Scholar]
- 123.Paumard P., Arselin G., Vaillier J., et al. Two ATP synthases can be linked through subunits i in the inner mitochondrial membrane of Saccharomyces cerevisiae . Biochemistry. 2002;41(33):10390–10396. doi: 10.1021/bi025923g. [DOI] [PubMed] [Google Scholar]
- 124.Habersetzer J., Ziani W., Larrieu I., et al. ATP synthase oligomerization: from the enzyme models to the mitochondrial morphology. International Journal of Biochemistry and Cell Biology. 2013;45(1):99–105. doi: 10.1016/j.biocel.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 125.Habersetzer J., Larrieu I., Priault M., et al. Human F1F0 ATP synthase, mitochondrial ultrastructure and OXPHOS impairment: a (super-)complex matter? PLoS ONE. 2013;8(10) doi: 10.1371/journal.pone.0075429.e75429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wittig I., Meyer B., Heide H., et al. Assembly and oligomerization of human ATP synthase lacking mitochondrial subunits a and A6L. Biochimica et Biophysica Acta—Bioenergetics. 2010;1797(6-7):1004–1011. doi: 10.1016/j.bbabio.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 127.Azzolin L., von Stockum S., Basso E., Petronilli V., Forte M. A., Bernardi P. The mitochondrial permeability transition from yeast to mammals. FEBS Letters. 2010;584(12):2504–2509. doi: 10.1016/j.febslet.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Belogrudov G. I., Tomich J. M., Hatefi Y. Membrane topography and near-neighbor relationships of the mitochondrial ATP synthase subunits e, f, and g. The Journal of Biological Chemistry. 1996;271(34):20340–20345. doi: 10.1074/jbc.271.34.20340. [DOI] [PubMed] [Google Scholar]
- 129.Joshi S., Hughes J. B. Inhibition of coupling factor B activity by cadmium ion, arsenite-2,3-dimercaptopropanol, and phenylarsine oxide, and preferential reactivation by dithiols. The Journal of Biological Chemistry. 1981;256(21):11112–11116. [PubMed] [Google Scholar]
- 130.Dabbeni-Sala F., Lippe G., Sorgato M. C. Structural and functional modifications induced by diamide on the F0 sector of the mammalian ATP synthase. FEBS Letters. 1991;281(1-2):47–50. doi: 10.1016/0014-5793(91)80355-7. [DOI] [PubMed] [Google Scholar]
- 131.Lippe G., Dabbeni Sala F., Sorgato M. C. ATP synthase complex from beef heart mitochondria. Role of the thiol group of the 25-kDa subunit of F(o) in the coupling mechanism between F(o) and F1. The Journal of Biological Chemistry. 1988;263(35):18627–18634. [PubMed] [Google Scholar]
- 132.Zanotti F., Guerrieri F., Capozza G., Fiermonte M., Berden J., Papa S. Role of F0 and F1 subunits in the gating and coupling function of mitochondrial H+-ATP synthase. The effect of dithiol reagents. European Journal of Biochemistry. 1992;208(1):9–16. doi: 10.1111/j.1432-1033.1992.tb17153.x. [DOI] [PubMed] [Google Scholar]
- 133.Baker L. A., Watt I. N., Runswick M. J., Walker J. E., Rubinstein J. L. Arrangement of subunits in intact mammalian mitochondrial ATP synthase determined by cryo-EM. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(29):11675–11680. doi: 10.1073/pnas.1204935109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Petronilli V., Šileikyte J., Zulian A., et al. Switch from inhibition to activation of the mitochondrial permeability transition during hematoporphyrin-mediated photooxidative stress. Unmasking pore-regulating external thiols. Biochimica et Biophysica Acta—Bioenergetics. 2009;1787(7):897–904. doi: 10.1016/j.bbabio.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 135.Kohr M. J., Aponte A. M., Sun J., et al. Characterization of potential S-nitrosylation sites in the myocardium. The American Journal of Physiology—Heart and Circulatory Physiology. 2011;300(4):H1327–H1335. doi: 10.1152/ajpheart.00997.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kohr M. J., Sun J., Aponte A., et al. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circulation Research. 2011;108(4):418–426. doi: 10.1161/CIRCRESAHA.110.232173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nguyen T. T., Stevens M. V., Kohr M., Steenbergen C., Sack M. N., Murphy E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. The Journal of Biological Chemistry. 2011;286(46):40184–40192. doi: 10.1074/jbc.m111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Linard D., Kandlbinder A., Degand H., Morsomme P., Dietz K.-J., Knoops B. Redox characterization of human cyclophilin D: identification of a new mammalian mitochondrial redox sensor? Archives of Biochemistry and Biophysics. 2009;491(1-2):39–45. doi: 10.1016/j.abb.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 139.Traba J., Del Arco A., Duchen M. R., Szabadkai G., Satrústegui J. SCaMC-1 promotes cancer cell survival by desensitizing mitochondrial permeability transition via ATP/ADP-mediated matrix Ca2+ buffering. Cell Death and Differentiation. 2012;19(4):650–660. doi: 10.1038/cdd.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chanoit G., Lee S., Xi J., et al. Exogenous zinc protects cardiac cells from reperfusion injury by targeting mitochondrial permeability transition pore through inactivation of glycogen synthase kinase-3β . The American Journal of Physiology—Heart and Circulatory Physiology. 2008;295(3):H1227–H1233. doi: 10.1152/ajpheart.00610.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Juhaszova M., Zorov D. B., Yaniv Y., Nuss H. B., Wang S., Sollott S. J. Role of glycogen synthase kinase-3β in cardioprotection. Circulation Research. 2009;104(11):1240–1252. doi: 10.1161/circresaha.109.197996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Miura T., Miki T. GSK-3β, a therapeutic target for cardiomyocyte protection. Circulation Journal. 2009;73(7):1184–1192. doi: 10.1253/circj.cj-09-0284. [DOI] [PubMed] [Google Scholar]
- 143.Miura T., Tanno M. Mitochondria and GSK-3β in cardioprotection against ischemia/reperfusion injury. Cardiovascular Drugs and Therapy. 2010;24(3):255–263. doi: 10.1007/s10557-010-6234-z. [DOI] [PubMed] [Google Scholar]
- 144.Ohori K., Miura T., Tanno M., et al. Ser9 phosphorylation of mitochondrial GSK-3β is a primary mechanism of cardiomyocyte protection by erythropoietin against oxidant-induced apoptosis. The American Journal of Physiology—Heart and Circulatory Physiology. 2008;295(5):H2079–H2086. doi: 10.1152/ajpheart.00092.2008. [DOI] [PubMed] [Google Scholar]
- 145.McCubrey J. A., Steelman L. S., Chappell W. H., et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochimica et Biophysica Acta—Molecular Cell Research. 2007;1773(8):1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.King T. D., Clodfelder-Miller B., Barksdale K. A., Bijur G. N. Unregulated mitochondrial GSK3β activity results in NADH: ubiquinone oxidoreductase deficiency. Neurotoxicity Research. 2008;14(4):367–382. doi: 10.1007/bf03033861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Petit-Paitel A., Brau F., Cazareth J., Chabry J. Involvment of cytosolic and mitochondrial GSK-3β in mitochondrial dysfunction and neuronal cell death of MPTP/MPP+-treated neurons. PLoS ONE. 2009;4(5) doi: 10.1371/journal.pone.0005491.e5491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shanmughapriya S., Rajan S., Hoffman N., et al. SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Molecular Cell. 2015;60(1):47–62. doi: 10.1016/j.molcel.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bernardi P., Di Lisa F., Fogolari F., Lippe G. From ATP to PTP and back: a dual function for the mitochondrial ATP synthase. Circulation Research. 2015;116(11):1850–1862. doi: 10.1161/circresaha.115.306557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Di Lisa F., Bernardi P. Modulation of mitochondrial permeability transition in ischemia-reperfusion injury of the heart. Advantages and limitations. Current Medicinal Chemistry. 2015;22(20):2480–2487. doi: 10.2174/0929867322666150530210005. [DOI] [PubMed] [Google Scholar]
- 151.Griffiths E. J., Halestrap A. P. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. Journal of Molecular and Cellular Cardiology. 1993;25(12):1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 152.Pan X., Liu J., Nguyen T., et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nature Cell Biology. 2013;15(12):1464–1472. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kwong J. Q., Lu X., Correll R. N., et al. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Reports. 2015;12(1):15–22. doi: 10.1016/j.celrep.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Luongo T. S., Lambert J. P., Yuan A., et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Reports. 2015;12(1):23–34. doi: 10.1016/j.celrep.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liu B., Tewari A. K., Zhang L., et al. Proteomic analysis of protein tyrosine nitration after ischemia reperfusion injury: mitochondria as the major target. Biochimica et Biophysica Acta. 2009;1794(3):476–485. doi: 10.1016/j.bbapap.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ristow M., Schmeisser K. Mitohormesis: promoting health and lifespan by increased levels of reactive oxygen species (ROS) Dose-Response. 2014;12(2):288–341. doi: 10.2203/dose-response.13-035.ristow. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yun J., Finkel T. Mitohormesis. Cell Metabolism. 2014;19(5):757–766. doi: 10.1016/j.cmet.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Otani H. Reactive oxygen species as mediators of signal transduction in ischemic preconditioning. Antioxidants and Redox Signaling. 2004;6(2):449–469. doi: 10.1089/152308604322899521. [DOI] [PubMed] [Google Scholar]
- 159.Tong G., Aponte A. M., Kohr M. J., Steenbergen C., Murphy E., Sun J. Postconditioning leads to an increase in protein S-nitrosylation. American Journal of Physiology—Heart and Circulatory Physiology. 2014;306(6):H825–H832. doi: 10.1152/ajpheart.00660.2013.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chung H. S., Wang S.-B., Venkatraman V., Murray C. I., Van Eyk J. E. Cysteine oxidative posttranslational modifications: emerging regulation in the cardiovascular system. Circulation Research. 2013;112(2):382–392. doi: 10.1161/circresaha.112.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sohal R. S., Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273(5271):59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Balaban R. S., Nemoto S., Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 163.Beal M. F., Hyman B. T., Koroshetz W. Do defects in mitochondrial energy metabolism underlie the pathology of neurodegenerative diseases? Trends in Neurosciences. 1993;16(4):125–131. doi: 10.1016/0166-2236(93)90117-5. [DOI] [PubMed] [Google Scholar]
- 164.Comelli M., Lippe G., Mavelli I. Differentiation potentiates oxidant injury to mitochondria by hydrogen peroxide in Friend's erythroleukemia cells. FEBS Letters. 1994;352(1):71–75. doi: 10.1016/0014-5793(94)00882-5. [DOI] [PubMed] [Google Scholar]
- 165.Zhang Y., Marcillat O., Giulivi C., Ernster L., Davies K. J. A. The oxidative inactivation of mitochondrial electron transport chain components and ATPase. Journal of Biological Chemistry. 1990;265(27):16330–16336. [PubMed] [Google Scholar]
- 166.Poon H. F., Calabrese V., Calvani M., Butterfield D. A. Proteomics analyses of specific protein oxidation and protein expression in aged rat brain and its modulation by l-acetylcarnitine: insights into the mechanisms of action of this proposed therapeutic agent for CNS disorders associated with oxidative stress. Antioxidants and Redox Signaling. 2006;8(3-4):381–394. doi: 10.1089/ars.2006.8.381. [DOI] [PubMed] [Google Scholar]
- 167.Sultana R., Poon H. F., Cai J., et al. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiology of Disease. 2006;22(1):76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 168.Groebe K., Krause F., Kunstmann B., et al. Differential proteomic profiling of mitochondria from Podospora anserina, rat and human reveals distinct patterns of age-related oxidative changes. Experimental Gerontology. 2007;42(9):887–898. doi: 10.1016/j.exger.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 169.Grosskreutz J., Van Den Bosch L., Keller B. U. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium. 2010;47(2):165–174. doi: 10.1016/j.ceca.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 170.Kawamata H., Manfredi G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mechanisms of Ageing and Development. 2010;131(7-8):517–526. doi: 10.1016/j.mad.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Beckman J. S., Carson M., Smith C. D., Koppenol W. H. ALS, SOD and peroxynitrite. Nature. 1993;364(6438):p. 584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- 172.Beal M. F., Ferrante R. J., Browne S. E., Matthews R. T., Kowall N. W., Brown R. H., Jr. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Annals of Neurology. 1997;42(4):644–654. doi: 10.1002/ana.410420416. [DOI] [PubMed] [Google Scholar]
- 173.Collard J.-F., Cote F., Julien J.-P. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature. 1995;375(6526):61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- 174.Damiano M., Starkov A. A., Petri S., et al. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. Journal of Neurochemistry. 2006;96(5):1349–1361. doi: 10.1111/j.1471-4159.2006.03619.x. [DOI] [PubMed] [Google Scholar]
- 175.Parone P. A., Da Cruz S., Han J. S., et al. Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. Journal of Neuroscience. 2013;33(11):4657–4671. doi: 10.1523/jneurosci.1119-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pasinelli P., Brown R. H. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nature Reviews Neuroscience. 2006;7(9):710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 177.Boillée S., Vande Velde C., Cleveland D. W. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52(1):39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 178.Haass C., Selkoe D. J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nature Reviews Molecular Cell Biology. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 179.Hansson Petersen C. A., Alikhani N., Behbahani H., et al. The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(35):13145–13150. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Smith M. A., Richey Harris P. L., Sayre L. M., Beckman J. S., Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. Journal of Neuroscience. 1997;17(8):2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hu J., Castets F., Guevara J. L., Van Eldiki L. J. S100β stimulates inducible nitric oxide synthase activity and mRNA levels in rat cortical astrocytes. The Journal of Biological Chemistry. 1996;271(5):2543–2547. doi: 10.1074/jbc.271.5.2543. [DOI] [PubMed] [Google Scholar]
- 182.Rossi F., Bianchini E. Synergistic induction of nitric oxide by β-amyloid and cytokines in astrocytes. Biochemical and Biophysical Research Communications. 1996;225(2):474–478. doi: 10.1006/bbrc.1996.1197. [DOI] [PubMed] [Google Scholar]
- 183.Wallace M. N., Geddes J. G., Farquhar D. A., Masson M. R. Nitric oxide synthase in reactive astrocytes adjacent to β-amyloid plaques. Experimental Neurology. 1997;144(2):266–272. doi: 10.1006/exnr.1996.6373. [DOI] [PubMed] [Google Scholar]
- 184.Akama K. T., Albanese C., Pestell R. G., Van Eldik L. J. Amyloid β-peptide stimulates nitric oxide production in astrocytes through an NFκB-dependent mechanism. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(10):5795–5800. doi: 10.1073/pnas.95.10.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Casley C. S., Canevari L., Land J. M., Clark J. B., Sharpe M. A. β-Amyloid inhibits integrated mitochondrial respiration and key enzyme activities. Journal of Neurochemistry. 2002;80(1):91–100. doi: 10.1046/j.0022-3042.2001.00681.x. [DOI] [PubMed] [Google Scholar]
- 186.Canevari L., Clark J. B., Bates T. E. β-Amyloid fragment 25-35 selectively decreases complex IV activity in isolated mitochondria. FEBS Letters. 1999;457(1):131–134. doi: 10.1016/s0014-5793(99)01028-5. [DOI] [PubMed] [Google Scholar]
- 187.Casley C. S., Land J. M., Sharpe M. A., Clark J. B., Duchen M. R., Canevari L. β-Amyloid fragment 25-35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiology of Disease. 2002;10(3):258–267. doi: 10.1006/nbdi.2002.0516. [DOI] [PubMed] [Google Scholar]
- 188.Demuro A., Parker I., Stutzmann G. E. Calcium signaling and amyloid toxicity in Alzheimer disease. The Journal of Biological Chemistry. 2010;285(17):12463–12468. doi: 10.1074/jbc.r109.080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Du H., Guo L., Fang F., et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nature Medicine. 2008;14(10):1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Boada M., Antúnez C., Ramírez-Lorca R., et al. ATP5H/KCTD2 locus is associated with Alzheimer's disease risk. Molecular Psychiatry. 2014;19(6):682–687. doi: 10.1038/mp.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Stys P. K., Zamponi G. W., van Minnen J., Geurts J. J. G. Will the real multiple sclerosis please stand up? Nature Reviews Neuroscience. 2012;13(7):507–514. doi: 10.1038/nrn3275. [DOI] [PubMed] [Google Scholar]
- 192.Frohman E. M., Racke M. K., Raine C. S. Multiple sclerosis—the plaque and its pathogenesis. The New England Journal of Medicine. 2006;354(9):942–955. doi: 10.1056/nejmra052130. [DOI] [PubMed] [Google Scholar]
- 193.Forte M., Gold B. G., Marracci G., et al. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(18):7558–7563. doi: 10.1073/pnas.0702228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Savino C., Pelicci P., Giorgio M. The P66Shc/mitochondrial permeability transition pore pathway determines neurodegeneration. Oxidative Medicine and Cellular Longevity. 2013;2013:7. doi: 10.1155/2013/719407.719407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Azzam S., Broadwater L., Li S., Freeman E. J., McDonough J., Gregory R. B. A SELDI mass spectrometry study of experimental autoimmune encephalomyelitis: sample preparation, reproducibility, and differential protein expression patterns. Proteome Science. 2013;11(1, article 19) doi: 10.1186/1477-5956-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ahari S. E., Houshmand M., Panahi M. S., Kasraie S., Moin M., Bahar M. A. Investigation on mitochondrial tRNA(Leu/Lys), NDI and ATPase 6/8 in Iranian multiple sclerosis patients. Cellular and Molecular Neurobiology. 2007;27(6):695–700. doi: 10.1007/s10571-007-9160-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Johnson A. W., Land J. M., Thompson E. J., Bolanos J. P., Clark J. E., Heales J. R. Evidence for increased nitric oxide production in multiple sclerosis. Journal of Neurology Neurosurgery and Psychiatry. 1995;58(1):p. 107. doi: 10.1136/jnnp.58.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Bö L., Dawson T. M., Wesselingh S., et al. Induction of nitric oxide synthase in demyelinating regions of multiple sclerosis brains. Annals of Neurology. 1994;36(5):778–786. doi: 10.1002/ana.410360515. [DOI] [PubMed] [Google Scholar]
- 199.Okuda Y., Nakatsuji Y., Fujimura H., et al. Expression of the inducible isoform of nitric oxide synthase in the central nervous system of mice correlates with the severity of actively induced experimental allergic encephalomyelitis. Journal of Neuroimmunology. 1995;62(1):103–112. doi: 10.1016/0165-5728(95)00114-h. [DOI] [PubMed] [Google Scholar]
- 200.Grzybicki D. M., Kwack K., Perlman S., Murphy S. P. Nitric oxide synthase type II expression by different cell types in MHV-JHM encephalitis suggests distinct roles for nitric oxide in acute versus persistent virus infection. Journal of Neuroimmunology. 1997;73(1-2):15–27. doi: 10.1016/s0165-5728(96)00159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bagasra O., Michaels F. H., Zheng Y. M., et al. Activation of the inducible form of nitric oxide synthase in the brains of patients with multiple sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(26):12041–12045. doi: 10.1073/pnas.92.26.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lu F., Selak M., O'Connor J., et al. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis. Journal of the Neurological Sciences. 2000;177(2):95–103. doi: 10.1016/s0022-510x(00)00343-9. [DOI] [PubMed] [Google Scholar]
- 203.Ding D., Enriquez-Algeciras M., Dave K. R., Perez-Pinzon M., Bhattacharya S. K. The role of deimination in ATP5b mRNA transport in a transgenic mouse model of multiple sclerosis. EMBO Reports. 2012;13(3):230–236. doi: 10.1038/embor.2011.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Büttner S., Habernig L., Broeskamp F., et al. Endonuclease G mediates α-synuclein cytotoxicity during Parkinson's disease. The EMBO Journal. 2013;32(23):3041–3054. doi: 10.1038/emboj.2013.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Luth E. S., Stavrovskaya I. G., Bartels T., Kristal B. S., Selkoe D. J. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. The Journal of Biological Chemistry. 2014;289(31):21490–21507. doi: 10.1074/jbc.m113.545749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Martin L. J., Semenkow S., Hanaford A., Wong M. The mitochondrial permeability transition pore regulates Parkinson's disease development in mutant α-synuclein transgenic mice. Neurobiology of Aging. 2014;35(5):1132–1152. doi: 10.1016/j.neurobiolaging.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Greenamyre J. T., Sherer T. B., Betarbet R., Panov A. V. Complex I and Parkinson's disease. IUBMB Life. 2002;52(3–5):135–141. doi: 10.1080/15216540152845939. [DOI] [PubMed] [Google Scholar]
- 208.Seaton T. A., Cooper J. M., Schapira A. H. V. Cyclosporin inhibition of apoptosis induced by mitochondrial complex I toxins. Brain Research. 1998;809(1):12–17. doi: 10.1016/S0006-8993(98)00790-2. [DOI] [PubMed] [Google Scholar]
- 209.Valente E. M., Abou-Sleiman P. M., Caputo V., et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304(5674):1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 210.Akundi R. S., Huang Z., Eason J., et al. Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0016038.e16038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gandhi S., Wood-Kaczmar A., Yao Z., et al. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Molecular Cell. 2009;33(5):627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Gautier C. A., Giaime E., Caballero E., et al. Regulation of mitochondrial permeability transition pore by PINK1. Molecular Neurodegeneration. 2012;7, article 22 doi: 10.1186/1750-1326-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kawajiri S., Saiki S., Sato S., Hattori N. Genetic mutations and functions of PINK1. Trends in Pharmacological Sciences. 2011;32(10):573–580. doi: 10.1016/j.tips.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 214.Brundin P., Li J.-Y., Holton J. L., Lindvall O., Revesz T. Research in motion: the enigma of Parkinson's disease pathology spread. Nature Reviews Neuroscience. 2008;9(10):741–745. doi: 10.1038/nrn2477. [DOI] [PubMed] [Google Scholar]
- 215.Palacino J. J., Sagi D., Goldberg M. S., et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. The Journal of Biological Chemistry. 2004;279(18):18614–18622. doi: 10.1074/jbc.m401135200. [DOI] [PubMed] [Google Scholar]
- 216.Hu W., Guan L.-S., Dang X.-B., Ren P.-Y., Zhang Y.-L. Small-molecule inhibitors at the PSD-95/nNOS interface attenuate MPP+-induced neuronal injury through Sirt3 mediated inhibition of mitochondrial dysfunction. Neurochemistry International. 2014;79:57–64. doi: 10.1016/j.neuint.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 217.Darios F., Corti O., Lücking C. B., et al. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Human Molecular Genetics. 2003;12(5):517–526. doi: 10.1093/hmg/ddg044. [DOI] [PubMed] [Google Scholar]