

Abstract
Retinoid-related orphan receptor gamma t (RORγt) is a master transcription factor central to type 17 immunity involving cells such as T helper 17, group 3 innate lymphoid cells or IL-17-producing γδ T cells. Here we show that the intracellular ion channel TMEM176B and its homologue TMEM176A are strongly expressed in these RORγt+ cells. We demonstrate that TMEM176A and B exhibit a similar cation channel activity and mainly colocalise in close proximity to the trans-Golgi network. Strikingly, in the mouse, the loss of Tmem176b is systematically associated with a strong upregulation of Tmem176a. While Tmem176b single-deficiency has no effect on the course of experimental autoimmune encephalomyelitis, T cell or DSS-induced colitis, it significantly reduces imiquimod-induced psoriasis-like skin inflammation. These findings shed light on a potentially novel specific process linked to post-Golgi trafficking for modulating the function of RORγt+ cells and indicate that both homologues should be simultaneously targeted to clearly elucidate the role of this intracellular ion flow.
CD4+ Th17 cells were definitely recognised as a distinct Th subset, along with Th1 and Th2, a decade ago owing to the identification of RORγt as their master transcription factor1. While Th1 and Th2 cells are required for the control of intracellular pathogens or extracellular worms respectively, Th17 cells appear essential for proper defence against extracellular bacteria and fungi2. Moreover, it is now established that deregulated IL-17 secretion by Th17 cells also contributes to the development of several immune-mediated inflammatory diseases (IMIDs)3, and a number of clinical trials aiming at evaluating the therapeutic value of IL-17 or IL-17R blockade have been conducted that led to both impressive and disappointing results, depending on the disease targeted4,5. Interestingly, RORγt function as a master regulator of transcription is not restricted to Th17 cells but also extends to group 3 innate lymphoid cells (ILC3s), which are regarded as their innate counterparts6. Additionally, RORγt+ expression is also detected in IL-17-producing γδ T cells that emerge as important players in inflammatory diseases as well7,8,9,10. Unveiling novel and specific aspects of RORγt+ lymphocytes beyond their cytokine production is thus important to better understand their actions during physiological and/or deregulated immune responses.
Tmem176b, initially named Torid (tolerance-related and induced), encodes a four-span transmembrane protein that we previously showed to be expressed in immature myeloid cells, including conventional dendritic cells (cDCs)11,12. More recently, we demonstrated that TMEM176B exerts major functions in the cross-presentation of antigens by tolerogenic DCs through acting as a non-selective cation channel that finely regulates the phagosomal pH13. Importantly, Tmem176b has a co-regulated homologue, namely Tmem176a, located within the same genomic locus and likely resulting from a recent duplication11,12,14. It is important to note that the co-expression of these two structurally similar genes is not restricted to myeloid cells but extends to other cell types including non-hematopoietic cells15,16,17,18, thus strongly suggesting other intracellular functions beyond antigen cross-presentation.
A recent study by Littman and colleagues19 revealed that, along with Il17a, Il17f, Il23r, Ccl20, Il1r1 or Ltb4r1, Tmem176a and b are part of the highly restricted group of 11 genes whose expression is directly dependent on RORγt in Th17 cells. Concordant with this, a significant upregulation of both homologues was detected in whole-blood samples of patients with multiple sclerosis20, an IMID in which the pathogenic role of type 17 immunity is strongly suspected21,22,23,24. More recently, as part of the Immunological Genome Project, Colonna and colleagues highlighted several ILC-specific genes including Tmem176a and b, whose expression was remarkably higher in ILC3 subsets than in group 1 and 2 ILCs25. These findings thus logically raise the question whether Tmem176a and b play a role in type 17 immunity-related RORγt+ lymphocytes, including Th17 cells and ILC3s, which is yet to be unveiled.
In the present study, we have characterised Tmem176a and b expression in RORγt+ lymphocytes at transcriptional and protein levels and present evidence that both genes exert a redundant ion channel function related to a colocalisation in close proximity to the Golgi apparatus.
Results
Tmem176a and Tmem176b are over-expressed in Th17 cells
We previously reported very low expression of Tmem176a and b mRNA in naive or anti-CD3/CD28 stimulated T cells11,12. However, whether these genes are upregulated in terminally differentiated T cell subsets has not been investigated. To this end, we took advantage of Foxp3EGFP reporter mice that specifically express GFP under the control of the Foxp3 promoter to purify Foxp3+ (GFP+) Tregs along with Foxp3− (GFP−) conventional T cells (Tconv) from the spleen in which most of the Tregs are thymically-derived (Nrp1+), and the gut lamina propria in which specific environmental factors strongly drives the differentiation of peripherally derived Tregs (Nrp1lo) specific for food and commensal antigens (Fig. 1a). Concordant with published microarray data suggesting a preferential expression of Tmem176a and b in peripherally derived Tregs26, we detected significantly higher mRNA levels of both homologues in intestinal Tregs as compared to splenic Tregs. However, the highest levels of expression were actually found specifically in intestinal Tconv cells (Fig. 1b), pointing to Th17 cells, the major T helper cell subset in the gut27, as another important population expressing Tmem176a and b.
Figure 1. Tmem176a and b mRNA expression in mouse and human T cells.

(a) Conventional GFP− (Foxp3− Tconv) or regulatory GFP+ (Foxp3+ Treg) CD4+ T cells were FACS-sorted from the spleen or intestinal lamina propria (small intestine and colon) of Foxp3EGFP mice. As expected, the population of Nrp1−/low “adaptive” peripherally Tregs is dominant in the intestines. Conversely, Nrp1+ “natural” thymically derived Tregs represent the major population of Tregs in spleen. (b) Expression of indicated genes was assessed by quantitative RT-PCR. Each dot represents an individual mouse (n = 6–7 in each group). Statistically significant differences between intestinal Tconv and Treg are indicated: *p < 0.05, **p < 0.01. (c) Mouse naive CD4+ T cells were stimulated with anti-CD3 and anti-CD28 under Th1, Th2, Th17 or iTreg polarising conditions for 3 days. Immature bone marrow-derived DCs (BMDC) were generated with GM-CSF. Expression of indicated genes was assessed by quantitative RT-PCR. Data show triplicates (mean ± SD) for each condition and are representative of three independent experiments. *indicates a statistically significant difference of the indicated population compared to all the other Th cells. (d) Human naive CD4+ T cells were stimulated with anti-CD3 and anti-CD28 under Th1 or Th17 polarising conditions for 7 days. Expression of indicated genes was assessed by quantitative RT-PCR by comparing Th1 and Th17 obtained from 4 independent healthy volunteers. Each donor is coded by a specific symbol.
The expression of Tmem176a and b was therefore assessed in in vitro polarised Th cell subsets. As shown in Fig. 1c, expression of both genes was markedly induced in Th17 cells and, to a much lower extent, in induced Tregs (iTregs) but not in Th1 or Th2 cells. Within the lymphoid lineage, this expression profile mirrors the ones of Rorc or Il17a and is consistent with the findings reported by Ciofani et al.19 showing that Tmem176a and b are direct targets of RORγt in Th17 cells. Importantly, these results hold true in human T cells as high levels of both TMEM176A and B human orthologues were also found in in vitro polarised Th17 cells (Fig. 1d).
Tmem176a and Tmem176b are strongly expressed in ILC3s
Similarly to Th17 cells, ILC3s require RORγt for their development6, thus suggesting that high levels of Tmem176a and b expression should also be detected in these cells. To test for this hypothesis, we took advantage of a RORγt-fate map mouse (Rorc (γt)-CreTG × Rosa26-tdRFP) in which cells expressing, or having previously expressed, RORγt are permanently marked by the tandem-dimer red fluorescent protein (RFP) and can be easily identified (Fig. 2a). We isolated lineage-negative (lin−) RFP+ ILC3s and ex-ILC3s from the gut lamina propria28, and compared them to spleen and gut lin− RFP− NK1.1+ ILC1s (including NK cells) for Tmem176a and b gene expression. Concordant with our hypothesis, the highest expression levels were indeed detected in the lin− RFP+ population (Fig. 2b). In fact, Tmem176a and b mRNA expression respectively reached or was higher than the levels found in CD11b/c+ myeloid cells that were previously considered as the strongest expressors of these two homologues among hematopoietic cells11,12. Thus, Tmem176a and b expression appears preferentially overexpressed in RORγt+ lymphocytes, at least in Th17 cells and ILC3s.
Figure 2. Tmem176a and b mRNA expression in intestinal ILC3s.

(a) Lymphocytes from intestinal lamina propria of RORγt-fate map mice (Rorc(γt)-CreTG × Rosa26-tdRFP) were isolated. Lineage-negative (CD11b−CD11c−CD19−TCRαβ−TCRγδ−) RFP+NK1.1− ILC3-enriched and RFP−NK1.1+ ILC1s were FACS-sorted. In parallel, CD11b/c+ and lineage-negative NK1.1+ conventional NK/ILC1s were FACS-sorted from the spleen. (b) Expression of indicated genes (mean ± SD) was assessed by quantitative RT-PCR in each population isolated from 3–4 independent mice. Statistically significant differences between ILC3s and the other populations are indicated: *p < 0.05, **p < 0.01, ***p < 0.001.
Tmem176a and Tmem176b share a similar cation channel function
Tmem176a and b genes are located within the same genomic locus in opposite direction (Fig. S1a), likely arising from the duplication of a common ancestral gene14, and are tightly co-regulated in various tissues11,12. Strikingly, we found that Tmem176a expression in Th17 cells was further increased in Tmem176b−/− compared to WT cells (mean fold change: 12.7 ± 3.5 (SD) from 5 independent experiments) whereas the expression of other Th17-specific genes such as Rorc, Il17a, Il17f, Ccl20 or Sgk129,30 were not altered (Fig. 3a). Consistently, IL-17A production revealed by intracellular FACS analysis remained unaltered (Fig. 3b). We12 and others31 have shown that TMEM176A and B are structurally similar transmembrane proteins that could physically interact. However, given that both homologues are also likely to function independently, Tmem176a upregulation might represent a physiological mechanism to compensate for the loss of Tmem176b and thus preserve, at least partially, their redundant and therefore presumably important ion channel activity.
Figure 3. Expression compensation and ion channel activity of Tmem176a and b.

(a) Naive CD4+ T cells from WT or Tmem176b−/− mice were stimulated with anti-CD3 and anti-CD28 under Th17 polarising conditions for 3 days. Expression of indicated genes was assessed by quantitative RT-PCR. Data show triplicates (mean ± SD) for each condition and are representative of five independent experiments. **p < 0.01. ND: Not detected. (b) Intracellular RORγt and IL-17A expression was assayed by FACS after PMA/ionomycin restimulation. (c) Xenopus oocytes were injected with Tmem176a or/and Tmem176b mRNA and currents were recorded in voltage-clamp 2–4 days later. Translocation of TMEM176A and TMEM176B to the plasma membrane was induced by a 30-min treatment with PMA. The currents were quantified 5–15 min after holding the extracellular pH at 5. Representative recordings are shown.
We recently demonstrated that Tmem176b encodes a non selective monovalent cation channel activated by acidification13. Protein sequence comparison across various mammalian species revealed that the most striking amino-acid conservations are concentrated within the three first transmembrane domains (Fig. S1b,c), strongly suggesting that both homologues could exert the same ion channel function. To test for this hypothesis, we expressed TMEM176A or TMEM176B proteins in Xenopus oocytes and recorded the electric activity under whole-cell patch clamp after PMA treatment in order to allow surface expression32. As shown in Fig. 3c, TMEM176A and B exhibited a similar ability to induce an inward current activated by acidification of the extracellular solution to pH 5, while concomitant expression resulted in the development of an even more intense current.
Taken together, these data show that both TMEM176A and B are cation channels, and suggest that each molecule has the potential to compensate for each other, either in terms of expression or function.
TMEM176B intracellular localisation is strongly associated with the Golgi apparatus
Having established the ion channel redundancy of TMEM176A and B, we next sought to determine their intracellular localisation in Th17 cells. We previously showed that, in DCs, TMEM176B is expressed in the endophagosomal membranes where its cationic conductance promotes V-ATPase activity and vesicular acidification13, a process referred to as counterion conductance. However, the localisation of TMEM176B in DCs is not restricted to the endophagosomes as we consistently observed TMEM176B expression in other intracellular vesicular compartments, notably in the perinuclear area11,13.
To determine the precise localisation of TMEM176B, we identified a polyclonal anti-human TMEM176B antibody whose specificity for indirect immunofluorescence application was checked on transiently transfected cells with an expression plasmid. Staining of in vitro-polarised human Th17 cells showed a predominant accumulation in a compact juxtanuclear compartment (Fig. 4a) while Th1 cells exhibited almost no staining (Fig. S2). This pattern was distinct from those observed with antibodies against the mitochondria (TOMM20), the endoplasmic reticulum (ER, calreticulin), the early endosomes (EEA1), the lysosomes (CD107a) or the T-cell receptor subunit CD3ε. In contrast, TMEM176B expression was closely apposed to GM130, a Golgi-resident protein, an observation confirmed by colocalisation measurement of several cells (Fig. 4b). Importantly, we found a similar pattern of expression in human monocyte-derived DCs (Fig. S3) as well as in HeLa cells (Fig. S4), strongly suggesting that TMEM176B association with the Golgi is a universal feature found in different types of cells. Of note, TMEM176B did not colocalise with autophagosomes in HeLa cells (Fig. S4).
Figure 4. Intracellular localisation of TMEM176A and TMEM176B.

(a) Human Th17 polarised cells from naive CD4+ T cells were coated on microscopy slides, fixed, permeabilised and co-stained for TMEM176B (red) and the indicated markers (green). DAPI was used for nuclear staining (blue). Arrows indicate TMEM176B colocalisation with the cis-Golgi protein GM130. Bar, 10 μm. (b) Pearson’s correlation coefficients of TMEM176B and the indicated markers (n = 10–15 in each group). (c) HeLa cells were treated or not with nocodazole for 4 hr and subsequently fixed, permeabilised and co-stained for TMEM176B (red), GM130 (cis-Golgi, green) and TGN46 (trans-Golgi and trans-Golgi network (TGN), blue). Bar, 10 μm. Insets represent higher magnifications of regions of interest. Linescans show fluorescence intensity along the lines overlaying the images. (d) HeLa cells were co-transfected with plasmids encoding TMEM176A-HA and TMEM176B-V5 fusion proteins and subsequently fixed, permeabilised and co-stained with HA (green) and V5 (red) monoclonal antibodies. DAPI was used for nuclear staining (blue). (e) Human Th17 polarised cells (as in A) were co-stained for GM130 (green), TMEM176A (purple) and TMEM176B (red). DAPI was used for nuclear staining (blue). Bar, 10 μm.
The Golgi apparatus consists of a collection of stack of cisternae and associated vesicles where proteins and lipids from the ER enter at its cis face and exit at its trans face. In mammal cells, several stacks concentrate to form a compact Golgi ribbon that precludes precise discrimination of distinct regions. To further define the localisation of TMEM176B in relation with the Golgi, we treated HeLa cells with the microtubule-disrupting agent nocodazole that induces spatial separation of the Golgi stacks33. We combined TMEM176B analysis with GM130 (cis-Golgi) and TGN46 that marks the trans-Golgi face and the trans-Golgi network (TGN). Whereas untreated cells showed an expected dense and overlapping expression profile of the three proteins, nocodazole treatment allowed fragmentation of the Golgi apparatus and identification of individual stacks in which cis (GM130) to trans (TGN46) polarisation was discernable (Fig. 4c). Interestingly, TMEM176B expression was consistently more associated with TGN46 but also appeared clearly beyond this marker. Thus, TMEM176B is probably not a Golgi-resident protein but concentrates in vesicles in close proximity to the TGN from where it could emanate to reach (or be recycled from) the endosomal system, a finding which is coherent with TMEM176B detection in the phagosomal membrane in DCs13,34.
TMEM176A and B are colocalised
We next sought to determine the intracellular localisation of TMEM176A relative to TMEM176B. Co-transfection of HeLa cells with plasmids encoding epitope-tagged fusion proteins TMEM176A-HA and TMEM176B-V5 showed a strong colocalisation of these two proteins in the perinuclear region reminiscent of the Golgi apparatus but also in punctate, vesicle-like structures dispersed throughout the cytoplasm (Fig. 4d). Of note, this latter pattern of expression revealed by transfection and clearly outside from the Golgi was also observed in untransfected Hela cells, Th17 cells and DCs by the TMEM176B-specific antibody, although to a lesser extent probably reflecting the detection limit of endogenous expression. In contrast to TMEM176B, we did not succeed in identifying a commercial antibody showing reproducible immunofluorescence specificity to human TMEM176A. We thus generated a rat polyclonal antibody directed against TMEM176A that could be combined with the rabbit antibody against TMEM176B and for which we checked the specificity on cells transiently expressing the fusion protein TMEM176A-HA. As shown in Fig. 4e, human Th17 cells displayed a strong colocalisation of TMEM176A and B along with GM130.
All together, these results show that TMEM176A and B are colocalised in intracellular vesicular structures that concentrate mainly in close association with the trans face of the Golgi apparatus, and more precisely the TGN.
Impact of Tmem176b deficiency in mouse models of IMIDs
High expression of Tmem176a and b in RORγt+ cells suggests that these homologues play a role in immune disorders involving protective or pathogenic actions of these cells. Given that mice deficient for both genes or for Tmem176a alone were not available, we chose to analyse the Tmem176b single-KO mice that we previously described13. We first examined the susceptibility of these mice to the EAE model induced by the MOG35–55 peptide, and which allowed the characterisation of RORγt as the master transcription factor for Th17 cells1. Tmem176b deficiency did not alter the susceptibility of mice to the disease, as we did not observe any significant difference between WT and Tmem176b−/− animals in terms of onset or severity of the disease (Fig. 5a). Second, we specifically assessed the role of Tmem176b in CD4+ T cells by using the transfer model of colitis, in which the intrinsic expression of RORγt in CD4+CD25−CD45RBhi T cells is known to be required to induce a severe colitis after injection into Rag1−/− recipients35. However, as shown in Fig. 5b, the absence of Tmem176b in transferred T cells did not alter the course of the disease. Next, to more specifically address the role of Tmem176b in innate cells, we moved to the dextran sulfate sodium (DSS) model of acute colitis. In this model, DSS rapidly induces epithelial damage leading to bacterial translocation and subsequent gut inflammation36,37. Previous work has demonstrated the crucial role of ILC3-derived IL-22 to restore epithelial barrier integrity and thus recovery of treated mice38,39. Again, in this model, Tmem176b−/− mice exhibited no difference with WT animals regarding colitis severity or duration (Fig. 5c).
Figure 5. Tmem176b single-deficient mouse susceptibility to the development of EAE, chronic and atute colitis.
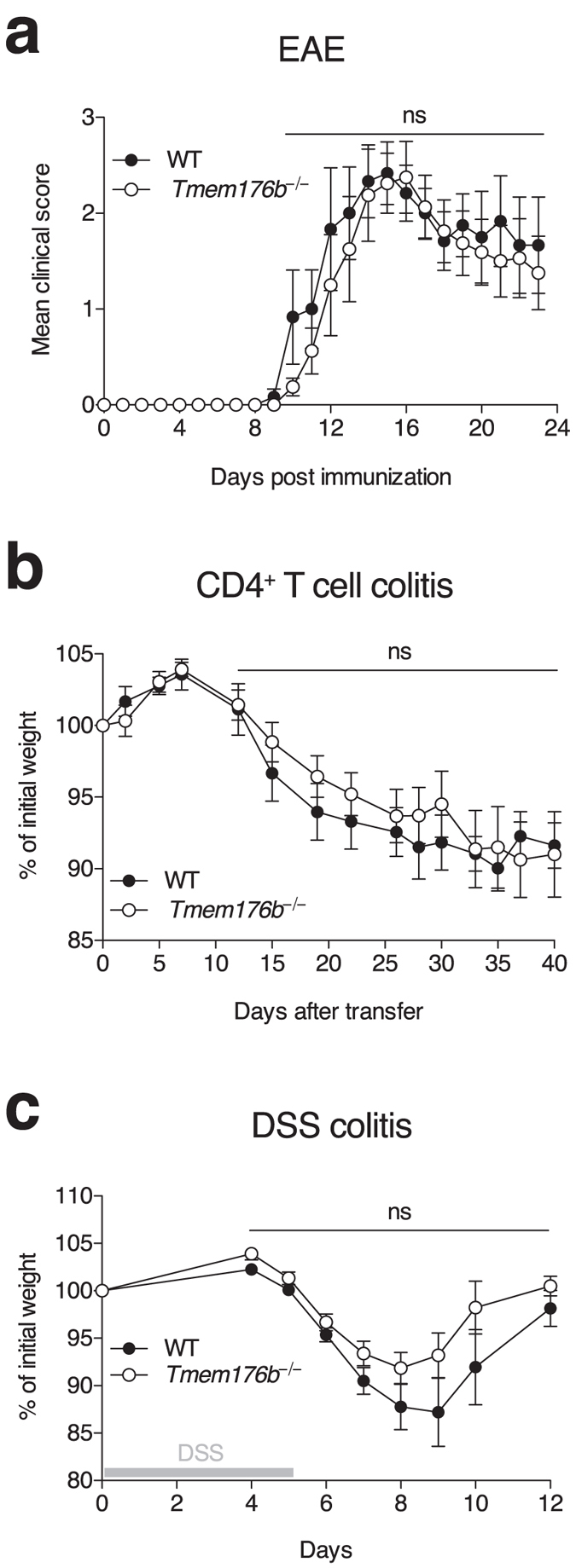
(a) EAE was induced in WT (n = 6) and Tmem176b−/− (n = 8) mice by immunisation (s.c.) with MOG peptide in CFA. Clinical course of disease is shown. (b) Chronic colitis was induced in Rag1−/− mice (n = 11–12 in each group) by adoptive transfer (i.v.) of FACS-sorted CD4+CD45RBhi T cells from WT or Tmem176b−/− mice. Data are presented as percent of initial weight. (c) Acute colitis was induced in WT (n = 6) and Tmem176b−/− (n = 8) mice with 3% DSS in drinking water for 5 consecutive days. Data are presented as percent of initial weight.
Lastly, we examined the model of psoriasis-like skin inflammation induced by topical application of Aldara cream (imiquimod, IMQ) where RORγt+ γδ T cells and ILC3s, but not αβ T cells, are the primary source of pathogenic IL-17A, IL-17F and IL-228,9,40. Importantly, γδ T cells isolated from the draining lymph nodes of treated mice (Fig. 6a) exhibited higher expression of Tmem176a and b than CD4+ T cells, similarly to Rorc or Il17a (Fig. 6b). Interestingly, although thickening was similar, Tmem176b−/− mice exhibited significantly reduced reddening and scaling of the skin compared to WT mice (Fig. 6c). Furthermore, Il17a, Il17f and Il22 as well as induced genes such as Lcn2 (lipocalin-2), Ngp (neutrophilic granule protein) or Cxcl3 mRNA levels of expression were decreased in the skin of treated Tmem176b−/− compared to WT mice but only with Cxcl3 reaching statistical significance (Fig. 6d).
Figure 6. Tmem176b single-deficient mouse susceptibility to the development of psoriasis-like dermatitis.

(a) Psoriasis-like dermatitis was induced in WT mice by topical application of imiquimod (IMQ) cream on the shaved back skin. At day 4, draining (inguinal) lymph nodes were harvested and TCRγδ+ and TCRβ+CD4+ T cells were FACS-sorted. (b) Expression of indicated genes was assessed by quantitative RT-PCR. Each dot represents an individual mouse (n = 6 in each group). Statistically significant differences are indicated: **p < 0.01. (c) Psoriasis-like dermatitis was induced in WT (n = 27) and Tmem176b−/− (n = 28) mice by topical application of imiquimod (IMQ) cream on the shaved back skin during 5 consecutive days. Mice were weighed and scored daily on a scale from 0 to 4 for skin thickening, erythema and scaling. *p < 0.05, ***p < 0.01. (d) Expression of indicated genes was assessed by quantitative RT-PCR in the skin of untreated or IMQ-treated WT and Tmem176b−/− mice (n = 4–6 in each group). Statistically significant differences between IMQ-treated WT and Tmem176b−/− mice are indicated: *p < 0.05.
Thus, Tmem176b single-deficiency can be associated with a significant although limited reduction of RORγt-dependent pathological inflammation that reinforces the possibility of compensatory mechanisms mediated by its homologue Tmem176a.
Discussion
The discovery of Th17 cells as a distinct differentiation of T cell has sparked an intense research on their role in host defence and IMIDs. RORγt+ lymphocytes comprise other cells than T cells and notably include ILC3s, the innate counterpart of Th17. Much attention has been given to these cells through the prism of key cytokines they produce, namely IL-17A, IL-17F, IL-22 or GM-CSF (CSF2), leading to mucosal defence and repair. In contrast to plasma membrane ion channels41, little is known about intracellular ion flows, especially if these channels are involved in specific immune cells. The high expression of TMEM176A and B cation channels in RORγt+ cells is intriguing and could represent a novel therapeutic entry point for treating IMIDs.
Here we demonstrate that the homologues Tmem176a and b are strongly expressed in Th17 as compared to other CD4+ T cells subsets both in mouse and human. These results extend the work of Ciofani et al. which revealed that, like Il17a and f, Tmem176a and b are direct targets of RORγt in Th17 cells19. In line with these results, the expression levels of Tmem176a and b were found significantly decreased after pharmacological treatment specifically targeting Th17 transcriptional program42. We also found that Foxp3+ Tregs exhibit a significant expression of Tmem176a and b, although to a lesser extent than Th17 cells, likely related to the presence of double positive Foxp3+RORγt+ cells that now emerge as regulators of specific helper T cell responses in the colon43,44. Interestingly, with regard to Tregs, Tmem176a and b have recently been shown to be preferentially expressed in amphiregulin-producing IL-10−IL-18R+ Tregs that are proficient in tissue repair45.
It is tempting to link the cation channel function of TMEM176A and B with the recent findings of the effect of sodium on Th17 cells29,30, Tregs46 or M2 macrophages47. Furthermore, TMEM176A and B mRNA upregulation in whole blood cells of MS patients20 raises the possibility of a causal connection with high dietary salt that has been associated with increased disease activity48.
Reasoning that Tmem176a and b may also be induced in other RORγt+ lymphocytes, we found high levels of expression in intestinal ILC3s. Of note, given that RFP+ cells from RORγt-fate map mouse contain a substantial proportion of RORγt− ex-ILC3s, this expression was likely underestimated, notably because intestinal lamina propria preparations included the colon which is particularly permissive for RORγt loss in ILC3s28. Retrospective analysis of published microarray data comparing lung ILC2 and spleen ILC3s49 revealed a clear differential expression of Tmem176a and b in ILC3s. However, this expression does not appear restricted to ILC3s since we found it substantially increased in ILC1s from the intestines compared to the spleen (conventional NK cells). In fact, Colonna and colleagues have recently shown that Tmem176a and b expression was also significantly increased in both ILC1s and ILC2s from the small intestine compared to conventional NK25. Thus, although Tmem176a and b highest levels of expression were found in ILC3, these genes could also play key roles in other types of ILCs, notably in the intestinal environment where local factors could favour their induction independently of RORγt.
Besides Th17 cells and ILC3s, we also found that IL-17-producing γδ T cells preferentially expressed Tmem176a and b. In fact, the development of psoriasis-like dermatitis in which these cells are central was partially reduced in Tmem176b single-deficient mice, suggesting an important intrinsic role of both homologues in these emerging players of type 17 immunity.
Based on the in vivo and in vitro data presented here, we propose that TMEM176A and B exert a redundant ion channel function at the interface of the endosomal network and the Golgi apparatus. It is then likely that pronounced phenotypes will be observed only when the two homologues are simultaneously targeted. To this end, we are currently generating a double KO mouse that will represent an invaluable tool to elucidate the role of these cation channels in RORγt+ cells or other cells such as selected DC subsets in which they are highly expressed50,51,52. Furthermore, conditional tissue-specific deletion will be instrumental to draw definitive conclusions on the contribution of Tmem176a and b in virtually any specific cell types.
To date, only one Golgi-resident ion channel (GPHR: Golgi pH regulator, Gpr89) have been reported to allow a pH gradient from the cis to the trans face of the Golgi apparatus that is crucial for optimal protein transport or glycosylation53. Counterion conductance generated by TMEM176B13 and presumably by TMEM176A could be required for further acidification control of organelles linked to the TGN (trans Golgi network) such as maturing endophagosomes or secretory vesicles54 and thus directly participate in the dynamic of the endocytic and exocytic system which is central for the cells to communicate with their surroundings. According to this hypothesis, we previously showed that non-physiological overexpression of Tmem176b in DCs altered their capacity to upregulate MHC II and CD86 molecules at the surface and to secrete IL-1211. Taking into consideration the pivotal role of RORγt+ cells in tissues that interface with the microbial environment, these cells could foster specific intracellular processes required for their sentinel function and in which TMEM176A and B are involved.
Methods
Mice
All mice in this study are on the C57BL/6 background. C57BL/6J mice were purshased from Centre d’élevage Janvier (France). Foxp3EGFP reporter mice55 were provided by Bernard Malissen. BAC transgenic Rorc(γt)-CreTG mice56 (provided by Gérard Eberl) and Rosa26-tdRFP reporter mice57 (generated by Hans Jörg Fehling and provided by Bernard Malissen) were crossed to obtain a RORγt-fate map mouse. Tmem176b−/− mice were previously described13 and backcrossed for 12 generations onto the C57BL/6J background. Rag1−/− were obtained from Institut Curie (Paris). All mice were kept under specific pathogen-free conditions and experimental procedures were carried out in strict accordance with the protocols approved by the Commitee on the Ethics of Animal Experiments of Pays de la Loire and authorized by the French Government’s Ministry of Higher Education and Research.
FACS analysis and cell sorting
Antibodies used in this study for FACS analysis and sorting are listed in Supplementary Table 1. Red blood cells were lysed with ammonium chloride. CD16/32 antibody (BD Biosciences) was used to block the non-specific binding to Fc receptors before all stainings. Dead cells were excluded using DAPI (Invitrogen). For Tconv and Treg isolation from Foxp3EGFP reporter mice, CD45+TCRβ+CD4+ T cells, respectively GFP− and GFP+, were FACS-sorted (BD FACS ARIATM IIu). Nrp1 staining was added to verify that the proportion of Nrp1−/low “adaptive” pTregs was substantially increased in the intestines compared to the spleen. For ILC3 isolation, CD45+/low lineage-negative (CD11b−CD11c−CD19−TCRαβ−TCRγδ−) RFP+NK1.1− cells were FACS-sorted from the intestines of RORγt-fate map mice (Rorc(γt)-CreTG × Rosa26-tdRFP). Lineage-negative NK1.1+ ILC1s were FACS-sorted from spleen or intestines. CD11b+CD11c+ myeloid cells were FACS-sorted from the spleen. TCRγδ+ and TCRβ+CD4+ T cells were FACS-sorted from inguinal lymph nodes of mice developing psoriasis-like dermatitis. All populations were isolated (>105 cells per mouse) to a purity of >98%.
Mouse in vitro Th polarisation
Spleens were harvested from adult mice and red blood cells were lysed with ammonium chloride. CD4+ T cells were enriched using magnetic-activated cell sorting (Miltenyi Biotec) and live (DAPI−) CD4+CD25−CD62L+CD44lo/− naive T cells were subsequently FACS-sorted to a purity of >98%. Cells were then cultured during 3 days in 48-well plates (5 × 105 cells per well) coated with 5 μg/mL anti CD3ε (145-2C11, BD Biosciences) in complete DMEM medium with GlutaMAX (Invitrogen) with 2 μg/mL soluble anti CD28 (37.51, BD Biosciences). Cultures were supplemented as follows. Th1: IL-12 (2 ng/mL, PeproTech) + neutralising anti IL-4 (2 μg/mL, eBioscience); Th2: IL-4 (2 ng/mL, eBioscience) + neutralising anti IFNγ (2 μg/mL, eBioscience); Th17: IL-6 (20 ng/mL, Sigma) + TGF-β (0.3 ng/mL, R&D systems) + neutralising anti IFNγ and anti IL-4; iTreg: TGF-β (5 ng/mL) + neutralising anti IFNγ and anti IL-4. Gene expression in Th1, Th2, Th17 and iTregs was compared to immature bone marrow-derived DCs generated as previously described13.
Human in vitro Th polarisation
Peripheral blood samples were collected from healthy donors upon written informed consent and approval by the Institutional Review Board (Etablissement Français du Sang Pays de la Loire, Nantes, France). This study was conducted in accordance with the approved guidelines by the Declaration of Helsinki. Live (DAPI−) CD3+CD4+CD45RO−CD45RA+ naive T cells were FACS-sorted to a purity of >98% and then cultured during 6 days in 96-well round-bottom plates (50,000 cells per well) coated with 5 μg/mL anti CD3ε (OKT3, prepared in the laboratory) in complete RPMI medium (Invitrogen) with 1 μg/mL soluble anti CD28 (CD28.2, prepared in the laboratory). Cultures were supplemented as follows. Th1: IL-12 (10 ng/mL, R&D systems); Th17: TGF-β (12.5 ng/mL, Peprotech) + IL-1β (5 ng/mL, Peprotech) + IL-6 (25 ng/mL, Peprotech) + IL-23 (25 ng/mL, Peprotech).
Quantitative RT-PCR
Total RNA from cells was isolated using RNeasy Mini Kit (Qiagen). Skin samples were powderised at low temperature before total RNA isolation using TRIzol® Reagent (Invitrogen, Carlsbad, CA). Reverse transcription was performed using M-MLV Reverse Transcriptase and random primers following manufacturer’s instructions (Invitrogen). Gene expression was assessed with the Fast SYBR Green Master Mix reagent (Applied Biosystems, Foster City, CA). Mouse and human-specific primers used in this study (listed in Supplementary Table 2) were all designed over different exons to prevent amplification of genomic DNA. Real-time PCR was performed using the ViiA 7 Real-Time PCR System (Applied Biosystems). For both mouse and human, gene expression was normalised to glyceraldehyde 3-phosphate dehydrogenase (Gapdh/GAPDH) and expressed in arbitrary units using the 2−ΔΔCt method.
Experimental Autoimmune-Encephalomyelitis (EAE) induced by immunisation with MOG35–55 peptide
Mice aged 8–12 weeks were immunised subcutaneously at the base of the tail and lower flanks with 200 μg of MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK, purity >85%, Genecust Europe, France) emulsified with complete Freund’s adjuvant supplemented with Mycobacterium tuberculosis H37Ra at 6 mg/mL (Difco Laboratories, Detroit, MI, USA). Pertussis toxin (300 ng) was injected intraperitoneally (i.p.) on the day of immunisation and 2 days later. (Calbiochem, Darmstadt, Germany). Mice were scored daily for EAE clinical signs on a scale of 0–5: 0, no disease; 1, complete limp tail; 2, limp tail with unilateral hindlimb paralysis; 3, bilateral hindlimb paralysis; 4, bilateral hindlimb paralysis and forelimb weakness (end point). The observer was blinded to the genotype during the scoring.
Chronic colitis induced by CD4+ T cell adoptive transfer
Naive CD4+CD25−CD45RBhi T cells from the spleen were FACS-sorted to a purity of >98% and 5 × 105 cells were injected intravenously into C57BL/6 Rag1−/− recipients aged 8–12 weeks. Mice were monitored and weighed daily.
Acute colitis induced by dextran sulfate sodium (DSS)
Mice aged 8–12 weeks were given 3% DSS (36,000–50,000 MW, MP Biomedical, Santa Ana, CA) in drinking water ad libitum as indicated for 5 days followed by a recovery period without DSS. Mice were monitored and weighed daily.
Psoriasis-like skin inflammation induced by imiquimod (IMQ) cream
Backs of mice mice aged 8–12 weeks were shaved with an electric clipper and then treated with depilatory cream (Veet) to remove hair. Two days later, mice received a daily topical dose of 62,5 mg of commercially available IMQ cream 5% (Aldara, 3M Pharmaceuticals) on the shaved back for 5 consecutive days, as previously described58. Mice were scored daily for skin inflammation. Dorsal skin thickening was determined by measuring double-skinfold thickness using a digital micrometer (Mitutoyo). Erythema and scaling were scored independently on a scale from 0 to 4: 0 = none, 1 = slight, 2 = moderate, 3 = marked, 4 = severe. For quantitative RT-PCR analysis, back skins were harvested from mice treated with IMQ at day 4 or from untreated mice.
Confocal microscopy analysis
Antibodies used in this study for confocal microscopy analysis are listed in supplementary Table 3. Th17 cells were were plated on poly-L-lysine-coated slides (Thermo Scientific) for 1, 5 hr in complete medium at 37 °C. The cells were then washed with PBS and fixed in 4% paraformaldehyde, 4% sucrose for 15 min at RT followed by 3 washes with PBS and permeabilisation in 0.05% Triton-X100 for 15 min ar RT. Stainings were then directly performed with primary antibodies (diluted in PBS) for 2 h at RT followed by 3 washes with PBS (5 min each at RT) and stainings with adapted secondary antibodies conjugated with fluorochromes (or biotin) for 30 min at RT. A step with fluorochrome-conjugated streptavidin was added when necessary. After 3 washes with PBS (5 min each at RT), DAPI (diluted in PBS) was incubated 10 min at RT. Slides were mounted with ProLong Gold antifade (Invitrogen). Images were obtained with A1 R Si Confocal microscope (Nikon, Champigny sur Marne, France) and analysed with Fiji software. Pearson’s correlation coefficients were calculated with Volocity software (PerkinElmer).
HeLa were seeded in 8-well μ-Slide (ibidi) and immunostaining was achieved as described above for Th17. HeLa cells were treated with 10 μM nocodazole (Sigma) for 4 hr prior fixation to disrupt the juxtanuclear Golgi ribbon. Linescan data were generated with Fiji software and plotted using Graphpad Prism software (La Jolla, CA). HeLa cells were transfected (Lipofectamine 2000, Invitrogen) with expression plasmids (pCI-neo, Promega) in which we cloned human TMEM176A (NM_018487.2) (codon optimisation was performed on a 5′ region of TMEM176A sequence to decreased G/C content) or TMEM176B (NM_014020.3) cDNA fused (C-term) to HA (YPYDVPDYA) or V5 (GKPIPNPLLGLDST) epitopes, respectively. After 48 hr, the cells were fixed, permeablized and co-stained with monoclonal antibodies against HA or V5 followed by secondary antibody staining. HA and V5 specificity was verified on cells separately transfected with TMEM176A-HA or TMEM176B-V5-encoding plasmids.
Statistical analysis
All statistical analyses were performed using Graphpad Prism software (La Jolla, CA) with two-tailed unpaired Student’s t test or one-way ANOVA followed by Tukey’s post hoc test. P values < 0.05 were considered significant.
Additional Information
How to cite this article: Drujont, L. et al. RORγt+ cells selectively express redundant cation channels linked to the Golgi apparatus. Sci. Rep. 6, 23682; doi: 10.1038/srep23682 (2016).
Supplementary Material
Acknowledgments
This work was supported by the Labex IGO project (n°ANR-11-LABX-0016-01) funded by the «Investissements d’Avenir» French Government programme, managed by the French National Research Agency (ANR). This work was realised in the context of the IHU-Cesti project which received French government financial support managed by the National Research Agency via the «Investment Into The Future programme» ANR-10-IBHU-005. The IHU-Cesti project is also supported by Nantes Métropole and région Pays de la Loire. LD was supported by an Inserm-Région Pays de la Loire fellowship. CL was supported by Fondation Progreffe. We are grateful to Philippe Hulin and Steven Nedellec from the MicroPICell imagery core facility (Nantes, France) for excellent assistance with confocal microscopy and to Claire Usal, Emmanuel Merieau and Chad Lemee for mouse housing and experimental help. We thank Gérard Eberl for providing Rorc(γt)-CreTG mice.
Footnotes
The authors declare no competing financial interests.
Author Contributions Conceptualization, C.L., A.M., E.C., J.C.M., R.J. and M.C.C.; Investigation, C.L., L.D., A.L., M.L., T.C., F.G., G.B., L.B.D., G.B., A.B.N. and P.C.; Resources, H.J.F.; Funding Acquisition, C.L. and M.C.C.; Writing–Original Draft, C.L.; Writing–Review & Editing, C.L., J.C.M. and R.J.
References
- Ivanov I. I. et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133, doi: 10.1016/j.cell.2006.07.035 (2006). [DOI] [PubMed] [Google Scholar]
- Korn T., Bettelli E., Oukka M. & Kuchroo V. K. IL-17 and Th17 Cells. Annu Rev Immunol 27, 485–517, doi: 10.1146/annurev.immunol.021908.132710 (2009). [DOI] [PubMed] [Google Scholar]
- Weaver C. T., Elson C. O., Fouser L. A. & Kolls J. K. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu Rev Pathol 8, 477–512, doi: 10.1146/annurev-pathol-011110-130318 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hueber W. et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61, 1693–1700, doi: 10.1136/gutjnl-2011-301668 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley R. G. et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med 371, 326–338, doi: 10.1056/NEJMoa1314258 (2014). [DOI] [PubMed] [Google Scholar]
- Eberl G., Colonna M., Di Santo J. P. & McKenzie A. N. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science 348, aaa6566, doi: 10.1126/science.aaa6566 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabuchi T., Takekoshi T. & Hwang S. T. Epidermal CCR6+ gammadelta T cells are major producers of IL-22 and IL-17 in a murine model of psoriasiform dermatitis. J Immunol 187, 5026–5031, doi: 10.4049/jimmunol.1101817 (2011). [DOI] [PubMed] [Google Scholar]
- Cai Y. et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity 35, 596–610, doi: 10.1016/j.immuni.2011.08.001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelyushin S. et al. Rorgammat+ innate lymphocytes and gammadelta T cells initiate psoriasiform plaque formation in mice. J Clin Invest 122, 2252–2256, doi: 10.1172/JCI61862 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. S. et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 43, 727–738, doi: 10.1016/j.immuni.2015.09.003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvet C. et al. Identification of a new member of the CD20/FcepsilonRIbeta family overexpressed in tolerated allografts. Am J Transplant 5, 2143–2153, doi: 10.1111/j.1600-6143.2005.01007.x (2005). [DOI] [PubMed] [Google Scholar]
- Condamine T. et al. Tmem176B and Tmem176A are associated with the immature state of dendritic cells. J Leukoc Biol 88, 507–515, doi: 10.1189/jlb.1109738 (2010). [DOI] [PubMed] [Google Scholar]
- Segovia M. et al. Autologous dendritic cells prolong allograft survival through Tmem176b-dependent antigen cross-presentation. Am J Transplant 14, 1021–1031, doi: 10.1111/ajt.12708 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccolo J. et al. Phylogenetic analysis of the MS4A and TMEM176 gene families. PLoS One 5, e9369, doi: 10.1371/journal.pone.0009369 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y. et al. Role of Clast1 in development of cerebellar granule cells. Brain Res 1104, 18–26, doi: 10.1016/j.brainres.2006.05.068 (2006). [DOI] [PubMed] [Google Scholar]
- Ryu S. H. et al. Oncogenic Ras-mediated downregulation of Clast1/LR8 is involved in Ras-mediated neoplastic transformation and tumorigenesis in NIH3T3 cells. Cancer Sci 101, 1990–1996, doi: 10.1111/j.1349-7006.2010.01626.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano M., Kawao N., Tamura Y., Okada K. & Kaji H. A novel factor, Tmem176b, induced by activin-like kinase 2 signal promotes the differentiation of myoblasts into osteoblasts. Exp Clin Endocrinol Diabetes 122, 7–14, doi: 10.1055/s-0033-1357129 (2014). [DOI] [PubMed] [Google Scholar]
- Otsubo T. et al. Identification of novel targets for antiangiogenic therapy by comparing the gene expressions of tumor and normal endothelial cells. Cancer Sci 105, 560–567, doi: 10.1111/cas.12394 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M. et al. A validated regulatory network for Th17 cell specification. Cell 151, 289–303, doi: 10.1016/j.cell.2012.09.016 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickles D. et al. Blood RNA profiling in a large cohort of multiple sclerosis patients and healthy controls. Hum Mol Genet 22, 4194–4205, doi: 10.1093/hmg/ddt267 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matusevicius D. et al. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler 5, 101–104 (1999). [DOI] [PubMed] [Google Scholar]
- Tzartos J. S. et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol 172, 146–155, doi: 10.2353/ajpath.2008.070690 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebir H. et al. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol 66, 390–402, doi: 10.1002/ana.21748 (2009). [DOI] [PubMed] [Google Scholar]
- Cao Y. et al. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci Transl Med 7, 287ra274, doi: 10.1126/scitranslmed.aaa8038 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinette M. L. et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol 16, 306–317, doi: 10.1038/ni.3094 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M. et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med 209, 1713–1722, S1711–1719, doi: 10.1084/jem.20120822 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K. et al. ATP drives lamina propria T(H)17 cell differentiation. Nature 455, 808–812, doi: 10.1038/nature07240 (2008). [DOI] [PubMed] [Google Scholar]
- Vonarbourg C. et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity 33, 736–751, doi: 10.1016/j.immuni.2010.10.017 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinewietfeld M. et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496, 518–522, doi: 10.1038/nature11868 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496, 513–517, doi: 10.1038/nature11984 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuajungco M. P. et al. Abnormal accumulation of human transmembrane (TMEM)-176A and 176B proteins is associated with cancer pathology. Acta Histochem 114, 705–712, doi: 10.1016/j.acthis.2011.12.006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J. Y. et al. Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci 4, 382–390, doi: 10.1038/86028 (2001). [DOI] [PubMed] [Google Scholar]
- Dejgaard S. Y., Murshid A., Dee K. M. & Presley J. F. Confocal microscopy-based linescan methodologies for intra-Golgi localization of proteins. J Histochem Cytochem 55, 709–719, doi: 10.1369/jhc.6A7090.2007 (2007). [DOI] [PubMed] [Google Scholar]
- Shui W. et al. Membrane proteomics of phagosomes suggests a connection to autophagy. Proc Natl Acad Sci USA 105, 16952–16957, doi: 10.1073/pnas.0809218105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppkes M. et al. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology 136, 257–267, doi: 10.1053/j.gastro.2008.10.018 (2009). [DOI] [PubMed] [Google Scholar]
- Chassaing B., Aitken J. D., Malleshappa M. & Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol 104, Unit 15 25, doi: 10.1002/0471142735.im1525s104 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J. C., Beriou G. & Josien R. Dextran Sulfate Sodium (DSS)-Induced Acute Colitis in the Rat. Methods Mol Biol 1371, 197–203, doi: 10.1007/978-1-4939-3139-2_12 (2016). [DOI] [PubMed] [Google Scholar]
- Sugimoto K. et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest 118, 534–544, doi: 10.1172/JCI33194 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickert G. et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med 206, 1465–1472, doi: 10.1084/jem.20082683 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Belle A. B. et al. IL-22 is required for imiquimod-induced psoriasiform skin inflammation in mice. J Immunol 188, 462–469, doi: 10.4049/jimmunol.1102224 (2012). [DOI] [PubMed] [Google Scholar]
- Feske S., Wulff H. & Skolnik E. Y. Ion channels in innate and adaptive immunity. Annu Rev Immunol 33, 291–353, doi: 10.1146/annurev-immunol-032414-112212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mele D. A. et al. BET bromodomain inhibition suppresses TH17-mediated pathology. J Exp Med 210, 2181–2190, doi: 10.1084/jem.20130376 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnmacht C. et al. MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORgammat(+) T cells. Science 349, 989–993, doi: 10.1126/science.aac4263 (2015). [DOI] [PubMed] [Google Scholar]
- Sefik E. et al. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science 349, 993–997, doi: 10.1126/science.aaa9420 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N. et al. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 162, 1078–1089, doi: 10.1016/j.cell.2015.08.021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez A. L. et al. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J Clin Invest 125, 4212–4222, doi: 10.1172/JCI81151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binger K. J. et al. High salt reduces the activation of IL-4- and IL-13-stimulated macrophages. J Clin Invest 125, 4223–4238, doi: 10.1172/JCI80919 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farez M. F., Fiol M. P., Gaitan M. I., Quintana F. J. & Correale J. Sodium intake is associated with increased disease activity in multiple sclerosis. J Neurol Neurosurg Psychiatry 86, 26–31, doi: 10.1136/jnnp-2014-307928 (2015). [DOI] [PubMed] [Google Scholar]
- Monticelli L. A. et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 12, 1045–1054, doi: 10.1031/ni.2131 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y. et al. Control of T helper 2 responses by transcription factor IRF4-dependent dendritic cells. Immunity 39, 722–732, doi: 10.1016/j.immuni.2013.08.028 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin D. A. et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779, doi: 10.1126/science.1247651 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandasabapathy N. et al. Classical Flt3L-dependent dendritic cells control immunity to protein vaccine. J Exp Med 211, 1875–1891, doi: 10.1084/jem.20131397 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y., Ide T., Koike M., Uchiyama Y. & Kinoshita T. GPHR is a novel anion channel critical for acidification and functions of the Golgi apparatus. Nat Cell Biol 10, 1135–1145, doi: 10.1038/ncb1773 (2008). [DOI] [PubMed] [Google Scholar]
- Casey J. R., Grinstein S. & Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol 11, 50–61, doi: 10.1038/nrm2820 (2010). [DOI] [PubMed] [Google Scholar]
- Wang Y. et al. Th2 lymphoproliferative disorder of LatY136F mutant mice unfolds independently of TCR-MHC engagement and is insensitive to the action of Foxp3+ regulatory T cells. J Immunol 180, 1565–1575 (2008). [DOI] [PubMed] [Google Scholar]
- Lochner M. et al. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORgamma t+ T cells. J Exp Med 205, 1381–1393, doi: 10.1084/jem.20080034 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luche H., Weber O., Nageswara Rao T., Blum C. & Fehling H. J. Faithful activation of an extra-bright red fluorescent protein in “knock-in” Cre-reporter mice ideally suited for lineage tracing studies. Eur J Immunol 37, 43–53, doi: 10.1002/eji.200636745 (2007). [DOI] [PubMed] [Google Scholar]
- van der Fits L. et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol 182, 5836–5845, doi: 10.4049/jimmunol.0802999 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.