

Abstract
Background
The study aimed to investigate the expression changes of genes in colorectal cancer (CRC) and screen the potential molecular targets.
Methods
The GSE37178 of mRNA expression profile including the CRC samples extracted by surgical resection and the paired normal samples was downloaded from Gene Expression Omnibus database. The genes whose expressions were changed at four different time points were screened and clustered using Mfuzz package. Then DAVID was used to perform the functional and pathway enrichment analysis for genes in different clusters. The protein–protein interaction (PPI) networks were constructed for genes in the clusters according to the STRING database. Furthermore, the related-transcription factors (TFs) and microRNAs (miRNAs) were obtained based on the resources in databases and then were combined with the PPI networks in each cluster to construct the integrated network containing genes, TFs and miRNAs.
Results
As a result, 314 genes were clustered into four groups. Genes in cluster 1 and cluster 2 showed a decreasing trend, while genes in cluster 3 and cluster 4 presented an increasing trend. Then 18 TFs (e.g., TCF4, MEF2C and FOS) and 18 miRNAs (e.g., miR-382, miR-217, miR-1184, miR-326 and miR-330-5p) were identified and three integrated networks for cluster 1, 3, and 4 were constructed.
Conclusions
The results implied that expression of PITX2, VSNL1, TCF4, MEF2C and FOS are time-related and associated with CRC development, accompanied by several miRNAs including miR-382, miR-217, miR-21, miR-1184, miR-326 and miR-330-5p. All of them might be used as potential diagnostic or therapeutic target molecules for CRC.
Keywords: Cluster, Colorectal cancer, MicroRNA, Target, Transcription factor
Background
Colorectal cancer (CRC) is the third most common cancer worldwide with over 1,000,000 new cases every year [1]. In China, CRC is ranked as the fourth leading cause of cancer death with a pronounced increasing incidence during recent years [2]. Reportedly, the main risk factors for CRC include dietary and lifestyle factors, such as diet, obesity, physical activity, smoking and alcohol abuse [3–5]. Moreover, accumulating evidence indicate that multiple molecules are involved in CRC [6, 7]. For instance, CARM1 has been linked to human CRC by modulating Wnt/β-catenin transcription and neoplastic transformation [8]. Several microRNAs (miRNAs) are also involved in the occurrence and development of CRC, such as miR-31, miR-126, miR-552, miR-592 and miR-224 [9, 10]. Moreover, p53-dependent expression of miR-34a is reported to suppress CRC progression by inhibiting an IL-6R/STAT3/miR-34a feedback loop [11]. More recently, utilizing the expression profile dataset of GSE44861, Wang et al. investigated the regulatory relationships between miRNAs and the target genes in CRC, and consequently identified a handful of crucial miRNAs such as miR-29 (with the putative target of COL11A1), miR-101 and miR-26 (both with the predicted targets of PTGS2 and ASPN) [12]. However, pathogenesis of the cancer remains obscure.
Currently, time course gene expression is an increasingly popular approach for researching a wide range of biological processes (BPs) [13]. Recently, Musella et al. [14] have analyzed the gene expression profiling in human normal and CRC tissues at four time points after routine surgical procedure. They have identified several time-dependent genes in tumor and normal samples, such as JUN, FOSB and ABL1. However, they put emphasis on determining the critical time point for tissue handling in colon before which the gene alterations are not detected. Gene alteration trends during the whole period are not investigated. Besides, the potential interactions of the genes and the regulatory relationships between these genes and the related transcription factors (TFs) or miRNAs are not explored.
Therefore, we further investigated the mRNA expression profile of patients with CRC at four time points by re-analyzing the data of Musella et al. [14] which were deposited in Gene Expression Omnibus (GEO) database. The genes whose expressions were changed during different periods were screened and clustered. Then the gene ontology (GO) and kyoto encyclopedia of genes and genomes (KEGG) enrichment analyses were performed for genes in different clusters. Furthermore, the protein–protein interaction (PPI) networks were constructed and the TFs and miRNAs were screened based on the information in the relevant databases. Moreover, an integrated PPI-TF-miRNA regulatory network was established in each cluster. Our study sought to explore the changes of the critical time-related genes in CRC with potential regulators such as miRNAs and TFs, and provide evidence for molecular targets therapy of CRC.
Methods
Microarray data
The mRNA expression profile of GSE37178 was downloaded from GEO database (http://www.ncbi.nlm.nih.gov/geo/) deposited by Musella et al. [14]. The paired tumor and normal specimens collecting from 14 patients who underwent surgical resection at the INT-MI (Fondazione IRCCS Istituto Nazionale dei Tumori) were utilized in our study. The tumor specimens were all classified as moderately differentiated colonic Adenocarcinomas NOS, namely grade G2 basing on the American Joint Committee on Cancer 2010 (http://www.cancerstaging.org/) by the histological routinely examination [14]. The RNAs were extracted from the above patients. Six fragments from each patient were acquired and were randomly left at room temperature at four time points as follows: three fragments at <20 min (T0), one fragment at 60 min (T1), one fragment at 180 min (T2) and one fragment at 360 min (T3). Time was measured starting from patient’s surgical excision and the first time point (T0) was processed and frozen within 20 min from surgery. The control samples were the matched normal tissue at four time points after surgery [14]. In Musella’s study, all patients signed an informed consent and the experiments were approved by the Independent Ethical Committee of the INT-MI.
Preprocessing of the raw data
Raw data were collected with Illumina Human HT-12 V3.0 expression beadchip (Illumina Inc, San Diego, CA, USA) and were preprocessed via background correction, quantile normalization and probe summarization using the LIMMA (Linear Models for Microarray Data, http://www.bioconductor.org/packages/release/bioc/html/limma.html) package [15] in R. Then the probe-level values in CEL files were converted into the mRNA expression values.
Clustering analysis
The noise-robust soft clustering was performed for the samples in four time groups using Mfuzz package (http://www.bioconductor.org/packages/release/bioc/html/Mfuzz.html, version 2.6.1) [16]. According to the changes of gene expression at four different time points, genes were clustered into different groups. The default parameters were as follows: minimum standard deviation = 0.3, acore = 0.6.
GO and KEGG pathway enrichment analysis
The DAVID (The Database for Annotation, Visualization and Integrated Discovery, http://david.abcc.Ncifcrf.gov/) online tool [17] was used to identify the over-represented GO categories in different BPs and the significant KEGG pathways for the genes in different clusters. The p value < 0.05 and gene count ≥ 2 were used as cut-off criteria for the selection of significant GO terms and KEGG pathways.
Screening of transcription factors (TFs) and tumor-associated genes (TAGs)
The TFs were identified amongst the clustered gene sets according to the TRANSFAC database (http://www.gene-regulation.com/pub/databases.html), which collects eukaryotic TFs and their binding sites, as well as the DNA binding profiles [18]. Furthermore, oncogenes and tumor suppressor genes (TSGs) were also obtained from the gene sets by using the TSGene database (http://bioinfo.mc.vanderbilt.edu/TSGene/) [19] and TAG database (http://www.binfo.ncku.edu.tw/TAG/) [20].
Network construction
The STRING (Search Tool for the Retrieval of Interacting Genes/Proteins, http://string-db.org/) database [21] was used to analyze PPIs for genes with the cut-off criterion of combined score >0.4 and the PPI network was then visualized by cytoscape (http://cytoscape.org/) software [22]. The protein product of a gene serves as a node in the PPI network, and the degree denotes the interplayed protein numbers of the specific protein. A node with high degrees is deemed as a hub node. By analyzing the connectivity degrees of the nodes in PPI networks, the hub proteins were obtained. Meanwhile, according to the miRNA-related databases such as miRanda (http://www.microrna.org/microrna/home.do), MirTarget2 (http://mirdb.org/miRDB/), PicTar (http://pictar.org/), PITA, TargetScan (http://www.targetscan.org/) and miRecords (http://mirecords.biolead.org/), the miRNAs which might regulate the genes were obtained with the cut-off criterion of p value <0.05. Then the miRNA-mRNA regulatory networks were constructed. Furthermore, based on the information in the ENCODE (encyclopedia of DNA elements) database (http://genome.ucsc.edu/ENCODE/), [23] the TF-target regulatory network was constructed. Finally, an integrated network combining the PPI network, miRNA-mRNA regulatory network and TF-target regulatory network was established for genes in each cluster.
Results
Clustering analysis
With the aforementioned cut-off criteria, four gene sets were obtained. Among which, a downward expression trend with the increasing time for genes in cluster 1 (35 genes) and cluster 2 (53 genes) was observed, while an upward expression trend was presented in cluster 3 (121 genes) and cluster 4 (105 genes) (Fig. 1). In cluster 1, expressions of the genes were almost stable from T0 to T2, while were downward obviously from T2 to T3. In cluster 2, the expressions tended to be stable from T0 to T1, decreasing from T1 to T2, and then leveling out from T2 to T3. In contrast to cluster 1, expressions of genes in cluster 3 were stable from T0 to T2, while increasing from T2 to T3. In cluster 4, the expressions kept stable from T0 to T1 and were then increasing remarkably from T1 to T3.
Fig. 1.
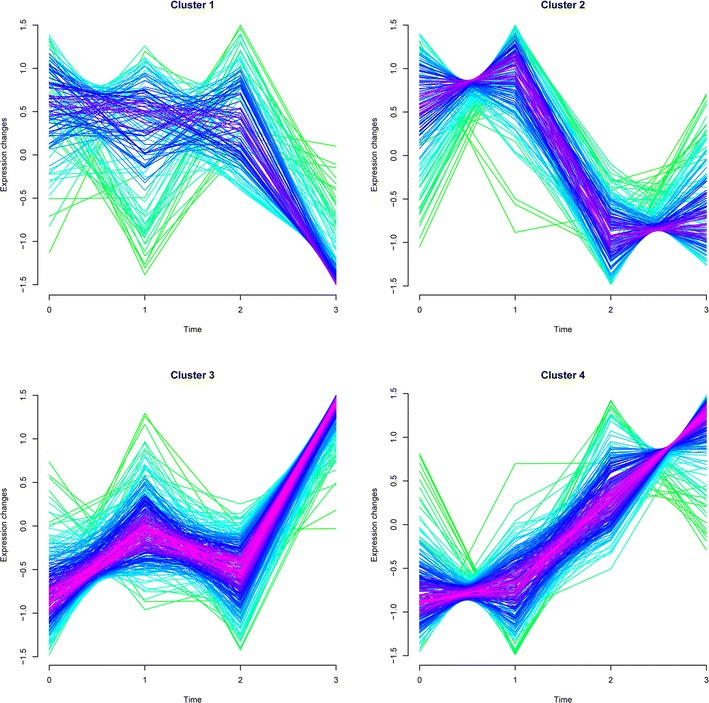
The gene expression changes in four clusters. The color varying from green to red represents that the trends of genes become more suitable to the changes of the cluster
GO and KEGG enrichment analysis
The GO enrichment results showed that the genes in four clusters were enriched in 13, 5, 133 and 119 GO terms, respectively. The top ten GO terms in each cluster are listed in Table 1. Meanwhile, the enriched KEGG pathways of the genes in cluster 1, 3, and 4 were shown in Table 2. The results demostrated that metabolism of xenobiotics by cytochrome P450 (P = 9.06E−02), intestinal immune network for IgA production (P = 2.24E−08) and systemic lupus erythematosus (P = 5.81E−07) were the predominant KEGG pathways for genes in cluster 1, 3 and 4, respectively. Genes in cluster 2 were not enriched in any significant pathway.
Table 1.
Enrichment analysis of top ten GO terms for genes in four clusters
| Term | Count | P value | Genes | |
|---|---|---|---|---|
| Cluster 1 | GO: 0006355~regulation of transcription, DNA-dependent | 8 | 4.53E−02 | PITX2, MSX2, DLX3, IRX3, ZNF181, SMAD9, DMRT2, ETV4 |
| GO: 0051252~regulation of RNA metabolic process | 8 | 5.03E−02 | PITX2, MSX2, DLX3, IRX3, ZNF181, SMAD9, DMRT2, ETV4 | |
| GO: 0016337~cell–cell adhesion | 3 | 9.79E−02 | REG3A, CLDN1, CDH3 | |
| GO: 0030574~collagen catabolic process | 2 | 3.78E−02 | KLK6, MMP7 | |
| GO: 0044243~multicellular organismal catabolic process | 2 | 4.88E−02 | KLK6, MMP7 | |
| GO: 0032963~collagen metabolic process | 2 | 5.25E−02 | KLK6, MMP7 | |
| GO: 0044259~multicellular organismal macromolecule metabolic process | 2 | 5.80E−02 | KLK6, MMP7 | |
| GO: 0044236~multicellular organismal metabolic process | 2 | 6.88E−02 | KLK6, MMP7 | |
| GO: 0042552~myelination | 2 | 7.06E−02 | KLK6, CLDN1 | |
| GO: 0007272~ensheathment of neurons | 2 | 7.77E−02 | KLK6, CLDN1 | |
| Cluster 2 | GO: 0007586~digestion | 3 | 2.69E−02 | CAPN9, TFF2, TFF1 |
| GO: 0002526~acute inflammatory response | 3 | 3.08E−02 | SERPINA1, C4BPA, LBP | |
| GO: 0010038~response to metal ion | 3 | 5.07E−02 | XDH, SERPINA1, TFF1 | |
| GO: 0045087~innate immune response | 3 | 5.72E−02 | C4BPA, LBP, DEFB1 | |
| GO: 0008544~epidermis development | 3 | 9.41E−02 | COL17A1, LAMA3, RBP2 | |
| Cluster 3 | GO: 0006955~immune response | 30 | 3.03E−15 | ITGAL, IL16, LY86 |
| GO: 0045321~leukocyte activation | 16 | 1.67E−10 | ITGAL, CD3G, TNFSF4 | |
| GO: 0001775~cell activation | 16 | 1.79E−09 | ITGAL, CD3G, TNFSF4 | |
| GO: 0006952~defense response | 16 | 3.00E−05 | ITGAL, TNFSF4, AIF1 | |
| GO: 0042110~T cell activation | 15 | 2.58E−13 | ITGAL, CD3G, TNFSF4 | |
| GO: 0046649~lymphocyte activation | 15 | 1.35E−10 | ITGAL, CD3G, TNFSF4 | |
| GO: 0008283~cell proliferation | 11 | 1.12E−03 | HHEX, STAT4, DOCK2 | |
| GO: 0042127~regulation of cell proliferation | 11 | 5.52E−02 | HHEX, CORO1A, TNFSF4 | |
| GO: 0007610~behavior | 10 | 6.45E−03 | CORO1A, DOCK2, IL16 | |
| GO: 0009611~response to wounding | 10 | 1.37E−02 | ITGAL, TNFSF4, AIF1 | |
| Cluster 4 | GO: 0006334~nucleosome assembly | 9 | 3.17E−08 | HIST2H2AA3, HIST4H4, HIST1H1E |
| GO: 0031497~chromatin assembly | 9 | 4.19E−08 | HIST2H2AA3, HIST4H4, HIST1H1E | |
| GO: 0065004~protein-DNA complex assembly | 9 | 5.98E−08 | HIST2H2AA3, HIST4H4, HIST1H1E | |
| GO: 0034728~nucleosome organization | 9 | 7.10E−08 | HIST2H2AA3, HIST4H4, HIST1H1E | |
| GO: 0006323~DNA packaging | 9 | 4.24E−07 | HIST2H2AA3, HIST4H4, HIST1H1E | |
| GO: 0007155~cell adhesion | 18 | 5.34E−07 | EMCN, NRP1, CCL2 | |
| GO: 0022610~biological adhesion | 18 | 5.45E−07 | EMCN, NRP1, CCL2 | |
| GO: 0006333~chromatin assembly or disassembly | 9 | 7.96E−07 | HIST2H2AA3, HIST4H4, HIST1H1E | |
| GO: 0009611~response to wounding | 14 | 1.26E−05 | C3AR1, CCL3, IL6 | |
| GO: 0001568~blood vessel development | 9 | 9.73E−05 | VEGFC, EMCN, NRP1 |
GO gene ontology
Table 2.
Enriched KEGG pathways for genes in four clusters
| Term | Count | P-value | Genes | |
|---|---|---|---|---|
| Cluster 1 | hsa00980: Metabolism of xenobiotics by cytochrome P450 | 2 | 9.06E−02 | CYP3A7, ALDH3B2 |
| hsa00982: Drug metabolism | 2 | 9.35E−02 | CYP3A7, ALDH3B2 | |
| Cluster 3 | hsa04672: Intestinal immune network for IgA production | 9 | 2.24E−08 | TNFSF13B, CXCR4, ICOS |
| hsa04514: Cell adhesion molecules (CAMs) | 9 | 4.80E−05 | ITGAL, SELL, ICOS | |
| hsa04060: Cytokine-cytokine receptor interaction | 9 | 4.66E−03 | IL2RB, TNFSF4, TNFSF13B | |
| hsa05416: Viral myocarditis | 8 | 6.22E−06 | ITGAL, MYH11, HLA-DPA1 | |
| hsa04062: Chemokine signaling pathway | 7 | 1.14E−02 | DOCK2, CCL21, CXCR4 | |
| hsa05340: Primary immunodeficiency | 6 | 2.37E−05 | CD3D, ICOS, TNFRSF13B | |
| hsa04640: Hematopoietic cell lineage | 6 | 1.71E−03 | CD37, CD3G, CD3D | |
| hsa04660: T cell receptor signaling pathway | 6 | 4.63E−03 | CD3G, CD3D, ICOS | |
| hsa05330: Allograft rejection | 5 | 4.50E−04 | HLA-DPA1, HLA-DPB1, HLA-DOA | |
| hsa05332: Graft-versus-host disease | 5 | 6.14E−04 | HLA-DPA1, HLA-DPB1, HLA-DOA | |
| hsa04940: Type I diabetes mellitus | 5 | 8.17E−04 | HLA-DPA1, HLA-DPB1, HLA-DOA | |
| hsa05320: Autoimmune thyroid disease | 5 | 1.70E−03 | HLA-DPA1, HLA-DPB1, HLA-DOA | |
| hsa04612: Antigen processing and presentation | 5 | 9.86E−03 | CD4, HLA-DPA1, HLA-DPB1 | |
| hsa05322: Systemic lupus erythematosus | 5 | 1.79E−02 | HLA-DPA1, HLA-DPB1, HLA-DOA | |
| hsa05310: Asthma | 4 | 3.06E−03 | HLA-DPA1, HLA-DPB1, HLA-DOA | |
| Cluster 4 | hsa05322: Systemic lupus erythematosus | 9 | 5.81E−07 | HIST2H2AA3, HIST4H4, HIST1H2BC |
| hsa04670: Leukocyte transendothelial migration | 6 | 1.89E−03 | HIST2H2AA3, HIST4H4, HIST1H2BC | |
| hsa04514: Cell adhesion molecules (CAMs) | 6 | 3.10E−03 | PECAM1, CLDN5, CLDN11 | |
| hsa04621: NOD-like receptor signaling pathway | 4 | 1.15E−02 | IL6, CCL2, NFKBIA, NLRP3 | |
| hsa04620: Toll-like receptor signaling pathway | 4 | 4.15E−02 | FOS, CCL3, IL6, NFKBIA |
KEGG Kyoto encyclopedia of genes and genomes
Identification of oncogenes, TSGs and TFs amongst the selected genes
Basing on the relevant information in the aforementioned databases, potential oncogenes, TSGs and TFs amongst the selected genes in four clusters were revealed. As shown in Table 3, in cluster 1, ETV4, MSX2, PITX2, SMAD9 were identified as TFs; CUL7 was as a TSG and KLK6 was an oncogene. In cluster 2, CEACAM7, DEFB1, MUC1 and SLC26A3 were considered as TSGs. In cluster 3, EOMES, HEY2, HHEX, MEF2C, POU2AF1, STAT4, TCF21 and TCF4 were TFs; AIM2, BEX2, MAP4K1, PEG3, PRICKLE1, PYHIN1 and TCF4 were TSGs. In cluster 4, ATF3, DBP, HAND1, KLF2, MAFB and SOX18 were TFs and FOS was an oncogene.
Table 3.
Transcription factors (TFs) and tumor associated genes (TAGs) for genes in four clusters
| TF | TSG | Oncogene | Other | |
|---|---|---|---|---|
| Cluster 1 | ETV4, MSX2, PITX2, SMAD9 | CUL7 | KLK6 | |
| Cluster 2 | CEACAM7, DEFB1, MUC1, SLC26A3 | |||
| Cluster 3 | EOMES, HEY2, HHEX, MEF2C, POU2AF1, STAT4, TCF21, TCF4 | AIM2, BEX2, MAP4K1, PEG3, PRICKLE1, PYHIN1, TCF4 | EVI2B | |
| Cluster 4 | ATF3, DBP, HAND1, KLF2, MAFB, SOX18 | ZFP36 | FOS | CCL2, MAFB, RGS2, RHOB |
TSG tumor suppress gene
Screening of miRNAs
With the predefined selection criteria, several miRNAs which could target the genes were predicted. The results indicated that three miRNAs were predicted to target genes in cluster 1: hsa-miR-382 could target CLDN1, DLX3 and IRX3; hsa-miR-217 could modeulate DMRT2, MSX2, SMAD9 and VSNL1; hsa-miR-21 could regulate PITX2 and VSNL1. In cluster 3, five miRNAs were obtained: EOMES and RELN were targeted by hsa-miR-935; HET2 and TCF4 were regulated by hsa-miR-1184; C16orf45, HLA-DOA, PKIA, PTGIS, SPOCK1 and TCF4 were targeted by hsa-miR-326. In cluster 4, ten miRNAs were obtained and the top three were hsa-miR-338-5p (the targets included EBF3, KLF2 and LDB2), hsa-miR-656 (the targets included COLEC12, DUSP1 and TEK) and hsa-miR-30d (the targets included CYYR1, EBF3 and EMCN) (Table 4).
Table 4.
The mircoRNAs (miRNAs) and their target genes
| MiRNA | Count | P-value | Gene | |
|---|---|---|---|---|
| Cluster 1 | hsa-miR-382 | 3 | 5.77E−03 | CLDN1, DLX3, IRX3 |
| hsa-miR-217 | 4 | 1.11E−02 | DMRT2, MSX2, SMAD9, VSNL1 | |
| hsa-miR-21 | 2 | 4.80E−02 | PITX2, VSNL1 | |
| Cluster 3 | hsa-miR-935 | 2 | 1.69E−02 | EOMES, RELN |
| hsa-miR-1184 | 2 | 2.90E−02 | TCF4, HEY2 | |
| hsa-miR-326 | 6 | 3.02E−02 | TCF4, C16orf45, HLA-DOA, PKIA, PTGIS, SPOCK1 | |
| hsa-miR-496 | 5 | 3.45E−02 | RASGRP3, ARHGEF6, MOXD1, PEG3, PTGIS | |
| hsa-miR-330-5p | 5 | 4.22E−02 | TCF4, C16orf45, HLA-DOA, PKIA, SPOCK1 | |
| Cluster 4 | hsa-miR-338-5p | 6 | 8.35E−04 | EBF3, KLF2, LDB2, NRP1, SLIT3, VEGFC |
| hsa-miR-656 | 4 | 6.96E−03 | COLEC12, DUSP1, TEK, ZFP36 | |
| hsa-miR-30d | 9 | 1.08E−02 | CYYR1, EBF3, EMCN, GFPT2, NRP1, RGS2, RHOB, RRAD, ZNF521 | |
| hsa-miR-30b | 9 | 1.10E−02 | CYYR1, EBF3, EMCN, GFPT2, NRP1, RGS2, RHOB, RRAD, ZNF521 | |
| hsa-miR-30a | 9 | 1.11E−02 | CYYR1, EBF3, EMCN, GFPT2,NRP1, RGS2, RHOB, RRAD, ZNF521 | |
| hsa-miR-30c | 9 | 1.12E−02 | CYYR1, EBF3, EMCN, GFPT2, NRP1, RGS2, RHOB, RRAD, ZNF521 | |
| hsa-miR-30e | 9 | 1.12E −02 | CYYR1, EBF3, EMCN, GFPT2, NRP1, RGS2, RHOB, RRAD, ZNF521 | |
| hsa-miR-300 | 10 | 2.79E−02 | EBF3, IER5, LDB2, MMRN1, NFKBIA, NRP1, SELE, TEK, ZFP36, ZNF521 | |
| hsa-miR-27a | 6 | 4.69E−02 | CDH5, EBF3, GEM, GFPT2, RGS1, VEGFC | |
| hsa-miR-27b | 6 | 4.73E−02 | CDH5, EBF3, GEM, GFPT2, RGS1, VEGFC |
Analysis of integrated networks
In order to explore the relationships among the genes, the PPI networks for genes in four clusters were constructed. As presented in Fig. 2, the network of cluster 1 contained 7 nodes and 4 PPIs; cluster 2 consisted of 11 nodes and 7 PPIs; cluster 3 contained 70 nodes and 219 PPIs; while cluster 4 comprised of 65 nodes and 187 PPIs. The hub nodes in four cluster were PITX2, PCYT1A, CD4 and FOS, respectively. Futhermore, the TF and miRNA regulatory networks were also constructed, respectively. The intergrated PPI-TF-miRNA networks were established for the genes in four clusters except in cluster 2, which did not contain the pronounced TFs and miRNAs. In the integrated network cluster (Int-c) 1, miR-217, miR-21, miR-382 and their respective target genes (e.g., PITX2, VSNL1 and DLX3) were interacted with the PPI network in cluster 1 (Fig. 3a). In the Int-c3 network, five miRNAs (e.g., miR-935, miR-1184 and miR-326) and two TFs (MEF2C and TCF4) were integrated with the PPI network in cluster 3. The Int-c3 network showed that MEF2C could regulate 17 genes, such as ITGAL, CORO1A and SGCA; TCF4 could modulate 15 genes, such as LAPTM5, FYB, and CYBRD1 (Fig. 3b). In the Int-c4 network, ten miRNAs (e.g., miR-338-5p, miR-656 and miR-30d) and two TFs (FOS and ATF3) were integrated with the PPI network in cluster 4. It was indicated that FOS could regulate 43 genes, such as ATF3, IL6 and HIST1H2BC; ATF3 could modulate three genes, including GADD45B, DUSP1 and RGS2 (Fig. 3c).
Fig. 2.
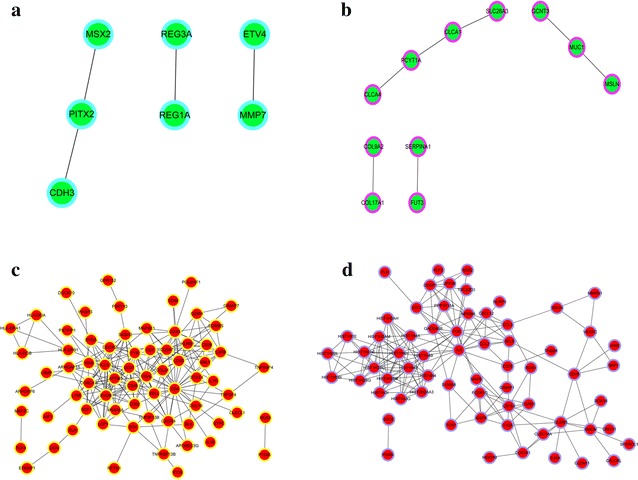
The protein–protein interaction (PPI) of genes in cluster 1 (a), cluster 2 (b), cluster 3 (c) and cluster 4 (d). Green and red nodes represent genes that were decreased and increased, respectively. The different outer rings indicate genes in different clusters
Fig. 3.
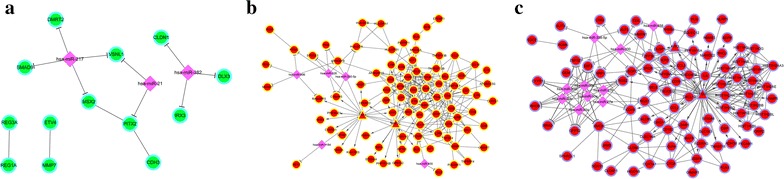
The integrated network of cluster 1 (a), cluster 3 (b) and cluster 4 (c). Green and red nodes represent genes that were decreased and increased, respectively. Purple diamonds represent miRNAs; triangles represent transcription factors
Discussion
In order to further investigate the pathogenesis and explore the molecular therapeutic methods for CRC, we re-analyzed the mRNA expression of CRC tissues extracted after surgical resection at four time points and screened a set of time-related genes, such as PITX2 (paired-like homeodomain 2), VSNL1 (visinin-like 1), TCF4 (transcription factor 4), MEF2C (myocyte enhancer factor 2C) and FOS (FBJ murine osteosarcoma viral oncogene homolog). Meanwhile, TCF4, MEF2C and FOS were found to be TFs that have many target genes. Besides, a set of miRNAs such as miR-382, miR-217, miR-21, miR-1184, miR-326 and miR-330-5p were obatined.
In this study, the screened genes were classified into four clusters according to the gene expression change trends. Genes in cluster 1 and cluster 2 showed a decreasing trend during the period from T2 to T3, and from T1 to T2, respectively. In Int-c1 network, miR-382, miR-217 and miR-21 were shown to target several genes, such as PITX2 and VSNL1.
PITX2, the hub node in PPI network 1, is shown to be involved in various functions such as tissue development by controlling cell growth in CRC [24]. Upregulation of VSNL1 is an indicator of lymph node metastasis and poor prognosis in patients with CRC [25]. Another study also detects the elevated PITX2 and VSNL1 in CRC samples [26]. MiR-21, which is upregulated in many tumours, was predicted to target PITX2 and VSNL1 in our study. It has been found that plasma miR-21 is a potential diagnostic marker of CRC [27, 28]. The results collectively suggested that PITX2 and VSNL1 might be both the targets of miR-21 in CRC, and it might be the suppression of miR-21 that result in the downregulation of the two genes in our present study. In other cancers such as breast cancer and non small cell lung cancer, PITX2 is also predicted as a target of miR-21, [29, 30] strengthening the reliability of our predictions and our faith to validate these targeting relationships in the follow-up studies using dual-luciferase reporter assay.
Besides, miR-382 was shown to target CLDN1 (claudin 1), DLX3 (distal-less homeobox 3) and IRX3 (iroquois homeobox 3); miR-217 could regulate VSNL1, MSX2 (msh homeobox 2), DMRT2 (doublesex and mab-3 related transcription factor 2) and SMAD9 (SMAD family member 9). Most of the targets were enriched in the GO terms of “regulation of transcription, DNA-dependent” and “regulation of RNA metabolic process”, indicating that the two BPs were mainly inhibited by miR-382 and miR-217.
Meanwhile, genes in cluster 3 and cluster 4 showed an increasing trend during the period from T2 to T3, and from T1 to T3, respectively. In cluster3, TCF4 was identified as an important TF, which regulated 15 genes, such as C16orf45 (chromosome 16 open reading frame 45) and RASGRP3 (RAS guanyl releasing protein 3 (calcium and DAG-regulated)). C16orf45 was also targeted by miR-326 and miR-330-5p, and RASGRP3 was regulated by miR-496. Besides, TCF4 was also the target gene of miR-1184, miR-326 and miR-330-5p. It is found that the β-catenin/TCF4 complex, through its control over c-MYC and p21 activity, inhibits the differentiation on CRC cells [31]. Li et al. [32] found that miR-1184 was down-regulated and might play an important role in rectal cancer. Chen et al [33] reported that miR-326 was abnormally expressed in CRC and could be used as a novel biomarker for diagnosis of the cancer. Lin et al [34] showed that miR-330-5p was dysregulated in CRC with liver metastasis. In addition, TCF4 also regulates another TF in Int-c3 network, MEF2C, which could modulate 17 genes in cluster 3. MEF2C is a TF in the MEF2 family and involved in cardiac morphogenesis, myogenesis and vascular development. MEF2 has been shown to have a significant role in angiogenesis [35], and proven to be over-expressed in hepatocellular carcinoma [36]. Although there was little research mentioned that C16orf45 and RASGRP3 were directly associated with CRC, our results suggested miR-1184, miR-326 and miR-330-5p were involve in CRC by interacting with TCF4, C16orf45 and RASGRP3. Therefore, it might be inferred that TCF4, C16orf45 and RASGRP3, mediated by these miRNAs, might play important roles in the CRC progression and be used as the therapeutic targets. The predicted regulation of TCF4 and three miRNAs, miR-1184, miR-326 and miR-330-5p, will be validated in our further studies.
FOS, the hub node in the PPI network 4, is proved to be a TF and regulate 43 genes in cluster 4. FOS is found to interact with JUN (jun proto-oncogene, c-JUN) to form the transcription factor AP-1 (activating protein 1) that is crucial for cell adaptation to environmental changes [37, 38]. Meanwhile, Musella et al. [14] also have found that JUN is a critical time-related gene in CRC. AP-1 regulates the expression of multiple genes essential for cell differentiation, proliferation, and apoptosis and plays an important role in various human diseases such as CRC [39]. Therefore, our results suggested that the increase of FOS may play important role in the period of CRC patients after surgery.
However, there are some limitations in our study. First, we mainly analyzed the relationships among genes and miRNAs in the same cluster. The regulatory relationships among molecules in different clusters and the interactions between clusters are needed further explored. Second, substantial experimental validations of the predicted miRNA-target relationships are warranted to confirm our results. Nevertheless, our results are still of great value to provide clues for further studies focusing on roles of several special miRNAs, such as miR-21, -1184, -330-5p and -326 in CRC progression.
Conclusions
In summary, a set of genes and the related TFs and miRNAs were screened by analyzing of time course gene expression in CRC tissues. Our findings suggested that expression of genes such as PITX2, VSNL1, TCF4, MEF2C and FOS, are time-related. Meanwhile, miRNAs such as miR-382, miR-217, miR-21, miR-1184, miR-326 and miR-330-5p might be involved in the progression of CRC. All the genes and miRNAs might be used as potential diagnostic and/or therapeutic target molecules for CRC.
Authors’ contributions
GC and NH participated in the design of this study, and performed the statistical analysis. QL and GL carried out the study, together with XL and GL collected important background information, and drafted the manuscript. GL and ZL conceived of this study, and participated in the design and helped to draft the manuscript. All authors read and approved the final manuscript.
Acknowledgements
None.
Competing interests
All authors declare that they have no competing interests.
Footnotes
Guoting Chen and Ning Han should be regarded as co-first authors
Contributor Information
Guoting Chen, Email: guotingchengtc@163.com.
Ning Han, Email: ninghannher@163.com.
Guofeng Li, Email: guofengligfler@163.com.
Xin Li, Email: xinlixler@163.com.
Guang Li, Email: guangligl@163.com.
Zengchun Li, Email: zengchunlizcl@163.com.
Qinchuan Li, Phone: +86- 021-38804518, Email: qinchuanliqcll@163.com.
References
- 1.Nagy VM. Updating the management of rectal cancer. J Gastrointestin Liver Dis. 2008;17(1):69–74. doi: 10.1007/s11749-006-0023-9. [DOI] [PubMed] [Google Scholar]
- 2.Zou X, Feng B, Dong T, Yan G, Tan B, Shen H, Huang A, Zhang X, Zhang M, Yang P, et al. Up-regulation of type I collagen during tumorigenesis of colorectal cancer revealed by quantitative proteomic analysis. J Proteomics. 2013;94:473–485. doi: 10.1016/j.jprot.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 3.Watson AJ, Collins PD. Colon cancer: a civilization disorder. Dig Dis. 2011;29(2):222–228. doi: 10.1159/000323926. [DOI] [PubMed] [Google Scholar]
- 4.Terry P, Giovannucci E, Michels KB, Bergkvist L, Hansen H, Holmberg L, Wolk A. Fruit, vegetables, dietary fiber, and risk of colorectal cancer. J Natl Cancer Inst. 2001;93(7):525–533. doi: 10.1093/jnci/93.7.525. [DOI] [PubMed] [Google Scholar]
- 5.Michels KB, Giovannucci E, Joshipura KJ, Rosner BA, Stampfer MJ, Fuchs CS, Colditz GA, Speizer FE, Willett WC. Prospective study of fruit and vegetable consumption and incidence of colon and rectal cancers. J Natl Cancer Inst. 2000;92(21):1740–1752. doi: 10.1093/jnci/92.21.1740. [DOI] [PubMed] [Google Scholar]
- 6.Han M, Liew CT, Zhang HW, Chao S, Zheng R, Yip KT, Song Z-Y, Li HM, Geng XP, Zhu LX. Novel blood-based, five-gene biomarker set for the detection of colorectal cancer. Clin Cancer Res. 2008;14(2):455–460. doi: 10.1158/1078-0432.CCR-07-1801. [DOI] [PubMed] [Google Scholar]
- 7.Engwegen JY, Alberts M, Knol JC, Jimenez CR, Depla AC, Tuynman H, van Heukelem HA, Snel P, Smits ME, Cats A. Influence of variations in sample handling on SELDI-TOF MS serum protein profiles for colorectal cancer. Proteomics Clin Appl. 2008;2(6):936–945. doi: 10.1002/prca.200780068. [DOI] [PubMed] [Google Scholar]
- 8.Ou C-Y, LaBonte MJ, Manegold PC, So AY-L, Ianculescu I, Gerke DS, Yamamoto KR, Ladner RD, Kahn M, Kim JH. A coactivator role of CARM1 in the dysregulation of β-catenin activity in colorectal cancer cell growth and gene expression. Mol Cancer Res. 2011;9(5):660–670. doi: 10.1158/1541-7786.MCR-10-0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oberg AL, French AJ, Sarver AL, Subramanian S, Morlan BW, Riska SM, Borralho PM, Cunningham JM, Boardman LA, Wang L. miRNA expression in colon polyps provides evidence for a multihit model of colon cancer. PLoS One. 2011;6(6):e20465. doi: 10.1371/journal.pone.0020465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou Y, Feng X, Liu Y-l, Ye S-C, Wang H, Tan W-K, Tian T, Qiu Y-M, Luo H-S. Down-regulation of miR-126 is associated with colorectal cancer cells proliferation, migration and invasion by targeting IRS-1 via the AKT and ERK1/2 signaling pathways. PLoS One. 2013;8(11):e81203. doi: 10.1371/journal.pone.0081203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rokavec M, Öner MG, Li H, Jackstadt R, Jiang L, Lodygin D, Kaller M, Horst D, Ziegler PK, Schwitalla S. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Investig. 2014;124(4):1853–1867. doi: 10.1172/JCI73531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Yu H, Ye L, Jin L, Yu M, Lv Y. Integrated regulatory mechanisms of miRNAs and targeted genes involved in colorectal cancer. Int J Clin Exp Patho. 2015;8(1):517. [PMC free article] [PubMed] [Google Scholar]
- 13.Ernst J, Nau GJ, Bar-Joseph Z. Clustering short time series gene expression data. Bioinformatics. 2005;21(suppl 1):i159–i168. doi: 10.1093/bioinformatics/bti1022. [DOI] [PubMed] [Google Scholar]
- 14.Musella V, Verderio P, Reid JF, Pizzamiglio S, Gariboldi M, Callari M, Massimo M, De Cecco L, Veneroni S, Pierotti MA. Effects of warm ischemic time on gene expression profiling in colorectal cancer tissues and normal mucosa. PLoS One. 2013;8(1):e53406. doi: 10.1371/journal.pone.0053406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smyth GK. Limma: linear models for microarray data. In: Bioinformatics and computational biology solutions using R and Bioconductor. Springer; 2005. p. 397–420.
- 16.Futschik ME, Carlisle B. Noise-robust soft clustering of gene expression time-course data. J Bioinform Comput Biol. 2005;3(04):965–988. doi: 10.1142/S0219720005001375. [DOI] [PubMed] [Google Scholar]
- 17.Alvord G, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wingender E, Dietze P, Karas H, Knüppel R. TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996;24(1):238–241. doi: 10.1093/nar/24.1.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao M, Sun J, Zhao Z. TSGene: a web resource for tumor suppressor genes. Nucleic Acids Res. 2013;41(D1):D970–D976. doi: 10.1093/nar/gks937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J-S, Hung W-S, Chan H-H, Tsai S-J, Sun HS. In silico identification of oncogenic potential of fyn-related kinase in hepatocellular carcinoma. Bioinformatics. 2013;29(4):420–427. doi: 10.1093/bioinformatics/bts715. [DOI] [PubMed] [Google Scholar]
- 21.Von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 2003;31(1):258–261. doi: 10.1093/nar/gkg034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kohl M, Wiese S, Warscheid B. Cytoscape: software for visualization and analysis of biological networks. In: Data mining in proteomics. Springer; 2011. p. 291–303. [DOI] [PubMed]
- 23.Raney BJ, Cline MS, Rosenbloom KR, Dreszer TR, Learned K, Barber GP, Meyer LR, Sloan CA, Malladi VS, Roskin KM. ENCODE whole-genome data in the UCSC genome browser (2011 update) Nucleic Acids Res. 2010;38:D620–D625. doi: 10.1093/nar/gkp961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirose H, Ishii H, Mimori K, Tanaka F, Takemasa I, Mizushima T, Ikeda M, Yamamoto H, Sekimoto M, Doki Y. The significance of PITX2 overexpression in human colorectal cancer. Ann Surg Oncol. 2011;18(10):3005–3012. doi: 10.1245/s10434-011-1653-z. [DOI] [PubMed] [Google Scholar]
- 25.Akagi T, Hijiya N, Inomata M, Shiraishi N, Moriyama M, Kitano S. Visinin-like protein-1 overexpression is an indicator of lymph node metastasis and poor prognosis in colorectal cancer patients. Int J Cancer. 2012;131(6):1307–1317. doi: 10.1002/ijc.27341. [DOI] [PubMed] [Google Scholar]
- 26.Khamas A, Ishikawa T, Shimokawa K, Mogushi K, Iida S, Ishiguro M, Mizushima H, Tanaka H, Uetake H, Sugihara K. Screening for epigenetically masked genes in colorectal cancer using 5-Aza-2′-deoxycytidine, microarray and gene expression profile. Cancer Genomics-Proteomics. 2012;9(2):67–75. [PubMed] [Google Scholar]
- 27.Toiyama Y, Takahashi M, Hur K, Nagasaka T, Tanaka K, Inoue Y, Kusunoki M, Boland CR, Goel A. Serum miR-21 as a diagnostic and prognostic biomarker in colorectal cancer. J Natl Cancer Inst. 2013;105(12):849–859. doi: 10.1093/jnci/djt101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toiyama Y, Takahashi M, Hur K, Tanaka K, Inoue Y, Nagasaka T, Kusunoki M, Boland CR, Goel A. Serum miR-21 is a promising biomarker for early detection and prognosis of colorectal cancer. Cancer Res. 2012;72(8 Supplement):4145. doi: 10.1158/1538-7445.AM2012-4145. [DOI] [Google Scholar]
- 29.Wu Q, Lu Z, Li H, Lu J, Guo L, Ge Q. Next-generation sequencing of microRNAs for breast cancer detection. BioMed Res Int. 2011;2011:597145. doi: 10.1155/2011/597145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Markou A, Sourvinou I, Vorkas P, Yousef G, Lianidou E. Clinical evaluation of microRNA expression profiling in non small cell lung cancer. Lung Cancer. 2013;81(3):388–396. doi: 10.1016/j.lungcan.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 31.Van De Wetering M, Sancho E, Verweij C, De Lau W, Oving I, Hurlstone A, Van Der Horn K, Batlle E, Coudreuse D, Haramis A-P. The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111(2):241–250. doi: 10.1016/S0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 32.Li X, Zhang G, Luo F, Ruan J, Huang D, Feng D, Xiao D, Zeng Z, Chen X, Wu W. Identification of aberrantly expressed miRNAs in rectal cancer. Oncol Rep. 2012;28(1):77. doi: 10.3892/or.2012.1769. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, Guo J, Zhang Y, Chen J, Guo X. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18(10):997–1006. doi: 10.1038/cr.2008.282. [DOI] [PubMed] [Google Scholar]
- 34.Lin M, Chen W, Huang J, Gao H, Ye Y, Song Z, Shen X. MicroRNA expression profiles in human colorectal cancers with liver metastases. Oncol Rep. 2011;25(3):739–747. doi: 10.3892/or.2010.1117. [DOI] [PubMed] [Google Scholar]
- 35.Liu G, Han J, Profirovic J, Strekalova E, Voyno-Yasenetskaya TA. Gα13 regulates MEF2-dependent gene transcription in endothelial cells: role in angiogenesis. Angiogenesis. 2009;12(1):1–15. doi: 10.1007/s10456-008-9123-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bai X, Wu L, Liang T, Liu Z, Li J, Li D, Xie H, Yin S, Yu J, Lin Q. Overexpression of myocyte enhancer factor 2 and histone hyperacetylation in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2008;134(1):83–91. doi: 10.1007/s00432-007-0252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bossis G, Malnou CE, Farras R, Andermarcher E, Hipskind R, Rodriguez M, Schmidt D, Muller S, Jariel-Encontre I, Piechaczyk M. Down-regulation of c-Fos/c-Jun AP-1 dimer activity by sumoylation. Mol Cell Biol. 2005;25(16):6964–6979. doi: 10.1128/MCB.25.16.6964-6979.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mahner S, Baasch C, Schwarz J, Hein S, Wolber L, Janicke F, Milde-Langosch K. C-Fos expression is a molecular predictor of progression and survival in epithelial ovarian carcinoma. Br J Cancer. 2008;99(8):1269–1275. doi: 10.1038/sj.bjc.6604650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ashida R, Tominaga K, Sasaki E, Watanabe T, Fujiwara Y, Oshitani N, Higuchi K, Mitsuyama S, Iwao H, Arakawa T. AP-1 and colorectal cancer. Inflammopharmacology. 2005;13(1–3):113–125. doi: 10.1163/156856005774423935. [DOI] [PubMed] [Google Scholar]