

Abstract
A variety of soil-dwelling bacteria produce polyhydroxybutyrate (PHB), which serves as a source of energy and carbon under nutrient deprivation. Bacteria belonging to the genus Pseudomonas do not generally produce PHB but are capable of using the PHB degradation product (R)-3-hydroxybutyrate [(R)-3-HB] as a growth substrate. Essential to this utilization is the NAD+-dependent dehydrogenase BdhA that converts (R)-3-HB into acetoacetate, a molecule that readily enters central metabolism. Apart from the numerous studies that had focused on the biochemical characterization of BdhA, there was nothing known about the assimilation of (R)-3-HB in Pseudomonas, including the genetic regulation of bdhA expression. This study aimed to define the regulatory factors that govern or dictate the expression of the bdhA gene and (R)-3-HB assimilation in Pseudomonas aeruginosa PAO1. Importantly, expression of the bdhA gene was found to be specifically induced by (R)-3-HB in a manner dependent on the alternative sigma factor RpoN and the enhancer-binding protein PA2005.This mode of regulation was essential for the utilization of (R)-3-HB as a sole source of energy and carbon. However, non-induced levels of bdhA expression were sufficient for P. aeruginosa PAO1 to grow on ( ± )-1,3-butanediol, which is catabolized through an (R)-3-HB intermediate. Because this is, we believe, the first report of an enhancer-binding protein that responds to (R)-3-HB, PA2005 was named HbcR for (R)-3-hydroxybutyrate catabolism regulator.
Introduction
Bacteria of the genus Pseudomonas are renowned for their versatile metabolism in that they can assimilate and break down a wide assortment of compounds to meet their nutritional demands. One compound that is not often affiliated with Pseudomonas metabolism is (R)-3-hydroxybutyrate [(R)-3-HB]. (R)-3-HB is commonly recognized for its role as a ketone body produced by mammalian cells when carbohydrate availability is limiting (Akram, 2013). However, there are a number of bacteria that biosynthesize CoA derivatives of (R)-3-HB and other (R)-3-hydroxy acids, which are polymerized into macromolecular structures called polyhydroxyalkanoates (PHAs) (Anderson & Dawes, 1990; Lu et al., 2009). Under starvation conditions, the PHA granule is degraded into its 3-hydroxy acid components, which can be used as sources of carbon and energy (Jendrossek & Handrick, 2002; Jendrossek et al., 1996).
Pseudomonas species do not biosynthesize nor incorporate (R)-3-HB into their PHA reserves (Huisman et al., 1989; Timm & Steinbüchel, 1990). Nonetheless, these bacteria possess an NAD+-dependent dehydrogenase (BdhA) that converts (R)-3-HB into acetoacetate, thereby allowing these bacteria to use (R)-3-HB as a growth substrate (Feller et al., 2006; Ito et al., 2006; Mountassif et al., 2010). BdhA dehydrogenases have been biochemically characterized for some species of Pseudomonas, including P. putida (Feller et al., 2006; Paithankar et al., 2007), P. fragi (Ito et al., 2006; Nakajima et al., 2005) and P. aeruginosa PAO1 (Mountassif et al., 2010). These studies have provided extensive information regarding catalytic properties, mechanisms and structural features of (R)-3-HB dehydrogenases. Despite the wealth of information regarding the biochemical properties of enzymes involved in (R)-3-HB metabolism, the current study is, we believe, the first to characterize the genetic regulation of bdhA expression and (R)-3-HB utilization for any given species of Pseudomonas.
The existence of BdhA dehydrogenases among Pseudomonas spp. indicates that these bacteria can assimilate (R)-3-HB from external sources. In an effort to understand this process, we focused on defining the genetic mechanisms surrounding (R)-3-HB utilization in P. aeruginosa PAO1. The bdhA (PA2003) gene is actually part of a bicistronic operon that starts with PA2004, encoding a putative transporter related to the H+ gluconate symporter family (Winsor et al., 2011). The PA2004-bdhA operon is preceded by a putative − 24/ − 12 promoter recognized by the alternative sigma factor σ54 or RpoN (Conway & Boddy, 2012). RpoN-RNA polymerase (RNAP) holoenzymes cannot spontaneously isomerize from a closed to open complex for transcription initiation (Buck & Cannon, 1992). Additional transcriptional regulators called enhancer-binding proteins (EBPs) interact with the RpoN-RNAP holoenzyme and use the energy of nucleotide hydrolysis to mediate formation of the open complex (Morett & Segovia, 1993; Studholme & Buck, 2000). Adjacent to the PA2004-bdhA operon is the PA2005 gene, which encodes a previously uncharacterized, putative EBP. The results of the current study show that RpoN and the EBP PA2005 are necessary for the induction of the PA2004-bdhA operon in response to (R)-3-HB.
Methods
Bacteria, plasmids and media
Bacteria and plasmids used in the study are given in Table 1. The P. aeruginosa strains obtained from the transposon mutant library (Jacobs et al., 2003) were verified using PCR as recommended by the library curators. Bacteria were grown in BD Difco Lennox broth (LB) or M63 minimal medium (Pardee et al., 1959). Unless otherwise stated, bacteria were grown in 50 ml medium (in 500 ml baffled shake flasks) in a rotary shaker at 37 °C, 200 r.p.m. Medium used for growth of the rpoN mutant (PAO6359) of P. aeruginosa was supplemented with 5 mM l-Gln (Heurlier et al., 2003). Plasmid selection was achieved using the following concentrations of antibiotics: kanamycin (Km) (50 μg ml− 1 for Escherichia coli), carbenicillin (Cb) (100 μg ml− 1 for E. coli or 200 μg ml− 1 for P. aeruginosa) and gentamicin (Gm) (20 μg ml− 1 for E. coli or 30 μg ml− 1 for P. aeruginosa).
Table 1. Bacteria, plasmids and oligonucleotides used in the current study.
| Bacterium, plasmid or oligonucleotide | Relevant characteristics | Source |
|---|---|---|
| P. aeruginosa | ||
| PAO1 M | WT | Jacobs et al. (2003) |
| PW4481 | PA2003-H08 : : ISphoA/hah derivative of PAO1 | Jacobs et al. (2003) |
| PW4483 | PA2004-E10 : : ISphoA/hah derivative of PAO1 | Jacobs et a. (2003) |
| PW4484 | PA2005-D07 : : ISphoA/hah derivative of PAO1 | Jacobs et al. (2003) |
| PAO6359 | rpoN : : Ω-Km derivative of PAO1 | Heurlier et al. (2003) |
| E. coli | ||
| BL21(DE3) | fhuA2 [lon] ompT gal (λ DE3) [dcm] ΔhsdS | EMD Millipore |
| Top10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 nupG recA1 araD139 Δ(ara-leu)7697 galE15 galK16 rpsL (StrR) endA1 λ− | Invitrogen |
| Plasmid | ||
| pCR-Blunt | Cloning plasmid; Kmr | Invitrogen |
| pJET1.2 | Cloning plasmid; Cbr | ThermoScientific |
| pTrc99a | Expression plasmid; Cbr | Pharmacia |
| pET28b | Expression plasmid; Kmr | EMD Millipore |
| pBBR1MCS-5 | Broad-host strain plasmid; Gmr | Kovach et al. (1995) |
| ΔPlac-pBBR1MCS-5 | pBBR1MCS-5 minus lac promoter; Gmr | Lundgren et al. (2014) |
| pBRL491 | bdhA gene in pCR-Blunt; Kmr | This study |
| pBRL492 | PA2004-lacZ in pCR-Blunt; Kmr | This study |
| pBRL494 | PA2004-bdhA genes in pCR-Blunt; Kmr | This study |
| pBRL496 | bdhA gene in pBBR1MCS-5; Gmr | This study |
| pBRL498 | PA2004-bdhA genes in pBBR1MCS-5; Gmr | This study |
| pBRL499 | PA2004-lacZ in ΔPlac-pBBR1MCS-5; Gmr | This study |
| pBRL500 | PA2004 gene in pCR-Blunt; Kmr | This study |
| pBRL501 | PA2004 gene in pBBR1MCS-5; Gmr | This study |
| pBRL505 | PA2004-lacZ with mutated RpoN promoter; Gmr | This study |
| pBRL510 | PA2005 ORF in pCR-Blunt; Kmr | This study |
| pBRL512 | PPA2004 probe in pJET1.2; Cbr | This study |
| pBRL516 | PA2005 ORF in pET28b; Kmr | This study |
| pBRL595 | PA2005 in pTrc99a with forwards orientation; Cbr | This study |
| pBRL596 | PA2005 in pTrc99a with backwards orientation; Cbr | This study |
| pJRH10 | PA2005 in pBBR1MCS-5 | This study |
| pZS406 | PgcvH2 probe in pJET1.2; Cbr | This study |
| Oligonucleotide | ||
| BL342.f | atgaccatgattacggattcactg | E. coli lacZ |
| BL342.r | gcagttatttttgacaccagaccaactggta | E. coli lacZ |
| BL442.f | gctggaggccgaattcttc | 5′ Regulatory region of PA2004 |
| BL442.r | gaatccgtaatcatggtcatcgggcatatctccacg | 5′ Regulatory region of PA2004 |
| BL443.f | gcatctagagccttccccatgcgaagc | PA2004 gene |
| BL443.r | gcagagctctcagaccaggccggtggc | PA2004 gene |
| BL444.f | gcatctagacatccagcgggagaaacagatg | bdhA gene |
| BL444.r | gcagagctcctactgcgccacccagcc | bdhA gene |
| BL445.f | cgcgattctggggcctaacacggttatcgcaacg | Mutation of RpoN promoter in 5′ regulatory region of PA2004 |
| BL445.r | cgttgcgataaccgtgttaggccccagaatcgcg | Mutation of RpoN promoter in 5′ regulatory region of PA2004 |
| BL446.f | gccatatgaacgacgccgacagcc | PA2005 ORF |
| BL446.r | gcgagctctcagttcgtctctatttcgagac | PA2005 ORF |
| BL447.f | gctcgattacgtctccattttg | PPA2004 probe |
| BL447.r | gaaaggcacgttgcgataacc | PPA2004 probe |
| JRH05.f | ggctcgagtttttcagcaagat | 5′-Labelled Cy5 pJET1.2 primer |
| JRH05.r | gaatattgtaggagatcttctagaaag | 5′-Labelled Cy5 pJET1.2 primer |
| ZS406.f | ggggctgtgccatcggctgtaacg | PgcvH2 probe |
| ZS406.r | gcggccccggccctgtcgccacgg | PgcvH2 probe |
Standard DNA procedures
DNA was purified using Promega nucleic acid purification kits. Restriction enzymes, ligases and polymerases were products of New England BioLabs. Oligonucleotides used for PCR applications were purchased from Integrated DNA Technologies and are listed in Table 1. Genomic DNA from P. aeruginosa PAO1 was used for all PCR applications. PCR products were gel-purified and cloned into pCR-Blunt (Invitrogen) according to the manufacturer's instructions. Cloned DNA was verified by sequencing (Genewiz).
Cloning of the bdhA (PA2003) and PA2004 genes
The bdhA and PA2004 genes were PCR amplified using the primers BL444.f/BL444.r and BL443.f/BL443.r, respectively. The PA2004-bdhA operon was amplified with primers BL443.f/BL444.r. All three PCR products were individually cloned into pCR-Blunt. The bdhA, PA2004 and PA2004-bdhA genes were then subcloned into the XbaI/SacI sites of pBBR1MCS-5 to give pBRL496, pBRL501 and pBRL498, respectively.
Cloning of the PA2005 ORF
The putative PA2005 ORF was PCR amplified with the primers BL446.f/BL446.r, and the resulting PCR product was cloned into pCR-Blunt to give pBRL510. The PA2005 ORF from pBRL510 was subcloned into the NdeI/SacI sites of pET28b (EMD Millipore) to give pBRL516. The pBRL516 plasmid was digested with XbaI/SacI to liberate the PA2005 ORF with a pET-derived RBS, which was cloned into the XbaI/SacI sites of pBBR1MCS-5 (Kovach et al., 1995) to yield pJRH010. Lastly, the PA2005 ORF from pBRL510 was cloned into the EcoRI site of pTrc99a (Pharmacia) with either a forward orientation (pBRL595) or reverse orientation (pBRL596) relative to the trc promoter.
Construction of the PA2004-lacZ reporter
The 1042 bp 5′ regulatory region positioned immediately upstream of the PA2004 ORF was PCR amplified with primers BL442.f/BL442.r. This amplicon was then fused to the lacZ ORF of E. coli MG1655 with primers BL442.f/BL342.r using PCR conditions as previously described (Lundgren et al., 2014). The PA2004-lacZ fusion PCR product was cloned into pCR-Blunt to give pBRL492. The PA2004-lacZ fusion in pBRL492 was cloned into the XbaI site of the promoterless ΔPlac-pBBR1MCS-5 plasmid (Lundgren et al., 2014) to yield pBRL499. The GG dinucleotide of the − 24 element of the RpoN promoter located 148 bp upstream of the lacZ ORF in pBRL499 was changed to an AA dinucleotide using QuikChange (Agilent Technologies) with the primers BL445.f/BL445.r. The resulting plasmid pBRL505 was sequenced to verify the desired mutation.
Growth studies of P. aeruginosa on (R,S)-3-HB
BdhA has been shown to be specific for (R)-3-HB (Ito et al., 2006), and the BdhA of P. aeruginosa PAO1 was observed to synthesize acetoacetate when given (R,S)-3-HB as a substrate (Mountassif et al., 2010). Therefore, the sodium salt, racemic (R,S)-3-HB (Sigma Aldrich) was used as the growth substrate, because it was more cost effective than enantiopure (R)-3-HB. All P. aeruginosa strains were grown in quadruplicate. For each replicate, M63 minimal medium that was supplemented with 30 mM (R,S)-3-HB or ( ± )-1,3-butanediol was inoculated to an initial OD600 of ∼0.1. Cultures were grown for 24 h and OD600 was periodically measured. For genetic complementation experiments, the medium was not supplemented with antibiotic for plasmid selection. The use of 30 mM (R,S)-3-HB or ( ± )-1,3-butanediol as the sole carbon source was sufficient for selection of recombinant strains.
β-Galactosidase (LacZ) assays
Each condition was tested in quadruplicate, and LacZ activity was determined using the Miller assay (Lundgren et al., 2013, 2014). To monitor the change in expression of PA2004-lacZ over time, P. aeruginosa strains harbouring pBRL499 or pBRL505 were grown in LB or M63 minimal medium, which was supplemented with 30 mM sodium acetate or sodium succinate, to an OD600 of 0.3. Cells were then challenged with 30 mM (R,S)-3-HB, and LacZ activity was measured at 1, 2, 3 and 4 h post-induction.
The abilities of compounds to induce expression of PA2004-lacZ were examined by growing P. aeruginosa PAO1 harbouring pBRL499 in M63 minimal medium supplemented with 30 mM sodium succinate to an OD600 of 0.3 and then adding (R,S)-3-HB, (R)-3-HB, (R)-2-hydroxybutyrate [(R)-2-HB], acetoacetate, (R)-lactate or (S)-carnitine to a final concentration of 1 mM. LacZ activity was then measured 30 min post-addition of substrate. For LacZ assays involving ( ± )-1,3-butanediol, P. aeruginosa strains possessing pBRL499 were grown in LB, which was supplemented with 30 mM ( ± )-1,3-butanediol or 1,4-butanediol. After a 16 h incubation period (37 °C, 200 r.p.m.), LacZ activity was measured for each sample.
LacZ assays involving E. coli were done using the lacZ-deficient E. coli strain Top10 (Invitrogen). E. coli Top10 was co-transformed with pBRL499 (or a PA2004-lacZ reporter derivative: ΔPlac-pBBR1MCS-5 or pBRL505) and pBRL595 (or a PA2005 expression plasmid derivative: pTrc99a or pBRL596). Recombinant strains were grown in LB supplemented with gentamicin and carbenicillin to an OD600 of 0.3. Subsequently, (R)-3-HB, (R,S)-3-HB, 2-(R)-HB or acetoacetate was added to a final concentration of 30 mM, and LacZ activity was measured at 2 h post-induction.
Measurement of BdhA activity
All conditions were tested in quadruplicate. Pseudomonas strains were grown in LB to an OD600 of 0.3 and subsequently challenged with (R,S)-3-HB at a final concentration of either 0 or 30 mM. After a 4 h induction period, cells were harvested, washed and suspended in 1.0 ml lysis buffer (100 mM potassium phosphate, 300 mM NaCl, 1 μg ml− 1 pepstatin A, 2 μg ml− 1 leupeptin and 3 mg ml− 1 lysozyme, pH 8.0). The cell suspensions were incubated at 25 °C for 30 min and then subjected to sonication on ice. Unlysed cells and cellular debris were removed via centrifugation. The cleared lysates were transferred to Amicon Microcon Centrifugal devices (molecular weight cut-off 10 000 Da) and washed twice with 0.5 ml lysis buffer (100 mM Tris, 300 mM NaCl, pH 8.0). Each lysate was concentrated to a final volume of 50 μl.
The Bradford assay (Pierce) was used to measure the protein concentrations of the lysates. BdhA activity was measured by the formation of NADH (extinction coefficient of 6220 M− 1 cm− 1) (Brashear & Cook, 1983). For each reaction, 2 μl (∼5 μg protein) lysate was added to 0.5 ml reaction buffer (100 mM Tris, 300 mM NaCl, 1.8 mM NAD+, 25 mM (R)-3-HB, pH 8.0). The reaction mixture was incubated at 25 °C and the increase in absorbance at 340 nM was recorded. Each sample exhibited a linear increase in the absorbance at 340 nm for the first 8 min. BdhA activities were reported as units (U) per mg of total protein where U was defined as the formation of 1 μmol NADH min− 1 at 25 °C, pH 8.0.
Purification and electrophoretic mobility shift assays (EMSAs) of His6-PA2005
The PA2005 ORF was cloned into pET28b, and the resulting plasmid pBRL516 encoded an N-terminal 6 × histidine-tagged PA2005 fusion protein (His6-PA2005). E. coli BL21(DE3) harbouring pBRL516 was grown in LB to an OD600 of ∼0.6 at 37 °C with shaking (200 r.p.m.). Protein expression was induced by the addition of 0.1 mM IPTG, and the induced cultures were incubated for 12 h at 16 °C (200 r.p.m.). Cells were harvested, suspended in buffer (100 mM sodium phosphate pH 8.0, 300 mM NaCl, 10 % v/v glycerol. 1 mg ml− 1 lysozyme, 5 U ml− 1 DNase I, 1 μg ml− 1 pepstatin, 1 μg ml− 1 leupeptin) and then lysed by sonication on ice. Unlysed cells and debris were removed via centrifugation. The His6-PA2005 protein was then purified from the clarified lysate using Ni-NTA Superflow resin (Qiagen). The His6-PA2005 protein was eluted off the resin using a step elution method with elution buffer (100 mM Tris, 300 mM NaCl, pH 8.0) containing 20, 100 and 250 mM imidazole. The purified His6-PA2005 protein was concentrated using Amicon Ultra centrifugal filter units (EMD Millipore). Protein expression and purification were monitored visually using SDS-PAGE. The concentration of purified protein was determined using the Bradford assay (Pierce).
His6-PA2005 was expected to bind to a region preceding the RpoN promoter positioned 148 bp upstream of the PA2004 ORF. Therefore, a 116 bp probe (PPA2004), which resembled the region 237–122 bp upstream of the PA2004 ORF, was PCR amplified with the primers BL447.f/BL447.r. For the non-specific probe, the 200 bp promoter region of gcvH2 (PgcvH2) (Lundgren et al., 2013) was PCR amplified with the primers ZS406.f/ZS406.r. The PPA2004 and PgcvH2 PCR products were individually cloned into pJET1.2 (ThermoScientific) to yield the plasmids pBRL512 and pZS406, respectively. The 5′-labelled Cy5 primers JRH05.f/JRH05.r were used to PCR amplify PPA2004 and PgcvH2 from pBRL512 and pZS406, respectively. The resulting 5′-labelled Cy5 PPA2004 and PgcvH2 probes were gel-purified and used in subsequent EMSAs.
For the first set of EMSA reactions, 500 nM His6-PA2005 was incubated with 1.0 nM 5′-labelled Cy5 PPA2004 probe (specific probe) or 1.0 nM 5′-labelled Cy5 PgcvH2 probe (non-specific probe) in EMSA buffer (25 mM Tris/acetate, 8.0 mM magnesium acetate, 10 mM KCl, 1.0 mM DTT, pH 8.0) for 30 min at 30 °C. In the second set of EMSA reactions, 1.0 nM 5′-labelled Cy5 PPA2004 probe was incubated with 0, 6.25, 12.5, 25, 50, 100, 200 or 400 nM PA2005 in EMSA buffer for 30 min at 30 °C. The samples were then analysed using PAGE in non-denaturing conditions and imaged using a Typhoon imager.
Results
Transposon insertions into the bdhA (PA2003), PA2004 or PA2005 gene hindered the growth of P. aeruginosa PAO1 on (R,S)-3-HB
The metabolism of (R)-3-HB was expected to be dependent on the PA2004-bdhA operon, because BdhA performs an essential function in that it oxidizes (R)-3-HB into acetoacetate. The role of PA2004 was less clear. Because PA2004 has homology to the H+ gluconate symporters (Winsor et al., 2011), it was speculated that PA2004 might function as an (R)-3-HB transporter. The PA2005 gene encodes a putative EBP, and because there is a − 24/ − 12 or RpoN promoter 148 bp upstream of the PA2004 ORF (Conway & Boddy, 2012), PA2005 was proposed to be the EBP that regulates expression of the PA2004-bdhA operon in response to (R)-3-HB.
Consistent with this hypothesis, a PA2005 transposon mutant (PA2005 : : Tn) could not use (R,S)-3-HB as a sole carbon source (Fig. 1). Growth of the PA2005 : : Tn mutant on (R,S)-3-HB was restored when the bdhA gene, the PA2004-bdhA operon or the PA2005 gene was expressed from the lac promoter on the broad-host-range pBBR1MCS-5 plasmid (Fig. 1a). This finding suggested that the diminished capacity of the PA2005 : : Tn mutant to grow on (R,S)-3-HB might be a result of insufficient expression of the bdhA gene. Not surprisingly, a bdhA transposon mutant (bdhA : : Tn) (Fig. 1b) and a PA2004 : : Tn mutant (Fig. 1c) also failed to utilize (R,S)-3-HB as a carbon source.
Fig. 1.

Transposon (Tn) insertions in the bdhA, PA2004 and PA2005 genes negatively affected the utilization of (R,S)-3-HB in P. aeruginosa PAO1.Growth deficiencies were observed for (a) PA2005 : : Tn, (b) bdhA : : Tn and (c) PA2004 : : Tn mutants of P. aeruginosa PAO1 on 30 mM (R,S)-3-HB. Expression of bdhA (bdhA+), the PA2004-bdhA operon [(PA2004-bdhA)+] or PA2005 (PA2005+) from the lac promoter of the pBBR1MCS-5 plasmid restored growth of the PA2005 : : Tn mutant on (R,S)-3-HB. The bdhA : : Tn and PA2004 : : Tn mutants were also complemented with plasmid-derived expression of bdhA (bdhA+) or the PA2004-bdhA operon [(PA2004-bdhA)+]. Data points represent mean values ± sd (n = 4). ANOVA was performed using a Dunnett's post-hoc test (α-value of 0.05) to identify significant changes (P < 0.0001).
Expression of a PA2004-lacZ construct was induced by (R)-3-HB
The expression of the PA2004-bdhA operon was expected to be regulated by PA2005 in response to (R)-3-HB availability. Therefore, it was first determined if the PA2004-bdhA operon was in fact inducible by (R)-3-HB. To achieve this goal, the 5′ regulatory region (1042 bp) located immediately upstream of the PA2004 start codon was fused to the lacZ ORF of E. coli MG1665, and the resulting PA2004-lacZ fusion was cloned into the promoterless plasmid ΔPlac-pBBR1MCS-5 (Lundgren et al., 2014). P. aeruginosa PAO1 harbouring the PA2004-lacZ reporter was grown in M63 minimal medium supplemented with 30 mM succinate. When the cells reached an OD600 of 0.3, they were challenged with various substrates added at a final concentration of 1 mM. Within 30 min of the addition of either (R)-3-HB or (R,S)-3-HB, PA2004-lacZ expression increased twofold (Fig. 2a). Other tested substrates, including (R)-2-HB, butyrate, acetoacetate, (R)-lactate and (S)-carnitine, did not induce expression of PA2004-lacZ (Fig. 2b). Increased expression of PA2004-lacZ was specific to (R)-3-HB, indicating that this molecule is an inducer of the PA2004-bdhA operon in P. aeruginosa PAO1.
Fig. 2.

(R)-3-HB induced expression of a PA2004-lacZ reporter in P. aeruginosa PAO1. P. aeruginosa PAO1 harbouring a PA2004-lacZ reporter was grown to an OD600 of 0.3 in M63 minimal medium supplemented with 30 mM succinate and subsequently challenged with (a) various concentrations of (R)-3-HB or (b) various substrates provided at final concentrations of 1.0 mM. LacZ activity was measured 30 min post-addition of substrate. As shown, racemic and enantiopure (R)-3-HB were the only compounds that induced expression of the PA2004-lacZ reporter (twofold increase in LacZ activity). Notably, a concentration of 500 μM (R)-3-HB was found to be sufficient to induce expression of PA2004-lacZ. Data points represent mean values ± sd (n = 4). ANOVA was performed using a Dunnett's post-hoc test (α-value of 0.05) to identify significant changes (P < 0.0001), which are marked with an asterisk.
RpoN was required for the induction of PA2004-lacZ by (R)-3-HB
There is a putative RpoN promoter located 148 bp upstream of the PA2004 ORF (Conway & Boddy, 2012). This RpoN promoter, T CACGGTTATC A, has the highly conserved ‘GG’ and ‘GC’ (underlined) nucleotides positioned at the − 24 and − 12 elements, respectively (Barrios et al., 1999). Since one of the hallmark attributes of RpoN is its role in the assimilation of various organic compounds, RpoN was considered to regulate expression of the PA2004-bdhA operon in response to (R)-3-HB. In support of this hypothesis, expression levels of the PA2004-lacZ reporter were fourfold lower in an rpoN mutant compared with WT P. aeruginosa PAO1 challenged with 30 mM (R,S)-3-HB (Fig. 3). In parallel, the conserved ‘GG’ nucleotides of the − 24 element were changed to ‘AA’ in the RpoN promoter of the PA2004-lacZ reporter. This ‘AA’ substitution at the − 24 element reduced the expression of PA2004-lacZ by more than fourfold (Fig. 3). Collectively, RpoN and its cognate − 24/ − 12 promoter were necessary for (R)-3-HB induction of PA2004-lacZ, increasing it from a background (non-induced) level of 200 MU to >800 MU.
Fig. 3.
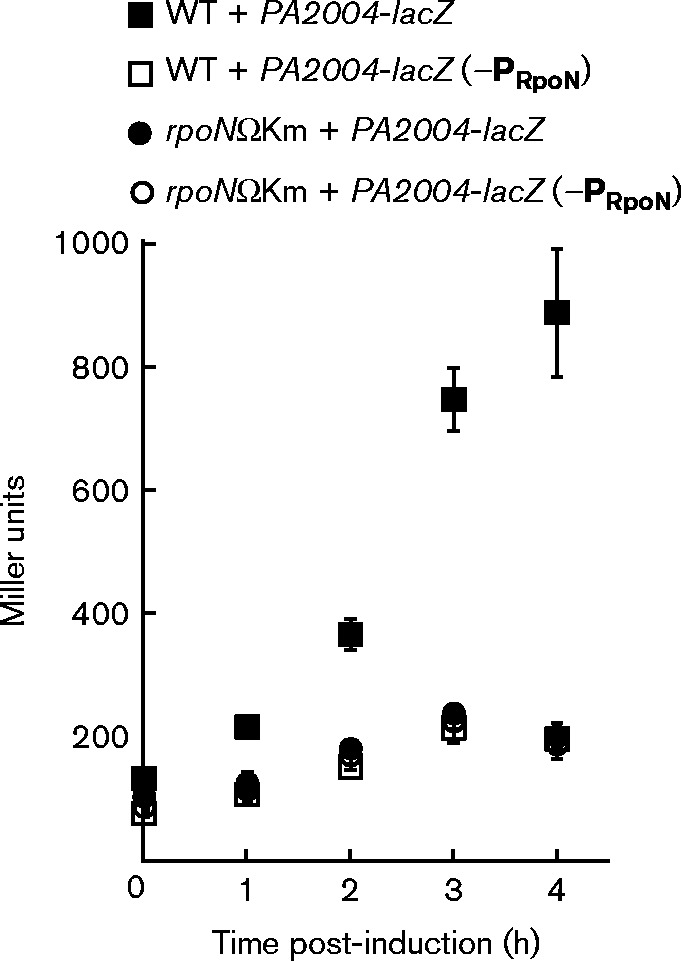
(R)-3-HB induction of PA2004-lacZ was RpoN dependent. There is a putative − 24/ − 12 or RpoN promoter located 148 bp upstream of the PA2004 ORF, suggesting that RpoN might be involved in the transcription of the PA2004-bdhA operon in response to (R)-3-HB. Addition of 30 mM (R,S)-3-HB did not induce expression of the PA2004-lacZ reporter in an rpoN mutant (rpoNΩKm) of P. aeruginosa PAO1. Furthermore, substitution of the conserved ‘GG’ nucleotides of the − 24 element with ‘AA’ in the RpoN promoter (–PRpoN) of the PA2004-lacZ reporter made it unresponsive to 30 mM (R,S)-3-HB. Cells were grown in LB supplemented with 5 mM l-Gln to an OD600 of 0.3 and then challenged with 30 mM (R,S)-3-HB. LacZ activity was determined 1, 2, 3 and 4 h post-induction. l-Gln was provided to support the growth of the rpoN mutant (Heurlier et al., 2003). Data points represent mean values ± sd (n = 4). ANOVA was performed using a Dunnett's post-hoc test (α-value of 0.05) to identify significant changes (P < 0.0001).
Induction of the PA2004-lacZ reporter was dependent on PA2005
(R)-3-HB and RpoN were observed to be crucial determinants for the induction of the PA2004-lacZ reporter in P. aeruginosa PAO1. These results in combination with the previous finding that a PA2005 : : Tn mutant had reduced growth on (R)-3-HB made PA2005 a prime candidate for being the EBP that participates with RpoN to activate transcription of the PA2004-bdhA operon in response to (R)-3-HB. Indeed, the addition of 30 mM (R,S)-3-HB did not induce expression of PA2004-lacZ in a PA2005 : : Tn mutant (Fig. 4). For bdhA : : Tn, PA2004 : : Tn and WT P. aeruginosa PAO1, PA2004-lacZ expression increased more than threefold with the addition of 30 mM (R,S)-3-HB (Fig. 4).
Fig. 4.

(R)-3-HB induction of PA2004-lacZ required the PA2005 gene. bdhA : : Tn, PA2004 : : Tn, PA2005 : : Tn and WT P. aeruginosa PAO1 harbouring the PA2004-lacZ reporter were grown in (a) LB or M63 minimal medium that was supplemented with either (b) 30 mM succinate or (c) 30 mM acetate. At an OD600 of 0.3, (R,S)-3-HB was added to a final concentration of 30 mM, and LacZ activity was determined 1, 2, 3 and 4 h post-induction. LacZ activity increased for bdhA : : Tn, PA2004 : : Tn and WT cells when challenged with (R,S)-3-HB. In contrast, (R,S)-3-HB did not induce LacZ activity in the PA2005 : : Tn mutant under any growth condition. Data points represent mean values ± sd (n = 4). ANOVA was performed using a Dunnett's post-hoc test (α-value of 0.05) to identify significant changes (P < 0.0001).
The LacZ findings were validated by assaying (R)-3-HB dehydrogenase activity for the bdhA : : Tn, PA2004 : : Tn and PA2005 : : Tn mutants. Cells were grown in LB to an OD600 of 0.3 and subsequently induced with either 0 or 30 mM (R,S)-3-HB. At 4 h post-induction, cells were harvested, washed, and lysed by sonication, and the resulting lysates were assayed for BdhA activity. For WT cells, BdhA activity increased from 93 ( ± 5.9) to 395 ( ± 17) U mg− 1 with the addition of (R,S)-3-HB. There was no detectable BdhA activity present in the bdhA : : Tn mutant under either condition. The PA2004 : : Tn mutants had a non-induced BdhA activity of 16 ( ± 0.7) U mg− 1, which marginally increased to 23 ( ± 2.4) U mg− 1 with the addition of (R,S)-3-HB. For the PA2005 : : Tn mutant, non-induced and induced cells had BdhA activities of 23 ( ± 0.9) and 19 ( ± 2.3) U mg− 1, respectively. These values were ∼20-fold lower than that of WT P. aeruginosa PAO1 challenged with (R,S)-3-HB. The measured BdhA activities are consistent with the LacZ data, indicating that PA2005 is necessary for the induction of BdhA expression in response to (R)-3-HB.
(R)-3-HB induced expression of PA2004-lacZ in E. coli Top10 that heterologously expressed PA2005
Expression of the PA2004-lacZ reporter was assayed in non-native E. coli Top10, which simultaneously expressed the PA2005 gene from pTrc99a. As shown in Fig. 5, the basal expression of PA2005 from the trc promoter of pTrc99a caused a >60-fold induction of PA2004-lacZ in E. coli Top10 when challenged with either (R)-3-HB or (R,S)-3-HB. LacZ activity increased from 50 MU to >3000 MU at 2 h post-addition of 30 mM (R)-3-HB or (R,S)-3-HB. (R)-3-HB induction was not observed in the absence of PA2005 or when the RpoN promoter in the PA2004-lacZ reporter was altered (Fig. 5a). As observed earlier, the addition of (R)-2-HB or acetoacetate did not induce expression of PA2004-lacZ (Fig. 5b). These findings demonstrate that, even in a non-native host, RpoN and PA2005 are sufficient and essential for the (R)-3-HB induction of the PA2004-lacZ reporter, and thus are key regulators of the PA2004-bdhA operon in P. aeruginosa PAO1.
Fig. 5.

PA2005 was essential for (R)-3-HB induction of PA2004-lacZ in E. coli. The lacZ-deficient E. coli Top10 strain was co-transformed with the PA2004-lacZ reporter and the PA2005 gene, which was carried on the pTrc99a expression plasmid. (a) Basal expression of the PA2005 gene from the trc promoter of pTrc99a was sufficient for (R)-3-HB induction of PA2004-lacZ (PA2004-lacZ + PA2005). Induction was not observed for the PA2004-lacZ reporter when (i) the PA2005 gene was cloned in a backwards orientation relative to the trc promoter in pTrc99a (PA2004-lacZ – PA2005), (ii) no PA2005 gene was present, only pTrc99a plasmid (PA2004-lacZ + pTrc99a), or (iii) the ‘GG’ nucleotides of the − 24 element of the RpoN promoter were substituted with ‘AA’ [PA2004-lacZ (–PRpoN) + PA2005]. (b) Induction of PA2004-lacZ was specific for (R)-3-HB. In total, these results argue that PA2005 is the EBP responsible for activating transcription from the RpoN promoter upstream of PA2004-bdhA in response to (R)-3-HB. Cells were grown in LB to an OD600 of 0.3 and then challenged with 30 mM of substrate. LacZ activity was measured 2 h post-induction. Data points represent mean values ± sd (n = 4). ANOVA was performed using a Dunnett's post-hoc test (α-value of 0.05) to identify significant changes (P < 0.0001), which are marked with an asterisk.
Promoter region of PA2004 was bound by purified His6-PA2005
EMSA was used to determine if PA2005 actually binds to the PA2004 promoter region. To this end, a 5′-labelled Cy5 probe, which spanned the region 237–122 bp upstream of the PA2004 ORF, was used in an EMSA with purified His6-PA2005. As shown in Fig. 6a, purified His6-PA2005 retarded the mobility of the PA2004 promoter probe but did not change the mobility of a non-specific target promoter, i.e. the promoter region of gcvH2, which encodes a glycine cleavage protein (Lundgren et al., 2013). When increasing concentrations of His6-PA2005 from 0 to 400 nM were incubated with the PA2004 promoter probe, a mobility shift was observed for His6-PA2005 concentrations as low as 12.5 nM and the shift intensity increased with the concentration of His6-PA2005 (Fig. 6b). These results indicate that PA2005 binds with high specificity and affinity to the PA2004 promoter region.
Fig. 6.

The promoter region of PA2004 was bound by His6-PA2005.EMSAs were performed with His6-PA2005 and 1.0 nM 5′-labelled Cy5 probe DNA unless specified otherwise. (a) PPA2004 (specific) or PgcvH2 (non-specific) were incubated in the absence (lanes 1 and 3, respectively) or presence (lanes 2 and 4, respectively) of 500 nM PA2005.The shift in the position of PPA2004 in lane 2 confirms that PA2005 binds to PPA2004. No shift corresponding to binding of the non-specific probe PgcvH2 by PA2005 was observed in lane 4. (b) His6-PA2005 (0 to 400 nM) was incubated with PPA2004. A shift in PPA2004 was observed for His6-PA2005 concentrations as low as 12.5 nM, indicating that His6-PA2005 binds with high affinity to PPA2004. The intensity of the shift increased with increasing His6-PA2005 concentration.
Assimilation of (±)-1,3-butanediol requires the bdhA gene
(R)-3-HB utilization was dependent on the bdhA gene and its regulator PA2005. However, it was not known whether BdhA and/or PA2005 were necessarily essential for the assimilation of other compounds that are metabolized through (R)-3-HB intermediates. The compound ( ± )-1,3-butanediol is a common industrial chemical that is converted into (R)-3-HB by both eukaryotic and bacterial cells (Kersters & De Ley, 1963; Tate et al., 1971; Ugwu et al., 2011). Nothing is known about the metabolism of ( ± )-1,3-butanediol in Pseudomonas except for a study that identified this molecule as a substrate for a quinoprotein alcohol dehydrogenase of P. putida (Toyama et al., 1995). We found that P. aeruginosa PAO1 could grow on ( ± )-1,3-butanediol when provided as a carbon source (Fig. 7). This metabolism was dependent on bdhA, as the bdhA : : Tn mutant was unable to grow on ( ± )-1,3-butanediol (Fig. 7a). Notably, this result reaffirms that (R)-3-HB is an intermediate of ( ± )-1,3-butanediol metabolism in P. aeruginosa PAO1. Disruptions of the PA2004 and PA2005 genes did not abolish growth of P. aeruginosa PAO1 on ( ± )-1,3-butanediol. The PA2004 : : Tn (Fig. 7b) and PA2005 : : Tn (Fig. 7c) mutants exhibited only reduced growth on this diol.
Fig. 7.

The bdhA gene was necessary for the growth of P. aeruginosa PAO1 on ( ± )-1,3-butanediol. (R)-3-HB is an intermediate of ( ± )-1,3-butanediol catabolism. (a) The bdhA : : Tn mutant failed to grow on ( ± )-1,3-butanediol as the sole carbon source. (b, c) The PA2004 : : Tn (b) and PA2005 : : Tn (c) mutants displayed reduced growth on ( ± )-1,3-butanediol, which suggested that the expression levels of the bdhA gene in these mutants were high enough to produce adequate amounts of (R)-3-HB dehydrogenase for converting intermediate (R)-3-HB into acetoacetate. Data points represent mean values ± sd (n = 4). ANOVA was performed using a Dunnett's post-hoc test (α-value of 0.05) to identify significant changes (P < 0.0001).
Expression of the PA2004-lacZ reporter remained unchanged (background levels) for PA2004 : : Tn, PA2005 : : Tn and WT P. aeruginosa PAO1 cells when grown on ( ± )-1,3-butanediol (Fig. S1, available in the online Supplementary Material). In contrast, expression of PA2004-lacZ increased twofold for the bdhA : : Tn mutant in the presence of ( ± )-1,3-butanediol. Only in the absence of bdhA does intermediate (R)-3-HB reach concentrations that induce expression of the PA2004-lacZ reporter. It would appear that the catabolism of ( ± )-1,3-butanediol does not generate sufficient levels of (R)-3-HB to trigger the induction of the PA2004-bdhA operon via RpoN-PA2005.
Discussion
The PA2004-bdhA operon is conserved among many Pseudomonas spp., suggesting that it is a core unit and regulatory site in (R)-3-HB catabolism. Analysis of a PA2004-lacZ reporter and BdhA enzymic activity identified several key factors involved in the expression of the PA2004-bdhA operon in P. aeruginosa PAO1. First, the expression of the PA2004-bdhA operon is inducible by (R)-3-HB (Fig. 2). This induction or transcriptional activation was completely dependent on the alternative sigma factor RpoN and its cognate − 24/ − 12 promoter positioned 148 bp upstream from the start codon of PA2004. (R)-3-HB did not induce expression of the PA2004-lacZ reporter in an rpoN mutant, while replacement of the conserved ‘GG’ motif of the − 24 element with ‘AA’ in the RpoN promoter of the PA2004-lacZ reporter rendered it unresponsive to exogenous (R)-3-HB (Fig. 3). Interestingly, the operons encoding the BdhA-PA2004 homologues in P. putida KT2440 (PP_3073-PP_3074) and Pseudomonas fluorescens SBW25 (PFLU2628-PFLU2629) are preceded by RpoN promoters at 65 and 72 bp upstream of PP_3074 and PFLU2629, respectively (Conway & Boddy, 2012). Additionally, adjacent to each of these operons is a gene (PP_3075, PFLU2630) encoding an EBP that is homologous to PA2005. EBPs are essential participants in RpoN-mediated transcription (Studholme & Buck, 2000), and therefore regulation of (R)-3-HB assimilation for some Pseudomonas spp. might be dependent on RpoN and an EBP homologous to PA2005.
The results of this study strongly suggest that the PA2005 gene encodes the EBP that interacts with RpoN to activate transcription of the PA2004-bdhA operon in response to (R)-3-HB. It was observed that the addition of exogenous (R)-3-HB did not induce expression of the PA2004-lacZ reporter nor did it lead to an increase in BdhA activity in a PA2005 : : Tn mutant compared with WT P. aeruginosa PAO1 (Fig. 4). We also found that (R)-3-HB induced the expression of the PA2004-lacZ reporter in E. coli, but only if the E. coli cells were expressing the PA2005 gene from the trc promoter of the plasmid pTrc99a (Fig. 5). The presence of PA2005 was essential for the (R)-3-HB induction of PA2004-lacZ in non-native E. coli. Lastly, purified PA2005 protein did bind to a probe resembling the promoter region of PA2004 (Fig. 6), which supports PA2005 being a direct regulator of the PA2004-bdhA operon.
The EBP PA2005 is not a homologue of any previously characterized EBPs. Pfam analysis of PA2005 identified an N-terminal PAS-4 domain (residues 25–116), the central RpoN-interaction domain (residues 158–326) and a C-terminal FIS-type helix–turn–helix (HTH) (residues 425–466). The PAS-4 domain most likely has a role in sensing intracellular (R)-3-HB, whereas the FIS-type HTH is expected to bind to a DNA motif upstream of the RpoN promoter of PA2004-bdhA. More in-depth EMSAs with PA2005 are expected to identify the DNA-binding sites for this transcriptional regulator. Based on the results in the current study, PA2005 was given the name HbcR for (R)-3- hydroxybutyrate catabolism regulator.
The assimilation of (R)-3-HB as a main carbon source was dependent on the induction of the PA2004-bdhA operon via RpoN-PA2005. In contrast, induction of the PA2004-bdhA operon was not observed when P. aeruginosa PAO1 was grown on ( ± )-1,3-butanediol even though (R)-3-HB is an intermediate in the breakdown of this diol (Fig. S1). This finding indicates that non-induced levels of BdhA are sufficient to convert any available (R)-3-HB into acetoacetate during the metabolism of ( ± )-1,3-butanediol. Importantly, non-induced levels of BdhA might support the growth of P. aeruginosa PAO1 on other molecules in which the formation of (R)-3-HB is a rate-limiting step in the catabolic process. For example, Pseudomonas spp. have been reported to hydrolyse PHB polyhydroxybutyrate (PHB) granules produced by other bacteria (Jendrossek et al., 1996), and subsequently use the liberated or free (R)-3-HB as a growth substrate. If the hydrolysis of the PHB granule is relatively slow then (R)-3-HB is not expected to accumulate to concentrations that lead to the induction of the PA2004-bdhA operon, and thus a similar situation to that described for ( ± )-1,3-butanediol metabolism will be observed. Whether P. aeruginosa PAO1 can degrade PHB granules remains to be determined, and what implications (R)-3-HB induction of PA2004-bdhA has for the assimilation of PHB granules in the environment is a subject worth exploring.
Acknowledgements
We acknowledge the grant NIH P30 DK089507 for funding the P. aeruginosa PAO1 transposon mutant used for our study. We also thank T. Duncan (SUNY Upstate) for access and use of the Typhoon Imager for EMSA experiments. This study was made possible by NIH R15 GM104880-01A1 and NSF CBET 1263905 awards to C. T. N. and funds from the Undergraduate Honours Program of SUNY-ESF.
Supplementary Data
Supplementary Data
Abbreviations:
- EMSA
electrophoretic mobility shift assay
- EBP
enhancer-binding protein
- PHA
polyhydroxyalkanoate
- PHB
polyhydroxybutyrate
- (R)-2-HB
(R)-2-hydroxybutyrate
- (R)-3-HB
(R)-3-hydroxybutyrate
References
- Akram M. (2013). A focused review of the role of ketone bodies in health and disease J Med Food 16 965–967 10.1089/jmf.2012.2592 . [DOI] [PubMed] [Google Scholar]
- Anderson A.J., Dawes E.A. (1990). Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates Microbiol Rev 54 450–472 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrios H., Valderrama B., Morett E. (1999). Compilation and analysis of σ54-dependent promoter sequences Nucleic Acids Res 27 4305–4313 10.1093/nar/27.22.4305 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brashear A., Cook G.A. (1983). A spectrophotometric, enzymatic assay for d-3-hydroxybutyrate that is not dependent on hydrazine Anal Biochem 131 478–482 10.1016/0003-2697(83)90201-4 . [DOI] [PubMed] [Google Scholar]
- Buck M., Cannon W. (1992). Specific binding of the transcription factor sigma-54 to promoter DNA Nature 358 422–424 10.1038/358422a0 . [DOI] [PubMed] [Google Scholar]
- Conway K., Boddy C.N. (2012). Sigma 54 Promoter Database. (www.sigma54.ca).
- Feller C., Günther R., Hofmann H.J., Grunow M. (2006). Molecular basis of substrate recognition in d-3-hydroxybutyrate dehydrogenase from Pseudomonas putida ChemBioChem 7 1410–1418 10.1002/cbic.200600167 . [DOI] [PubMed] [Google Scholar]
- Heurlier K., Dénervaud V., Pessi G., Reimmann C., Haas D. (2003). Negative control of quorum sensing by RpoN (σ54) in Pseudomonas aeruginosa PAO1 J Bacteriol 185 2227–2235 10.1128/JB.185.7.2227-2235.2003 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman G.W., de Leeuw O., Eggink G., Witholt B. (1989). Synthesis of poly-3-hydroxyalkanoates is a common feature of fluorescent pseudomonads Appl Environ Microbiol 55 1949–1954 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Nakajima Y., Ichihara E., Ogawa K., Katayama N., Nakashima K., Yoshimoto T. (2006). D-3-hydroxybutyrate dehydrogenase from Pseudomonas fragi: molecular cloning of the enzyme gene and crystal structure of the enzyme J Mol Biol 355 722–733 10.1016/j.jmb.2005.10.072 . [DOI] [PubMed] [Google Scholar]
- Jacobs M.A., Alwood A., Thaipisuttikul I., Spencer D., Haugen E., Ernst S., Will O., Kaul R., Raymond C., other authors (2003). Comprehensive transposon mutant library of Pseudomonas aeruginosa Proc Natl Acad Sci U S A 100 14339–14344 10.1073/pnas.2036282100 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendrossek D., Handrick R. (2002). Microbial degradation of polyhydroxyalkanoates Annu Rev Microbiol 56 403–432 10.1146/annurev.micro.56.012302.160838 . [DOI] [PubMed] [Google Scholar]
- Jendrossek D., Schirmer A., Schlegel H.G. (1996). Biodegradation of polyhydroxyalkanoic acids Appl Microbiol Biotechnol 46 451–463 10.1007/s002530050844 . [DOI] [PubMed] [Google Scholar]
- Kersters K., De Ley J. (1963). The oxidation of glycols by acetic acid bacteria Biochim Biophys Acta 71 311–331 10.1016/0006-3002(63)91086-2 . [DOI] [PubMed] [Google Scholar]
- Kovach M.E., Elzer P.H., Hill D.S., Robertson G.T., Farris M.A., Roop R.M., II, Peterson K.M. (1995). Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes Gene 166 175–176 10.1016/0378-1119(95)00584-1 . [DOI] [PubMed] [Google Scholar]
- Lu J.N., Tappel R.C., Nomura C.T. (2009). Mini review: biosynthesis of poly(hydroxyalkanoates) Polym Rev (Phila Pa) 49 226–248 10.1080/15583720903048243. [DOI] [Google Scholar]
- Lundgren B.R., Thornton W., Dornan M.H., Villegas-Peñaranda L.R., Boddy C.N., Nomura C.T. (2013). Gene PA2449 is essential for glycine metabolism and pyocyanin biosynthesis in Pseudomonas aeruginosa PAO1 J Bacteriol 195 2087–2100 10.1128/JB.02205-12 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren B.R., Villegas-Peñaranda L.R., Harris J.R., Mottern A.M., Dunn D.M., Boddy C.N., Nomura C.T. (2014). Genetic analysis of the assimilation of C5-dicarboxylic acids in Pseudomonas aeruginosa PAO1 J Bacteriol 196 2543–2551 10.1128/JB.01615-14 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morett E., Segovia L. (1993). The sigma 54 bacterial enhancer-binding protein family: mechanism of action and phylogenetic relationship of their functional domains J Bacteriol 175 6067–6074 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountassif D., Andreoletti P., Cherkaoui-Malki M., Latruffe N., El Kebbaj M.S. (2010). Structural and catalytic properties of the d-3-hydroxybutyrate dehydrogenase from Pseudomonas aeruginosa Curr Microbiol 61 7–12 10.1007/s00284-009-9568-7 . [DOI] [PubMed] [Google Scholar]
- Nakajima Y., Ito K., Ichihara E., Ogawa K., Egawa T., Xu Y., Yoshimoto T. (2005). Crystallization and preliminary X-ray characterization of d-3-hydroxybutyrate dehydrogenase from Pseudomonas fragi Acta Crystallogr F Struct Biol Cryst Commun 61 36–38 10.1107/S1744309104024741 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paithankar K.S., Feller C., Kuettner E.B., Keim A., Grunow M., Sträter N. (2007). Cosubstrate-induced dynamics of d-3-hydroxybutyrate dehydrogenase from Pseudomonas putida FEBS J 274 5767–5779 10.1111/j.1742-4658.2007.06102.x . [DOI] [PubMed] [Google Scholar]
- Pardee A.B., Jacob F., Monod J. (1959). The genetic control and cytoplasmic expression of inducibility in the synthesis of β-galactosidase by E. coli J Mol Biol 1 165–178 10.1016/S0022-2836(59)80045-0. [DOI] [Google Scholar]
- Studholme D.J., Buck M. (2000). The biology of enhancer-dependent transcriptional regulation in bacteria: insights from genome sequences FEMS Microbiol Lett 186 1–9 10.1111/j.1574-6968.2000.tb09074.x . [DOI] [PubMed] [Google Scholar]
- Tate R.L., Mehlman M.A., Tobin R.B. (1971). Metabolic fate of 1,3-butanediol in the rat: conversion to β-hydroxybutyrate J Nutr 101 1719–1726 . [DOI] [PubMed] [Google Scholar]
- Timm A., Steinbüchel A. (1990). Formation of polyesters consisting of medium-chain-length 3-hydroxyalkanoic acids from gluconate by Pseudomonas aeruginosa and other fluorescent pseudomonads Appl Environ Microbiol 56 3360–3367 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama H., Fujii A., Matsushita K., Shinagawa E., Ameyama M., Adachi O. (1995). Three distinct quinoprotein alcohol dehydrogenases are expressed when Pseudomonas putida is grown on different alcohols J Bacteriol 177 2442–2450 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugwu C.U., Tokiwa Y., Ichiba T. (2011). Production of (R)-3-hydroxybutyric acid by fermentation and bioconversion processes with Azohydromonas lata Bioresour Technol 102 6766–6768 10.1016/j.biortech.2011.03.073 . [DOI] [PubMed] [Google Scholar]
- Winsor G.L., Lam D.K., Fleming L., Lo R., Whiteside M.D., Yu N.Y., Hancock R.E., Brinkman F.S. (2011). Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes Nucleic Acids Res 39 D596–D600 10.1093/nar/gkq86920929876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data