

Abstract
Red blood cell (RBC) alloimmunization can be a life-threatening complication for patients with sickle cell disease (SCD) receiving therapeutic transfusions. Despite provision of extended antigen-matched donor RBCs, patients continue to develop antibodies due to high degree of polymorphisms in the immunogenic antigens in individuals of African ancestry. Identification of biomarkers of alloimmunization in this patient population is therefore of great interest and will help to identify in advance patients most likely to make antibodies in response to transfusion. We have recently identified altered T cell responses and innate immune abnormalities in alloimmunized SCD patients. In this paper, we summarize this work and propose our working model of how innate immune abnormalities can contribute to pathogenic T cell responses in alloimmunized SCD patients. We believe that unravelling the basis of such altered interactions at the cellular and molecular level will help future identification of biomarkers of alloimmunization with the goal that this information will ultimately help guide therapy in these patients.
Keywords: Sickle cell, Alloimmunization, Heme, Heme oxygenase, Tregs, Th1, Transfusions
1. Introduction
SCD results from a mutation in the β-globin gene which causes hemoglobin (Hb) S to polymerize when deoxygenated, forming rigid polymers within RBCs. Hb S containing RBCs are deformed in sickle-prone conditions, resulting in chronic hemolytic anemia, shortened RBC survival in circulation, increased reticulocytosis, periodic painful vaso-occlusive crises (VOCs), and end organ damage due to persistent tissue hypoxia [1]. RBC transfusions remain a very important modality of treatment for patients with SCD with majority (60–90%) of patients receiving RBC transfusions in their lifetime [2]. Its use has been in primary and secondary stroke prophylaxis and treatment, acute chest syndrome treatment and preoperative prophylaxis [3]. Despite its benefits, RBC transfusion results in alloimmunization in approximately 20–50% of patients with SCD [4]. Alloantibodies can cause delayed hemolytic transfusion reactions which in SCD patients can trigger hyperhemolysis, a life-threatening poorly understood phenomena in which the transfused and the patient’s own RBCs are destroyed [4]. In addition, finding compatible units for patients with alloantibodies can be difficult and identifying and characterizing the antibodies can be costly, time-consuming and laborious, causing transfusion delays. As such, alloimmunization is associated with morbidity and mortality in chronically transfused SCD patients. However for the indications described above, few additional/alternative therapeutic options besides transfusion exist. Differences in minor (non-ABO) blood group antigens between mostly Caucasian blood donors and transfusion recipients who are of African descent are mainly responsible for formation of alloantibodies [5]. Even with provision of Rh-D, -C, and -E antigen-matched donor RBCs, patients continue to develop Rh antibodies, which may in part be due to genetic diversity of the RH locus in donors of African ancestry; many of these antibodies are considered clinically significant [6]. This highlights the need for better characterization of triggers of alloimmunization and identification of risk factors in this highly vulnerable population. Genetic as well as acquired patient-related factors are likely to influence the process of alloimmunization. In a small study of chronically transfused patients with SCD, we recently reported reduced peripheral regulatory T cell (Treg) suppressive function (in the absence of accessory cells) and altered Th responses with higher circulating IFN-α (Th1 cytokine) but lower IL-10 (anti-inflammatory) cytokine levels in antibody responders as compared to non-responders [7]. These data are consistent with a model in which a generalized immune dysregulation exists in SCD alloimmunized patients with an imbalance between the regulatory (Tregs) and effector (Th) cells, possibly due to an underlying inflammatory state [8]. As a result, the model predicts that the likelihood of antibody production is increased (Fig. 1) since Tregs can suppress B cells either directly [9,10] or indirectly through inhibition of activation/expansion of effector Th cells which in turn control IgG antibody responses. Understanding how Treg/Th differentiation and expansion are controlled is thus likely to provide an explanation of how alloimmunization may ensue.
Fig. 1.
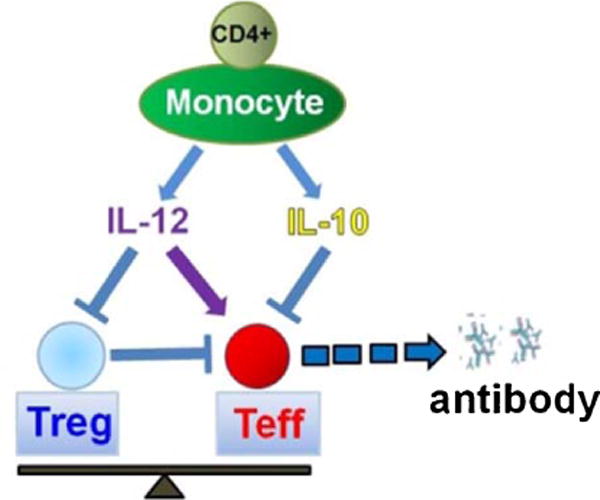
Working model of monocyte control of T cells that leads to antibody production by B cells. Balance between Tregs and effector T cell (Teff) is dictated by cytokines secreted by T cell-monocyte interactions.
2. Heme and heme oxygenase I
Heme oxygenase 1 (HO-1) is expressed in various cell types and its expression can be induced in response to its substrate heme as well as acute stress stimuli [11]. Through its enzymatic activity, HO-1 breaks down the pro-oxidant heme into iron, bilirubin and carbon monoxide, thereby conferring cytoprotective and anti-inflammatory effects via heme breakdown products as well as by reducing intracellular heme availability [12–17]. Deficiency of HO-1 in mice, and in the one reported case in human is associated with chronic inflammatory state [18]. HO-1 is upregulated in SCD [19–21]. Furthermore, modulation of HO-1 expression in mouse models appears to affect vascular inflammation and vaso-occlusion with high HO-1 levels increasing microvasculature blood flow whereas attenuated HO-1 levels associated with increased red blood stasis [12,15,22]. In non-SCD setting, HO-1 is considered immunosuppressive as it was shown to inhibit T lymphocyte proliferation [11], block maturation of dendritic cells (DCs) and inhibit proinflammatory and allogeneic immune responses [23,24]. In myeloid derived cells (specifically monocyte/macrophage/DCs), HO-1 expression inhibits inflammatory cytokine secretion (IL-6, IL-12, TNFα, IL-1β) and increases regulatory cytokine (IL-10) expression [25–27] HO-1 levels/activity in response to its substrate (e.g. hemin) can thus be thought of as a critical parameter to switch the proinflammatory activity of monocyte/macrophages into an immunoregulatory one.
3. T cell responses to hemin in SCD alloimmunization
Human monocytes which are generally regarded as precursors of tissue macrophages and dendritic cells (DCs) [28], are increasingly recognized for their ability to trigger and polarize Th responses [29,30] as well as to both stimulate and suppress T-cell responses [30,31]. Such T cell-monocyte interactions are likely to occur in secondary lymphoid organs such as the spleen, but also in inflamed tissues [30]. In a group of patients with or without a history of alloimmunization, we found differences in monocyte control of Treg/Th cells in alloimmunized vs non-alloimmunized SCD patients in part due to altered secretion/responsiveness to IL-12 [32]. Surprisingly, baseline HO-1 levels in CD16+ monocyte subset from alloimmunized patients were lower as compared to non-alloimmunized patients [32]. More importantly, in response to hemin, a surrogate marker for transfused RBC breakdown products, HO-1 levels were still lower and alloimmunized monocytes were unable to dampen Th1 proliferation and were less effective in increasing Treg expansion as compared to non-alloimmunized group [32]. Since the monocyte/macrophage system of the spleen and liver is responsible for extravascular clearance of transfused RBCs, as a working model, we hypothesize that inadequate levels/activity of HO-1 in SCD monocytes in these tissues is associated with alloimmunization. Thus, low baseline HO-1 in SCD alloimmunized monocyte/macrophage of liver/spleen may lead to ineffective removal of heme following RBC transfusion, resulting in a proinflammatory state (high IL-12) that drives effector T cell proliferation while inhibiting Treg development, thus increasing the likelihood of developing B cell responses against allogeneic determinants on transfused RBCs i.e. alloimmunization (Fig. 2). In contrast, effective removal of heme (in non-alloimmunized) ensures an anti-inflammatory (low IL-12), immunoregulatory state (high Treg/low Teff) that is less conducive to alloimmunization. A goal of our research program is to better understand the immune etiology of alloimmunization in SCD as a means to identify novel pathways (e.g. HO-1) for averting alloimmunization in SCD, and perhaps other conditions.
Fig. 2.
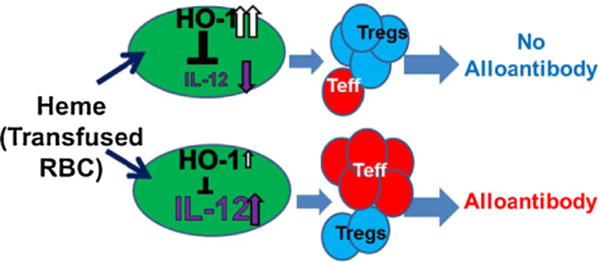
Working model HO-1 basal levels in monocytes is critical in T cell polarization in response to hemin/transfused RBCs. Secretion of pro-inflammatory (IL-12) by monocytes in response to transfused RBCs/hemin will depend on the baseline HO-1 levels. Higher Treg/T effector ratios will suppress T cell activation and B cell humoral immunity whereas lower Treg/T effector ratios will trigger alloimmunization.
4. Conclusions and future directions
In conclusion, our data indicate that in alloimmunized patients with SCD, monocyte subsets, which are increasingly recognized as key regulators of T cell development, are altered in their ability to control T cell expansion and that their ability to develop an anti-inflammatory response to hemin is impaired due to their lower baseline monocyte HO-1 levels. Our overall hypothesis is that inadequate levels/activity of HO-1 alters the anti-inflammatory state of the sickle innate immune cells following RBC transfusion, resulting in pathogenic T cell responses against RBCs and alloimmunization. Our future studies will determine the functional consequence of culturing monocytes, ex vivo, from SCD patients with or without alloantibodies in the absence or presence of hemin. This simple in vitro model of T cell-monocyte interactions which we envision occur in vivo in SCD spleen (remnants) and liver where transfused RBCs are cleared, allows us to examine at the molecular level the direct functional consequence of the action of sickle monocytes and the role of heme/HO-1 on T cell function. As biomarkers for responder/non-responders in human SCD are lacking, our belief is that combining mechanistic and prospective longitudinal studies should produce valuable information to meet this diagnostic and intellectual void.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (R01HL121415; Yazdanbakhsh), and an American Heart Association grant (14GRNT20480109; Yazdanbakhsh).
Footnotes
Cette communication s’est déroulée dans le cadre du XXVIIe Congrès de la Société française de transfusion sanguine (SFTS), qui s’est tenu à Montpellier, du 15 au 17 septembre 2015.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
References
- 1.Frenette PS, Atweh GF. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest. 2007;117:850–8. doi: 10.1172/JCI30920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walter PB, Harmatz P, Vichinsky E. Iron metabolism and iron chelation in sickle cell disease. Acta Haematol. 2009;122:174–83. doi: 10.1159/000243802. [DOI] [PubMed] [Google Scholar]
- 3.Josephson CD, Su LL, Hillyer KL, Hillyer CD. Transfusion in the patient with sickle cell disease: a critical review of the literature and transfusion guidelines. Transfus Med Rev. 2007;21:118–33. doi: 10.1016/j.tmrv.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Yazdanbakhsh K, Ware RE, Noizat-Pirenne F. Red blood cell alloimmunization in sickle cell disease: pathophysiology, risk factors, and transfusion management. Blood. 2012;120:528–37. doi: 10.1182/blood-2011-11-327361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vichinsky EP. Current issues with blood transfusions in sickle cell disease. Semin Hematol. 2001;38:14–22. doi: 10.1016/s0037-1963(01)90056-3. [DOI] [PubMed] [Google Scholar]
- 6.Chou ST, Jackson T, Vege S, Smith-Whitley K, Friedman DF, Westhoff CM. High prevalence of red blood cell alloimmunization in sickle cell disease despite transfusion from Rh-matched minority donors. Blood. 2013;122:1062–71. doi: 10.1182/blood-2013-03-490623. [DOI] [PubMed] [Google Scholar]
- 7.Bao W, Zhong H, Li X, Lee MT, Schwartz J, Sheth S, et al. Immune regulation in chronically transfused allo-antibody responder and nonresponder patients with sickle cell disease and beta-thalassemia major. Am J Hematol. 2011;86:1001–6. doi: 10.1002/ajh.22167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Platt OS. Sickle cell anemia as an inflammatory disease. J Clin Invest. 2000;106:337–8. doi: 10.1172/JCI10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim HW, Hillsamer P, Banham AH, Kim CH. Cutting edge: direct suppression of B cells by CD4+ CD25+ regulatory T cells. J Immunol. 2005;175:4180–3. doi: 10.4049/jimmunol.175.7.4180. [DOI] [PubMed] [Google Scholar]
- 10.Iikuni N, Lourenco EV, Hahn BH, La CA. Cutting edge: regulatory T cells directly suppress B cells in systemic lupus erythematosus. J Immunol. 2009;183:1518–22. doi: 10.4049/jimmunol.0901163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 12.Loboda A, Jazwa A, Grochot-Przeczek A, Rutkowski AJ, Cisowski J, Agarwal A, et al. Heme oxygenase-1 and the vascular bed: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2008;10:1767–812. doi: 10.1089/ars.2008.2043. [DOI] [PubMed] [Google Scholar]
- 13.Sedlak TW, Saleh M, Higginson DS, Paul BD, Juluri KR, Snyder SH. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc Natl Acad Sci USA. 2009;106:5171–6. doi: 10.1073/pnas.0813132106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minetti M, Mallozzi C, Di Stasi AM, Pietraforte D. Bilirubin is an effective antioxidant of peroxynitrite-mediated protein oxidation in human blood plasma. Arch Biochem Biophys. 1998;352:165–74. doi: 10.1006/abbi.1998.0584. [DOI] [PubMed] [Google Scholar]
- 15.Belcher JD, Beckman JD, Balla G, Balla J, Vercellotti G. Heme degradation and vascular injury. Antioxid Redox Signal. 2010;12:233–48. doi: 10.1089/ars.2009.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willis D, Moore AR, Frederick R, Willoughby DA. Heme oxygenase: a novel target for the modulation of the inflammatory response. Nat Med. 1996;2:87–90. doi: 10.1038/nm0196-87. [DOI] [PubMed] [Google Scholar]
- 17.Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, et al. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest. 1992;90:267–70. doi: 10.1172/JCI115847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci USA. 1997;94:10919–24. doi: 10.1073/pnas.94.20.10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nath KA, Grande JP, Haggard JJ, Croatt AJ, Katusic ZS, Solovey A, et al. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am J Pathol. 2001;158:893–903. doi: 10.1016/S0002-9440(10)64037-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jison ML, Munson PJ, Barb JJ, Suffredini AF, Talwar S, Logun C, et al. Blood mononuclear cell gene expression profiles characterize the oxidant, hemolytic, and inflammatory stress of sickle cell disease. Blood. 2004;104:270–80. doi: 10.1182/blood-2003-08-2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lanaro C, Franco-Penteado CF, Albuqueque DM, Saad ST, Conran N, Costa FF. Altered levels of cytokines and inflammatory mediators in plasma and leukocytes of sickle cell anemia patients and effects of hydroxyurea therapy. J Leukoc Biol. 2009;85:235–42. doi: 10.1189/jlb.0708445. [DOI] [PubMed] [Google Scholar]
- 22.Beckman JD, Belcher JD, Vineyard JV, Chen C, Nguyen J, Nwaneri MO, et al. Inhaled carbon monoxide reduces leukocytosis in a murine model of sickle cell disease. Am J Physiol Heart Circ Physiol. 2009;297:H1243–53. doi: 10.1152/ajpheart.00327.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chauveau C, Remy S, Royer PJ, Hill M, Tanguy-Royer S, Hubert FX, et al. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood. 2005;106:1694–702. doi: 10.1182/blood-2005-02-0494. [DOI] [PubMed] [Google Scholar]
- 24.Remy S, Blancou P, Tesson L, Tardif V, Brion R, Royer PJ, et al. Carbon monoxide inhibits TLR-induced dendritic cell immunogenicity. J Immunol. 2009;182:1877–84. doi: 10.4049/jimmunol.0802436. [DOI] [PubMed] [Google Scholar]
- 25.Ma JL, Yang PY, Rui YC, Lu L, Kang H, Zhang J. Hemin modulates cytokine expressions in macrophage-derived foam cells via heme oxygenase-1 induction. J Pharmacol Sci. 2007;103:261–6. doi: 10.1254/jphs.fp0060270. [DOI] [PubMed] [Google Scholar]
- 26.Kapturczak MH, Wasserfall C, Brusko T, Campbell-Thompson M, Ellis TM, Atkinson MA, et al. Heme oxygenase-1 modulates early inflammatory responses: evidence from the heme oxygenase-1-deficient mouse. Am J Pathol. 2004;165:1045–53. doi: 10.1016/S0002-9440(10)63365-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Listopad J, Asadullah K, Sievers C, Ritter T, Meisel C, Sabat R, et al. Heme oxygenase-1 inhibits T cell-dependent skin inflammation and differentiation and function of antigen-presenting cells. Exp Dermatol. 2007;16:661–70. doi: 10.1111/j.1600-0625.2007.00581.x. [DOI] [PubMed] [Google Scholar]
- 28.Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Ann Rev Immunol. 2009;27:669–92. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- 29.Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Ann Rev Immunol. 2008;26:421–52. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans HG, Gullick NJ, Kelly S, Pitzalis C, Lord GM, Kirkham BW, et al. In vivo activated monocytes from the site of inflammation in humans specifically promote Th17 responses. Proc Natl Acad Sci U S A. 2009;106:6232–7. doi: 10.1073/pnas.0808144106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–44. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 32.Zhong H, Bao W, Friedman D, Yazdanbakhsh K. Hemin controls T cell polarization in sickle cell alloimmunization. J Immunol. 2014;193:102–10. doi: 10.4049/jimmunol.1400105. [DOI] [PMC free article] [PubMed] [Google Scholar]