

Abstract
Purpose
Pancreatic cancer is the fourth leading cause of cancer-related deaths, in which the 5-year survival rate is less than 5%. Current standard of care therapies offer little selectivity and high toxicity. Novel, tumor-selective approaches are desperately needed. Although prior work suggested that β-lapachone (β-lap) could be used for the treatment of pancreatic cancers, the lack of knowledge of the compound’s mechanism of action prevented optimal use of this agent.
Experimental Design
We examined the role of NAD(P)H:quinone oxidoreductase-1 (NQO1) in β-lap–mediated antitumor activity, using a series of MIA PaCa-2 pancreatic cancer clones varying in NQO1 levels by stable shRNA knockdown. The antitumor efficacy of β-lap was determined using an optimal hydroxypropyl-β-cyclodextran (HPβ-CD) vehicle formulation in metastatic pancreatic cancer models.
Results
β-lap–mediated cell death required ~90 enzymatic units of NQO1. Essential downstream mediators of lethality were as follows: (i) reactive oxygen species (ROS); (ii) single-strand DNA breaks induced by ROS; (iii) poly(ADP-ribose)polymerase-1 (PARP1) hyperactivation; (iv) dramatic NAD+/ATP depletion; and (v) programmed necrosis. We showed that 1 regimen of β-lap therapy (5 treatments every other day) efficaciously regressed and reduced human pancreatic tumor burden and dramatically extended the survival of athymic mice, using metastatic pancreatic cancer models.
Conclusions
Because NQO1 enzyme activities are easily measured and commonly overexpressed (i.e., >70%) in pancreatic cancers 5- to 10-fold above normal tissue, strategies using β-lap to efficaciously treat pancreatic cancers are indicated. On the basis of optimal drug formulation and efficacious antitumor efficacy, such a therapy should be extremely safe and not accompanied with normal tissue toxicity or hemolytic anemia.
Introduction
Pancreatic cancer is the fourth leading cause of cancer-related death in the United States. (1). Current standard therapies for these patients include surgery, often in com bination with radiotherapy and/or chemotherapy, and offer a 5-year survival rate of less than 5%, due to the aggressive and invasive nature of this disease and considerable normal tissue toxicity in patients. Most pancreatic cancer patients are not candidates for surgical intervention. They present with locally advanced or metastatic disease and have a median survival of 6–10 or 3–6 months, respectively (2, 3). Thus, new approaches based on tumor-selective targets are desperately needed to efficaciously treat pancreatic cancer. As a result, considerable resources have been invested in the development of novel therapies that target molecular aberrations in pancreatic cancers (3–5), with the hope of exploiting specific markers that are elevated in pancreatic cancer.
One “marker” commonly elevated in various human cancers (6, 7), and particularly in pancreatic tumors (8–11), is NAD(P)H:quinone oxidoreductase-1 (NQO1; E.C.1.6.99.2). NQO1 was overexpressed greater than 10-fold versus associated normal tissue in more than 70% of patients (8–11). It detoxifies most quinones by catalyzing a 2-electron reduction using NADH or NADPH (12), converting them to hydroquinones, bypassing the normally unstable and highly reactive semiquinone intermediates. Hydroquinones are then typically and readily conjugated with glutathione (GSH) via glutathione-S-transferase and excreted, constituting a protective mechanism against quinone-mediated toxicity (13). Dicoumarol [3-3′-methylene-bis(4-hydroxy-coumarin); DIC] is a fairly specific NQO1 inhibitor that competes with NADH/NADPH substrate binding.
NQO1 can, however, catalyze certain quinones to more reactive DNA-damaging agents. These “bio-activation” reactions result in cytotoxic alkylating and/or intercalating quinones and include mitomycin C (MMC), streptonigrin (STN), and 3-hydroxy-5-aziridinyl-1-methyl-2-[1H-indole-4,7-dione]prop-β-en-α-ol (EO9; ref. 14). Exposure to these 3 agents results in DNA lesions in direct linear proportion to NQO1 levels in tumor and normal tissue (6, 15). Unfortunately, the clinical efficacy of these agents was greatly limited due to resistance caused by tumor-related DNA repair processes (16) and normal tissue toxicity (17).
β-Lapachone (β-lap, 3,4-dihydro-2,2-dimethyl-2H-naphtho[1,2-b]pyran-5-6-dione) also requires NQO1 activity for effective killing of NQO1-overexpressing cancer cells, while causing minimal effects to neighboring “normal” cells that lack, or have low levels of, the enzyme (18–21). However, the mechanism of action of β-lap in vitro was significantly different from that of MMC, STN, or E09. β-lap undergoes an NQO1-dependent “futile cycle,” wherein ~60 moles of NAD(P)H are used per mole drug in 5 minutes (18, 22). As a result, dramatic elevation of reactive oxygen species (ROS) and released Ca2+ from endoplasmic reticulum (ER) stores were noted and are required for cell death (19–21). Unlike E09, STN, or MMC, β-lap does not seem to cause alkylation of DNA. β-lap (i.e., clinically formulated as ARQ 501) has completed phase I trials in pancreatic cancer patients (23). Although tumor responses were noted, the studies were limited by hemolysis caused by incorrect regimen application and hydroxylpropyl-β-cyclodextran (HPβ-CD) vehicle formulation.
We previously showed antitumor activities and mechanism of action of β-lap–mediated cell death in vitro in endogenous NQO1-expressing pancreatic (24, 25), and forced NQO1-overexpressing prostate, breast, and non– small cell lung cancer (NSCLC) cells (18, 19, 21, 26). A relationship between NQO1 levels and lethality in vitro using endogenous knockdown has not been established for this drug. Here, pancreatic cancer clones, whose endogenous NQO1 levels were stably knocked down in a gradient and ordered manner, were used to explore NQO1 levels required for β-lap–induced lethality. We show that a threshold level of NQO1 enzyme activity is required for β-lap–induced cell death responses and define key reactions essential for β-lap–induced cytotoxicity. We show that β-lap is an extremely efficacious agent against pancreatic cancers with endogenously elevated NQO1 levels, with extensive reductions in tumor burden, dramatic tumor growth delays, and extended survival in a metastatic pancreatic cancer animal model. Minimal to no toxicity to normal tissue or hemolytic anemia was noted using an optimal regimen.
Our current studies show that pancreatic tumors expressing NQO1 levels of at least 90 U would be responsive to β-lap therapy with minimal to no side-effects. Development of novel delivery vehicles, for example, using nanoparticles, to increase tumor-selective delivery should further augment the antitumor efficacy of β-lap.
Materials and Methods
Reagents and chemicals
β-lap was synthesized and purified by us (21). DIC, hydrogen peroxide (H2O2), staurosporine, Hoechst 33258, bovine serum albumin, cytochrome c, and propidium iodide were from Sigma-Aldrich. 5-(and 6-)Chloromethyl-2,7-dichlorodihydrofluorescein diacetate (DCFDA) was purchased from Invitrogen Life Technologies. HPβ-CD (>98% purity) was obtained from Cyclodextrin Technologies Development, Inc. β-lap–HPβ-CD was prepared as previously described (27).
Cell culture and transfections
MIA PaCa-2 cells were obtained from Dr. Joseph J. Cullen (University of Iowa). Human pancreatic cancer cell lines ASPC1, BXPC3, CFPAC-1, HS766T, Capan 1, and Capan 2 were obtained from the ATCC. All cells were grown in Dulbecco’s modified Eagle medium (Invitrogen) as described (25) and were mycoplasma free. The human shRNA-NQO1 (RHS1764-9691437: 5′-TGCTGTTGACAGTGAGCGCGGGATGAGACACCACT GTATTTAGTGAAGCCACAGATGTAAATACAGTGGTGTCTCATCCCATGCCTAC-TGCCTCGGA-3′) retroviral vector was purchased from Open Biosystems. Stable shRNA knockdown clones (KD17-1, KD17-3, and KD17-7) were generated by infecting MIA PaCa-2 cells with polybrene-supplemented medium obtained from Phoenix packaging cells transfected with the human retrovirus vector targeting NQO1 as described earlier (28). Cells containing stable scrambled controls (shRNA-SCR) were also generated and were referred to as nonsilencing (NS) cells. Individual clones were isolated by limiting dilution in media containing puromycin (1 μg/mL) and screened for NQO1 expression levels individually as indicated. All experiments were carried out without antibiotics. For bioluminescence (BLI) analyses, MIA PaCa-2 cells were infected with a lentiviral construct that expresses the luciferase (Luc) gene under the control of a strong promoter, the cytomegalovirus (CMV-Luc) promoter, as described (29).
NQO1 enzyme assays
NQO1 enzyme levels (Fig. 1F Table; Supplementary Table 1) were determined from triplicate S9 whole-cell extracts, using NADH (200 μmol/L) as an immediate electron donor and menadione (10 μmol/L) as an intermediate electron acceptor as described (18, 30). Enzyme units (U) of NQO1 were calculated as nmol of cytochrome c reduced/min/μg of protein, based on initial rate of change in absorbance at 550 nm.
Figure 1.
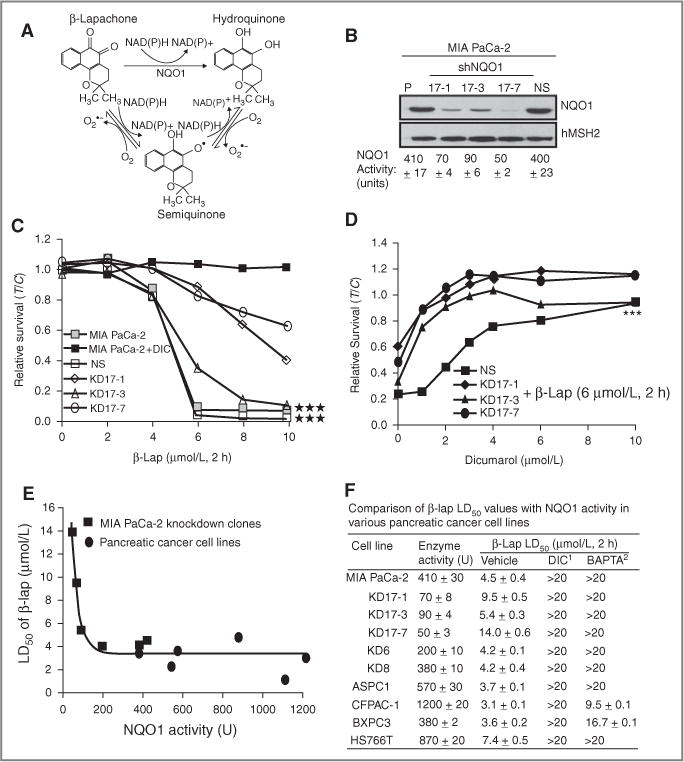
Functional shRNA-NQO1 knockdown protects from β-lap–induced cell death. A, model depicting the futile cycling of β-lap by NQO1; B, Western blot and enzymatic analyses for NQO1 in MIA PaCa-2 parental cells, shRNA-NQO1 knockdown clones (KD17-1, KD17-3, and KD17-7) and nonsilencing control clone (NS, MIA PaCa-2 shSCR). NQO1 protein expression and enzyme levels were assessed as in “Material and Methods.” hMSH2 levels were monitored for loading. C, relative survival assays in MIA PaCa-2 cells, NS- and NQO1-knockdown clones (KD17-1, KD17-3, KD17-7) were mock-treated or exposed to β-lap at the indicated doses for 2 hours, with or without DIC (50 μmol/L). Data are means ± SE for 3 independent experiments done in triplicate (***P < 0.001). D, relative survival of β-lap (6 μmol/L, 2 hours)-exposed, NS- or NQO1-knockdown MIA PaCa-2 cells titrated with increasing doses of DIC. Statistical significance (***P < 0.001) for relative survivals (C and D) were evaluated using ANCOVA as in “Materials and Methods.” E, LD50 of β-lap in MIA PaCa-2 knockdown clones and pancreatic cancer cell lines with varying NQO1 levels [BXPC3, Capan1 (NQO1 enzymatic activity: 540 ± 8 U; β-lap LD50: 2.6 ± 0.5 μmol/L, 2 hours; β-lap + DIC LD50: >20 μmol/L, 2 hours), ASPC1, HS766T, Capan2 (NQO1 enzymatic activity: 1,100 ± 3; β-lap LD50: 1.4 ± 0.2 μmol/L, 2 hours; β-lap + DIC LD50: >20 μmol/L, 2 hours), and CFPAC-1)]; R2 = 0.9053. F, analyses of β-lap lethality in various pancreatic cancer cells with or without DIC (50 μmol/L) or BAPTA-AM (5 μmol/L, 1 hour) pretreatments. LD50 calculations for various human pancreatic cancer cell lines were done by relative survival assays as described in “Materials and Methods.” NQO1 activity units ± SE were assessed and defined in “Materials and Methods.” DIC1, LD50 values for cells treated with various doses of β-lap for 2 hours in the presence of DIC (50 μmol/L, 2 hours). BAPTA2, LD50 values for cells pretreated with 5 μmol/L of BAPTA-AM for 1 hour followed by treatment with various doses of β-lap for 2 hours. All experiments were done 3 times in triplicate. Shown are means ± SE.
Relative survival assays
Relative cell survival levels from pancreatic cancer cells seeded at 5 × 103 per well of 48-well plates were determined using 7-day DNA assays as previously described (18). Results were reported as means ± standard error (SE) from at least 3 independent experiments done in sextuplicate.
ROS analyses
Disulfide glutathione (GSSG) and total GSH levels using spectrophotometric recycling assays and % GSSG normalized to protein content measured by Lowry assays (31) were assessed as previously described (32). ROS formation was further quantified by assessing digital images of the conversion of nonfluorescent 5 μmol/L of DCFDA to its fluorescent derivative at indicated times with a fluorescent microscope (Leica Microsystems) as described (33). All data were graphed as means ± SE for experiments performed 3 times, each in triplicate.
Alkaline comet assays
DNA single- and double-strand breaks (SSB and DSB, respectively) and base damage were assessed using alkaline comet assays (TREVIGEN). Digital photomicrographs of comet tail lengths were quantified from experiments done 3 or more times, each in duplicate (21).
Nucleotide analyses
Changes in intracellular NAD+ levels were measured and expressed as percentage treated divided by control (%T/C, ± SE) from at least 3 individual experiments, each in duplicate (21). ATP levels were analyzed from whole-cell extracts by using CellTier-Glo luminescent cell viability assays (Promega). Data were graphed as means ± SE of experiments done 3 or more times in triplicate for ATP, or 3 or more independent experiments for NAD+.
Flow cytometry
Cell-cycle distribution and apoptotic populations using TUNEL staining were analyzed as previously described (28, 34) using a FACS Calibur flow cytometer (BD Biosciences) and CellQuest software. Results from experiments repeated at least 3 times, each in duplicate, are presented. Apoptosis was observed from all phases of the cell cycle as described (35).
Immunoblot analyses
Whole-cell extracts and Western immunoblots were prepared and developed as described (28). For Western blots, primary antibodies and dilutions were as follows: poly (ADP-ribose) or PAR (BD Pharmingen), 1:1,000; human MutS homolog-2 (hMSH2, Ab-1; Oncogene), 1:500; p53 (DO-1; Santa Cruz Biotechnology), 1:5,000; and β-actin (Santa Cruz Biotechnology), 1:20,000. An a-human NQO1 antibody (1:5,000 dilution) was kindly provided by Dr. David Ross (University of Colorado Health Science Center). Results shown are representative of experiments performed at least 3 times.
Antitumor efficacy
A metastatic (spleen to liver tumor burden) pancreatic tumor model was used to evaluate β-lap efficacy. Pancreas tumors were generated by inoculating 2.5 × 105 Luc-tagged MIA PaCa-2 cells into the spleens of female mice weighing 20 to 25 g as described (36). Mice were randomly distributed so that average group (5 mice/group) body weights were not statistically different. Four weeks after implantation, and after random imaging, mice were treated with tail-vein injections of β-lap–HPβ-CD or HPβ-CD at various doses every other day for 5 treatments (i.e., 1 regimen). Mouse body weights were measured thrice weekly. Mice were sacrificed when metastatic tumor burden reached 20% of initial body weight. Survival and body weight data were graphed from 3 separate studies. Separately, ex vivo Luc-tumor volumes (luciferase levels) of excised spleens were evaluated using a bioluminescent imager (Xenogen Vivovision IVIS Lumina) for tumor burden and reported as means ± SE from 3 separate experiments. Spleen, liver, and pancreatic tissues were removed for histologic examination; tissues were snapfrozen in liquid nitrogen for Western blot analyses to confirm NQO1 expression. In another series of studies, the spleens of athymic mice were inoculated with 2.5 × 105 Luc-tagged Pan-GFP murine pancreatic tumor cells expressing human NQO1 (hNQ2) by lentiviral infection and similar spleen to liver tumor burden metastatic analyses performed (Supplementary Figs. 2 and 3).
Statistical analyses
Log-rank tests were applied to survival analyses (Kaplan–Meier curves). All in vivo statistical analyses were conducted using GraphPad Prism software. Statistical analyses of survival data in vitro were conducted using linear regression analyses. Analyses of covariance (ANCOVA) and nonlinear regression models and overall P values were simultaneously calculated, comparing intercepts and slopes for different treatments. In nonlinear regression models, β-lap LD50 doses (denoted as Y) were fitted with NQO1 activities (denoted as X) by Y = a exp(−bX) + c, where a, b, and c were fitting constants. P values were 2-sided, and statistical calculations were carried out by SAS (Service Pack 4) for Windows and SigmaPlot.
Results
NQO1 was a key determinant of β-lap cytotoxicity in human pancreatic cancer cells
Prior studies from our laboratory used forced NQO1 overexpression via CMV-directed cDNA transfection into NQO1*2 polymorphic breast, prostate, or NSCLC cells to elucidate the mechanism of action of β-lap. Because forced NQO1 overexpression could result in the localization of the enzyme into compartments that are not physiologic, we knocked down NQO1 levels in pancreatic cancer cells that have naturally elevated levels of this 2-electron oxidoreductase. This allowed us to determine the threshold of NQO1 level (in enzymatic units) at which β-lap treatments were no longer effective, especially given the mechanism of action of this drug (Fig. 1A). MIA PaCa-2 cells that have ~400 NQO1 enzymatic units (U) were infected with retroviral shRNA specific to the 5′-untranslated region (UTR) of NQO1 mRNA or with an identical shRNA lentiviral vector driving nonspecific (NS) siRNA expression as defined in “Materials and Methods.” Cell lines containing between 400 (clone NS) to 50 (clone 17-7) NQO1 units were isolated (Fig. 1B). Exposure of parental (410 U), NS (400 U), and clone 17-3 (~90 U) cells with varying doses of β-lap resulted in statistically similar lethalities (indicating LD50 values of 4.5 μmol/L; Fig. 1C) that were blocked with DIC (Fig. 1C). Interestingly, clones 17-1 and 17-7, with 70 and 50 U, respectively, resulted in significant resistance to β-lap under the same treatment conditions (Fig. 1C), with LD50 values of 10 to 12 μmol/L, respectively. To dissect the responses of the clones further, cells were treated with a fixed dose of β-lap (6 μmol/L, 2 hours), while varying DIC concentrations. Unlike the NS clone, in which ~5 μmol/L DIC was required for complete protection from β-lap, all 3 knockdown clones required significantly less DIC (~1–2 μmol/L) for complete protection against β-lap (Fig. 1D). When LD50 values of NS and knockdown clones (closed squares), and a series of available pancreatic cancer cells [Fig. 1E (open circles) and Fig. 1F Table), were graphed against expressed NQO1 levels (U), a clear biphasic curve was noted with a lethality inflection response to β-lap noted between 90 and 100 enzymatic units of NQO1 (Fig. 1E). These data strongly suggested that NQO1 levels were critical to β-lap lethality but that a mere 90 to 100 enzymatic units were required for lethality (Fig. 1E and F Table) due to NQO1 recycling of β-lap. In cells with higher NQO1 enzymatic activity, NAD(P)H (electron donor) most likely became rate-limiting, in which case higher NQO1 levels did not confer enhanced lethality and lowered LD50 values for β-lap treatments in pancreatic cancer cells (Fig. 1E).
β-lap lethality required released calcium (Ca2+) from ER stores
We previously showed in human breast, NSCLC, and prostate cancer cells that NQO1-dependent lethality caused by β-lap depended on Ca2+ release from ER stores (19–21, 37). Pretreatment of such NQO1 endogenously overexpressed cancer cells with BAPTA-AM, a specific intracellular Ca2+ chelator, spared β-lap lethality without affecting DNA damage caused by this agent (19–21, 37). In a similar manner, 60-minute pretreatment of various pancreatic cancer cells with nontoxic doses of 5 μmol/L of BAPTA-AM prevented cell death, showing the universal requirement of released intracellular ER Ca2+ in β-lap lethality (Fig 1F Table).
β-lap–induced ROS formation, DNA damage, poly (ADP-ribose)polymerase-1 hyperactivation, and nucleotide depletions were NQO1-dependent
NQO1-knockdown MIA PaCa-2 clones were then used to delineate the mechanism by which β-lap induced cell death. The futile cycling of β-lap by NQO1, specifically its back-reaction, in which the hydroquinone is converted to a semiquinone and then back to β-lap (Fig. 1A), caused dramatic formation of ROS. Significantly reduced ROS formation in β-lap–treated NQO1-knockdown MIA PaCa-2 clones were noted by lowered % GSSG (Fig. 2A) and DCFDA fluorescence (Fig. 2B) in direct proportion to NQO1 enzymatic activities. Concomitant decreased DNA lesion formation monitored by alkaline comet assays (Fig. 2C and D) was also noted in the knockdown clones versus NS cells; interestingly, no DNA damage was apparent when cells were analyzed by neutral comet assays, as previously reported in endogenously NQO1-overexpressed breast, prostate, and NSCLC cells (19, 21). Thus, DNA lesions other than DSBs are formed in response to β-lap exposures. DIC coaddition completely blocked NQO1-mediated, β-lap–induced SSBs and base damage.
Figure 2.
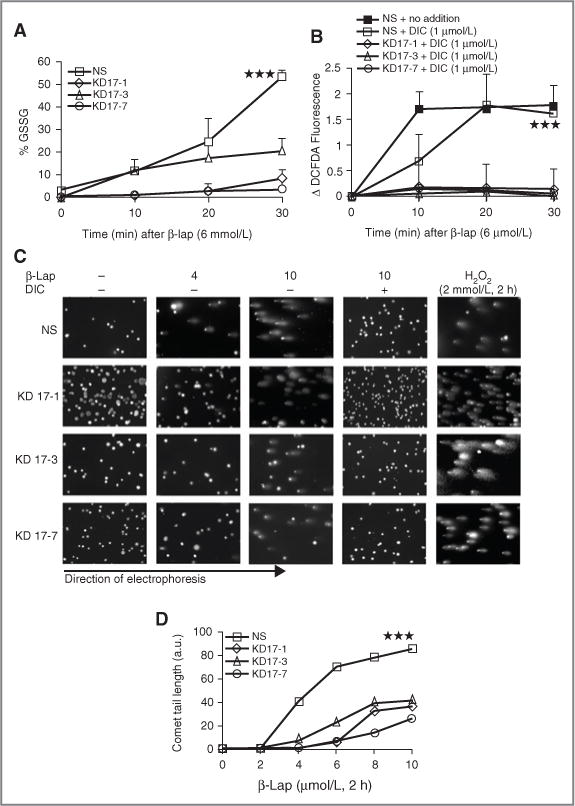
β-lap–induced oxidative stress and DNA damage was reduced in NQO1-knockdown MIA PaCa-2 cells. A, β-lap–induced oxidative stress in NS- and shRNA-NQO1– knockdown MIA PaCa-2 cells was evaluated by changes in % GSSG after β-lap (6 μmol/L) exposure at indicated times. B, ROS formation was monitored by DCFDA staining in NS- and NQO1-knockdown MIA PaCa-2 clones after β-lap (6 μmol/L) treatment, with or without DIC (1 μmol/L). DIC of 1μmol/L was used in these experiments to delineate responses between NS and NQO1 knockdown clones as determined in Fig 1D. C, NS- and shRNA-NQO1–knockdown clones (KD17-1, KD17-3, and KD17-7) were exposed to various doses of β-lap with or without 50 μmol/L of DIC for 2 hours as indicated and assessed for DNA damage by alkaline comet assays at the end of the treatment period. Exposures to H2O2 (2 mmol/L, 2 hours) served as NQO1 independent positive controls. Shown are representative micrographs of studies conducted at least 3 times. D, comet tail lengths were measured using NIH Image J software. a.u., arbitrary unit. Graphed are means ± SE from 3 experiments. Student’s t tests were done (***P < 0.001).
We previously reported that β-lap induced programmed necrosis involving poly(ADP-ribose)polymerase-1 (PARP1) hyperactivation as a result of released ER Ca2+ and extensive, threshold-reaching DNA damage events, particularly SSBs and base damages (19, 21, 38). Exposure of MIA PaCa-2 cells that endogenously overexpressed NQO1 with β-lap resulted in extensive PARP1 hyperactivation as measured by PAR formation within 10 to 90 minutes, which was completely blocked by DIC (Fig. 3A). As a positive control, exposure of cells to supra-lethal H2O2 doses caused extensive SSBs and base damages and hyperactivation of PARP1. Decreased PARP1 hyperactivation was noted in direct correlation to loss of NQO1 enzymatic activity in β-lap–exposed MIA PaCa-2 knockdown clones (compare NS clone with ordered NQO1-knockdown MIA PaCa-2 clones; Fig. 3B and C).
Figure 3.
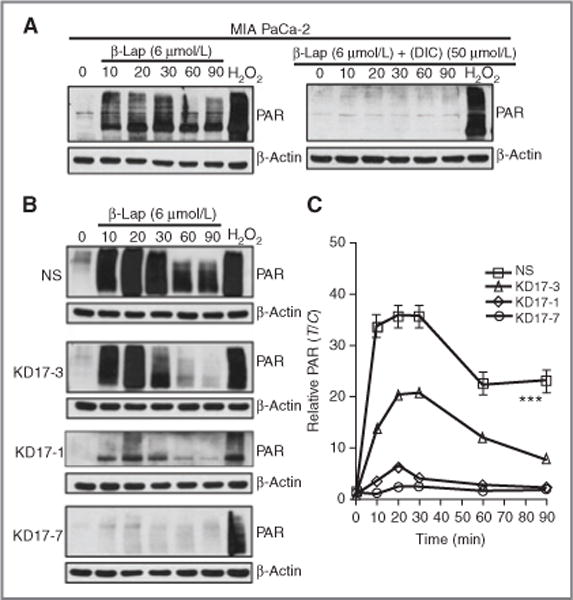
Abrogation of β-lap–induced PARP1 hyperactivation by shRNA-NQO1 knockdown. A, Western blot analyses of PAR formation in MIA PaCa-2 cells treated with 6 μmol/L of β-lap, with or without DIC (50 μmol/L). B, Western blot analyses of PAR formation in NS- and NQO1-knockdown cells treated with β-lap (6 μmol/L) alone. C, relative PAR levels were calculated by densitometry using NIH Image J software and β-actin as the loading control. Student’s t tests were done (***P < 0.001).
β-lap is one of only 3 agents (others are supra-lethal doses of MNNG or H2O2) known to cause hyperactivation of PARP1 and dramatic losses of total intracellular NAD+ levels, ultimately resulting in corresponding dramatic losses of ATP in exposed cells (19, 21, 39). However, β-lap is the only known agent that induces PARP1 hyper-activation with concomitant NAD+/ATP losses at doses achievable under preclinical and clinical conditions (19, 21). β-lap is the only known tumor-selective agent acting in an NQO1-dependent manner, whereby DIC prevented these responses (Fig. 4A and C). Consistent with decreased PARP1 hyperactivation in NQO1-knockdown MIA PaCa-2 clones (Fig. 3B and C), ordered loss of NAD+ (Fig. 4B) and ATP (Fig. 4D) was inversely noted with NQO1 activities. The more extensive NQO1 levels were knocked down in MIA PaCa-2 clones, the weaker the losses of NAD+ and ATP, consistent with spared lethality of these clones after β-lap treatment (Fig. 1E, closed squares represent genetically matched MIA PaCa-2 clones). We then examined a series of pancreatic cancer cell lines from various sources whose NQO1 levels ranged from ~390 to 1,200 U, yet their LD50 values were all fairly similar, ranging from 1.8 to 4.2 μmol/L (Fig. 1E, open circles).
Figure 4.
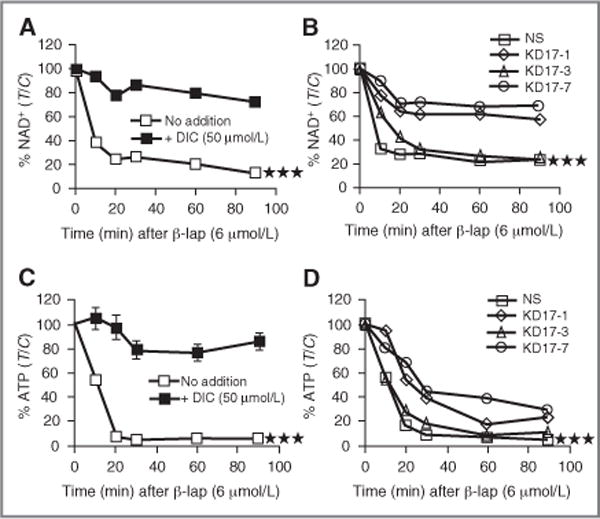
Abrogation of β-lap–induced nucleotide depletion by DIC or shRNA-NQO1 knockdown. A and C, MIA PaCa-2 cells were exposed to 6 μmol/L of β-lap with or without DIC (50 μmol/L) as indicated and analyzed for NAD+ or ATP levels. B and D, ATP and NAD+ levels were assessed in shRNA-NQO1–knockdown MIA PaCa-2 clones (KD17-1, KD17-3, and KD17-7) or NS control cells after 6 μmol/L of β-lap at indicated times. Results are means ± SE for studies conducted 3 or more times. Student’s t tests were done (***P < 0.001).
β-lap induced programmed necrosis in pancreatic cancer cells with endogenous NQO1 overexpression
Cell death induced by β-lap occurred via a unique programmed necrotic mechanism, in which cells lost energy (NAD+/ATP) but activated a default cysteine proteolytic apoptotic response. This cell death response is mediated by μ-calpain and exposed cells undergo dramatic nuclear condensation and apoptotic responses (20, 37). Importantly, cells undergoing this response showed diagnostic μ-calpain–dependent p53 proteolytic cleavage events (20, 37). MIA PaCa-2 cells responded in an identical manner to β-lap as breast, prostate, and NSCLC cancers that exhibit endogenous NQO1 overexpression (19, 21, 26), inducing dramatic apoptotic (TUNEL+ cells, Fig. 5A and B) responses and atypical proteolysis of p53 (see ~43-kDa p53 cleavage fragment, Fig. 5C) at doses corresponding to β-lap cytotoxicity (Fig. 1). Significant decreases in apoptotic responses (TUNEL+ cells, Fig. 5B) and downstream atypical p53 cleavage stimulated by activated μ-calpain (Fig. 5C; refs. 20, 37) were noted in NQO1-knockdown MIA PaCa-2 clones that correlated well with their resistance to β-lap cytotoxicity (Fig. 1).
Figure 5.
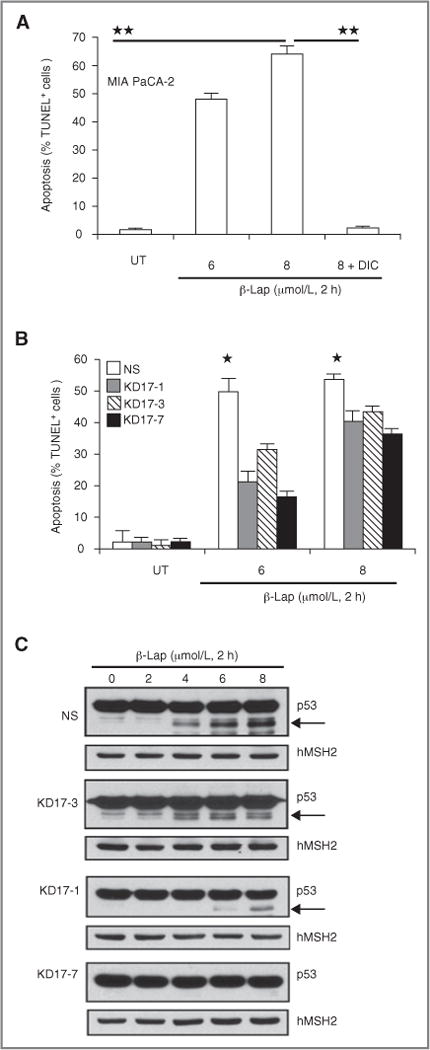
NQO1 expression knockdown protects cells from β-lap-induced apoptosis. A, MIA PaCa-2 cells were treated with β-lap (6 or 8 μmol/L) with or without DIC (50 μmol/L) for 2 hours. Cells were collected at 96 hours, and apoptosis was monitored by apoptotic reactions. Apoptotic (% TUNNEL+ cells) data are means ± SE from 3 independent experiments. Student’s t tests were done (**P < 0.01). B, NS- and shRNA-NQO1– knockdown MIA PaCa-2 cells were treated with β-lap (6 or 8 μmol/L, 2 hours) and apoptosis was monitored by TUNEL assay at 96 hours. Data are means ± SE from 3 independent experiments as in Figure 5A. Student’s t tests were done (*P < 0.05) C, NS- and NQO1-knockdown MIA PaCa-2 cells were treated with β-lap (μmol/L, 2 hours) and harvested at 96 hours for Western blot analyses of atypical p53 cleavage (arrows, ≈ 40-kDa p53 cleavage fragments). hMSH2 levels served for loading.
β-lap was an efficacious agent for the treatment of pancreatic cancer
The overexpression of NQO1 in pancreatic cancers compared with associated normal tissues (8–11), and the ability of β-lap to kill cells expressing this enzyme, strongly suggested its use for the treatment of pancreatic cancer. Although β-lap can kill pancreatic cancers in vitro, the lack of an adequate delivery vehicle to make the drug bioavailable for therapy in vivo limited use of this agent until recently. A version of HPβ-CD complexed with β-lap (β-lap–HPβ-CD) that increased solubility and bioavailability of β-lap 400-fold (27) was used in clinical trials as ARQ 501 (23). In our study, β-lap–HPβ-CD was used in a spleen to liver metastatic pancreatic cancer model to test the efficacy of this drug against MIA PaCa-2 tumors. Tumor growth assessed as body weight increases due to MIA PaCa-2 tumor burden was significantly repressed in animals treated with 20 or 30 mg/kg β-lap–HPβ-CD versus HPβ-CD alone (Fig. 6A). Reduction of tumor burden in MIA PaCa-2-containing mice treated with 20 or 30 mg/kg β-lap– HPβ-CD correlated well with overall survival (P< 0.0001) versus mice treated with HPβ-CD alone (Fig. 6B). In fact, 100% (10/10) of mice treated with β-lap–HPβ-CD were alive at 11 weeks posttreatment and were “apparently cured” compared with 100% tumor-driven lethality of mice bearing metastatic MIA PaCa-2 tumors exposed to HPβ-CD. These findings were corroborated in parallel studies evaluating tumor volumes using BLI, in which significant tumor regression without regrowth was noted after drug treatment in metastatic pancreatic human/murine tumor model (Fig. 6C and D; Supplementary Figs. 2 and 3). Morbidity, monitored by weight loss, was not noted at these optimal doses of β-lap–HPβ-CD.
Figure 6.
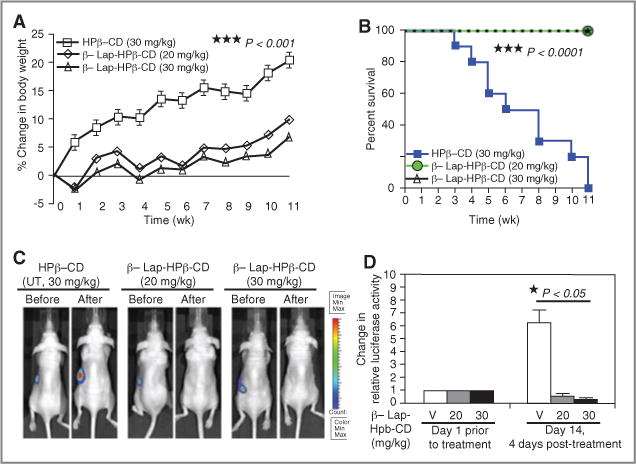
β-lap has significant antitumor efficacy in MIA PaCa-2 tumor xenograft models. A, body weight changes of MIA PaCa-2 tumor-bearing mice. Mice bearing spleen-implanted, NQO1-overexpressing MIA PaCa-2 cells were intravenously (iv) treated by tail-vein injections with vehicle alone (HPβ-CD) or β-lap–HPβ-CD at 20 or 30 mg/kg every other day for 5 treatments (Methods and Materials). Results (means ± SE) are representative of 3 similar experiments. Student’s t tests (***P < 0.001) were conducted comparing treated versus control groups. B, Kaplan–Meier survival for pancreatic antitumor efficacy experiments described as in (A); log-rank analyses were conducted comparing survival curves (***P < 0.0001) for HPβ-CD versus β-lap–HPβ-CD at 20 or 30 mg/kg iv. Results are combined survival data from 3 similar experiments. C, BLI images of mice bearing spleen-implanted pancreas cancers before and after treatment with HPβ-CD or β-lap–HPβ-CD at 20 or 30 mg/kg iv. D, quantification of pancreatic tumor burden. BLI (photons per second) was determined before and 12 days posttherapy with HPβ-CD or β-lap–HPβ-CD at 20 or 30 mg/kg iv. Results are means ± SE (n = 5). Student’s t tests (* P < 0.05).) were conducted comparing HPβ-CD versus β-lap–HPβ-CD at 20 or 30 mg/kg iv. V, vehicle (HPβ-CD).
Discussion
Numerous studies reported elevated levels of NQO1 in various human cancers, linking overexpression of this enzyme for prognostic and predictive values (12, 40). We previously reported the NQO1-mediated antitumor activity in vitro of β-lap, using endogenous and exogenous NQO1-overexpressing pancreatic, breast, prostate, and NSCLC models (19, 21, 25, 26). Prior to this work, genetic demonstration of the role of NQO1 in β-lap–induced lethality was confined to forced overexpression of NQO1 in rare polymorphic cancer cells. These studies had the limitation of using potentially nonphysiologic NQO1 levels (due to forced overexpression), in which the enzyme may be improperly localized and/or overexpressed in specific compartments at nonphysiologic levels. We show for the first time that the antitumor activity of β-lap can be progressively abrogated in MIA PaCa-2 clones ordered for NQO1 knockdown. More importantly, these knockdown clones were used to delineate specific nodal points of metabolism critical for β-lap–induced lethality. These β-lap–induced events included: (i) ROS formation and intracellular Ca2+ release from ER stores, where preloading cells with BAPTA-AM significantly spared lethality (Fig. 1F Table); (ii) SSBs and base damage induced by ROS by alkaline comet assays (Fig. 2); (iii) PARP1 hyperactivation and PAR formation (Fig. 3); (iv) NAD+/ATP depletion (Fig. 4); and (v) programmed necrosis (Fig. 5). Further analyses showed that an optimal threshold level of NQO1 (~90 U, Fig. 1E) was necessary to trigger β-lap–induced cell death.
Our detailed understanding of metabolic changes occurring in NQO1-overexpressing compared with knockdown cells during β-lap–induced cytotoxicity will further improve the development of an efficacious therapeutic regimen. Sound, exploitable, tumor-selective approaches based on drugable, tumor-specific protein/enzyme expression, such as NQO1, are desperately needed for clinical trials against pancreatic cancer. Our studies highlight that NQO1 is more than a “biomarker,” but an exploitable, tumor-selective target, whose expression is not cell-cycle regulated, nor affected by alterations in common tumor suppressors, such as p53 or Rb. Importantly, we show that NQO1 is exploitable using β-lap, whose unique futile cycle metabolism and broad metabolic changes induced downstream make the drug extremely efficacious for treatment of pancreatic cancer.
The current “standard of care” for treating advanced pancreatic cancer is gemcitabine (Gemzar), dideoxyfluorocytidine (ddFC; ref. 41). However, gemcitabine has proven palliative at best, offering only modest extension of long-term survival for patients with advanced disease (2). A major problem is that ddFC, a prodrug that requires phosphorylation by deoxycytidine kinase for activation, does not target a tumor-specific enzyme that distinguishes pancreatic cancers from normal pancreatic tissue (42). ddFC phosphorylation results in the accumulation of ddFC-triphosphate that competes with dCTP for DNA incorporation, leading to chain termination by DNA polymerase-alpha and replication stalling. These events cause unrecognized DNA lesions, preventing immediate DNA break detection by repair enzymes (42). Thus, the antitumor activity of gemcitabine is S-phase–dependent, whose efficacy is limited by the growth state of tumors (43). Importantly, ddFC antitumor activity has not been shown to be tumor selective, occurring in both normal and tumor tissues alike. Thus, nonselective cytotoxicity is a major disadvantage of this therapy. In contrast to gemcitabine, β-lap kills in a tumor-selective manner, eliciting essential oxidative stress and intracellular Ca2+ release from ER stores generated in an NQO1-dependent manner (19, 21). These intracellular events cause irreversible DNA damage, as a consequence of the unique hyperactivation of PARP1, which leads to a cascade of events culminating in a unique cell death termed “programmed necrosis” or “necroptosis.” β-lap–HPβ-CD (ARQ 501) is the only clinically used drug that exploits this pathway of cell death. Our current studies show that cells from patients with pancreatic tumors expressing NQO1 levels of at least 90 U would be indicated for β-lap therapy. Development of novel delivery vehicles, for example, using nanoparticles (29) to increase tumor-selective delivery, should further augment the antitumor efficacy of β-lap.
In addition to gemcitabine monotherapy, radiotherapy as a single agent, or in combination with gemcitabine, is often used to treat advanced stage pancreatic cancer patients (44). Gemcitabine has been reported to be a potent radiosensitizer with dose enhancement ratios of 1.6 to 2.0 (40). Gemcitabine (10 nmol/L, 24 hours) was reported to enhance human colon cancer cell lethality when administered immediately following ionizing radiation (IR) treatments (45). The mechanism of this combination therapy involves depletion of dATP, as well as dTTP pools. Our laboratory also reported that the mechanism of β-lap toxicity involves nucleotide depletion. However, in contrast to gemcitabine, β-lap depletion of nucleotide pools (NAD+/ATP) is tumor specific, because it occurs in NQO1-expressing tumor tissue and not in cells with low enzymatic levels (e.g., normal tissue). Thus, an added therapeutic advantage with β-lap therapy when treating pancreatic cancer patients is anticipated to be the ability to avoid systemic toxicity due to nonselective normal tissue cell death. β-lap synergizes with IR (46, 47), and we are currently exploring its ability to enhance the radiation lethality responses of pancreatic cancers. Our group found that the combination of sublethal doses of IR and β-lap kills prostate cancer cells expressing elevated endogenous NQO1 levels as a result of extensive ROS formation, intracellular ER Ca2+ release, massive DNA damage, PARP1 hyperactivation, and dramatic NAD+/ATP depletion (48). Because β-lap–induced PARP1 hyperactivation utilized nucleotide pools (i.e., caused dramatic depletion of NAD+ and ATP as a direct consequence of NADH and NAD+ losses) for its activity, DNA repair responses were blocked, including base excision repair and all other DNA repair pathways requiring ATP. Thus, β-lap greatly enhanced the lethal effects of radiotherapy in vivo, as reported in vitro (46, 47).
Our data clearly showed that NQO1 was a key determinant in antitumor efficacy of human and mouse pancreatic cancers. The activity of NQO1 was elevated greater than 10-fold in pancreatic cancer patients compared with associated normal tissues in more than 70% of patients (8–11). Therefore, monitoring the enzymatic activity of NQO1 and treating NQO1-overexpressing patients with β-lap should result in maximal therapeutic efficacy, with minimal side-effects. Application of site-specific drug delivery, such as using β-lap–encapsulated millirods (49) or β-lap–loaded micelles, was shown as an efficient solution to solubility and delivery problems that enhanced β-lap–induced anticancer efficacy (29). Because combined treatment of sublethal doses of IR with β-lap resulted in synergistic therapeutic efficacy in NQO1-overexpressing prostate cancers (48), combination of β-lap micelles with IR or any other DNA damaging agents (e.g., gemcitabine) may be an ideal therapeutic strategy to treat NQO1-overexpressing pancreatic cancer patients.
In summary, we theorize that β-lap, in an optimal formulation (and/or possibly enhanced when combined with DNA damaging agents), is ideally suited for treating pancreatic cancer patients. Our studies suggest that the antitumor efficacy of β-lap could be further enhanced with knowledge of key principal determinants of its tumor selectivity identified in our NQO1-knockdown clones. Our data specifically define the mechanism of PARP1 hyperactivation, strongly suggesting that agents that cause ROS or SSBs, such as IR, should be potentiated by β-lap because of a lowering of threshold of DNA lesions required for PARP1 hyperactivation. β-lap therapy alone or in combination with other DNA damaging agents, therefore, represents an exploitable and clinically available therapy for pancreatic cancer, as well as other specific cancers (e.g., breast, prostate, and NSCLC) in which NQO1 levels are endogenously elevated.
Supplementary Material
Translational Relevance.
Pancreatic cancer is the fourth leading cause of cancer-related deaths in the United States. Current standard therapies for these patients include surgery, often in combination with adjuvant radiotherapy and/or chemotherapy, and offer a 5-year survival rate of less than 5%. β-Lapachone (β-lap; clinically formulated as ARQ 501) has completed phase I trials in pancreatic cancer patients. Although tumor responses were noted, the studies were limited by hemolysis caused by incorrect regimen application and hydroxylpropyl-β-cyclodextran (HPβ-CD) vehicle formulation. However, our in vitro as well as in vivo data clearly show that β-lap, in an optimal formulation, is ideally suited for treating pancreatic cancer.
Acknowledgments
We are grateful to Dr. James K.V. Willson for his helpful discussions.
Grant Support
This research was supported by NIH/NCI grant, CA102792-01, and from the Robert A. and Virginia Payne endowment to D.A.B.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
This is manuscript CSCN 059 from the “Program in Cell Stress and Cancer Nanomedicine” (CSCN).
Disclosure of Potential Conflicts of Interest:
No potential conflicts of interest were disclosed.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Chua YJ, Zalcberg JR. Pancreatic cancer—is the wall crumbling? Ann Oncol. 2008;19:1224–30. doi: 10.1093/annonc/mdn063. [DOI] [PubMed] [Google Scholar]
- 3.Pliarchopoulou K, Pectasides D. Pancreatic cancer: current and future treatment strategies. Cancer Treat Rev. 2009;35:431–6. doi: 10.1016/j.ctrv.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Wong HH, Lemoine NR. Pancreatic cancer: molecular pathogenesis and new therapeutic targets. Nat Rev Gastroenterol Hepatol. 2009;6:412–22. doi: 10.1038/nrgastro.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y, Sun X, LaMont JT, Pardee AB, Li CJ. Selective killing of cancer cells by beta-lapachone: direct checkpoint activation as a strategy against cancer. Proc Natl Acad Sci U S A. 2003;100:2674–8. doi: 10.1073/pnas.0538044100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belinsky M, Jaiswal AK. NAD(P)H:quinone oxidoreductase1 (DT-diaphorase) expression in normal and tumor tissues. Cancer Metastasis Rev. 1993;12:103–17. doi: 10.1007/BF00689804. [DOI] [PubMed] [Google Scholar]
- 7.Siegel D, Ross D. Immunodetection of NAD(P)H: quinone oxidoreductase 1 (NQO1) in human tissues. Free Radic Biol Med. 2000;29:246–53. doi: 10.1016/s0891-5849(00)00310-5. [DOI] [PubMed] [Google Scholar]
- 8.Lyn-Cook BD, Yan-Sanders Y, Moore S, Taylor S, Word B, Hammons GJ. Increased levels of NAD(P)H: quinone oxidoreductase 1 (NQO1) in pancreatic tissues from smokers and pancreatic adenocarcinomas: A potential biomarker of early damage in the pancreas. Cell Biol Toxicol. 2006;22:73–80. doi: 10.1007/s10565-006-0156-3. [DOI] [PubMed] [Google Scholar]
- 9.Lewis AM, Ough M, Hinkhouse MM, Tsao MS, Oberley LW, Cullen JJ. Targeting NAD(P)H:quinone oxidoreductase (NQO1) in pancreatic cancer. Mol Carcinog. 2005;43:215–24. doi: 10.1002/mc.20107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Logsdon CD, Simeone DM, Binkley C, Arumugam T, Greenson JK, Giordano TJ, et al. Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Res. 2003;63:2649–57. [PubMed] [Google Scholar]
- 11.Awadallah NS, Dehn D, Shah RJ, Russell Nash S, Chen YK, Ross D, et al. NQO1 expression in pancreatic cancer and its potential use as a biomarker. Appl Immunohistochem Mol Morphol. 2008;16:24–31. doi: 10.1097/PAI.0b013e31802e91d0. [DOI] [PubMed] [Google Scholar]
- 12.Ross D, Siegel D. NAD(P)H: quinone oxidoreductase 1 (NQO1, DT-diaphorase), functions and pharmacogenetics. Methods Enzymol. 2004;382:115–44. doi: 10.1016/S0076-6879(04)82008-1. [DOI] [PubMed] [Google Scholar]
- 13.Radjendirane V, Joseph P, Lee YH, Kimura S, Klein-Szanto AJ, Gonzalez FJ, et al. Disruption of the DT diaphorase (NQO1) gene in mice leads to increased menadione toxicity. J Biol Chem. 1998;273:7382–9. doi: 10.1074/jbc.273.13.7382. [DOI] [PubMed] [Google Scholar]
- 14.Ross D, Kepa JK, Winski SL, Beall HD, Anwar A, Siegel D. NAD(P)H: quinone oxidoreductase 1 (NQO1): chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem Biol Interact. 2000;129:77–97. doi: 10.1016/s0009-2797(00)00199-x. [DOI] [PubMed] [Google Scholar]
- 15.Lown JW, Begleiter A, Johnson D, Morgan AR. Studies related to antitumor antibiotics. Part V. Reactions of mitomycin C with DNA examined by ethidium fluorescence assay. Can J Biochem. 1976;54:110–9. doi: 10.1139/o76-018. [DOI] [PubMed] [Google Scholar]
- 16.Moynahan ME, Cui TY, Jasin M. Homology-directed DNA repair, mitomycin-C resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001;61:4842–50. [PubMed] [Google Scholar]
- 17.Cheung RY, Rauth AM, Yu Wu X. In vivo efficacy and toxicity of intratumorally delivered mitomycin C and its combination with doxorubicin using microsphere formulations. Anticancer Drugs. 2005;16:423–33. doi: 10.1097/00001813-200504000-00009. [DOI] [PubMed] [Google Scholar]
- 18.Pink JJ, Planchon SM, Tagliarino C, Varnes ME, Siegel D, Boothman DA. NAD(P)H:quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J Biol Chem. 2000;275:5416–24. doi: 10.1074/jbc.275.8.5416. [DOI] [PubMed] [Google Scholar]
- 19.Bentle MS, Reinicke KE, Bey EA, Spitz DR, Boothman DA. Calcium-dependent modulation of poly(ADP-ribose) polymerase-1 alters cellular metabolism and DNA repair. J Biol Chem. 2006;281:33684–96. doi: 10.1074/jbc.M603678200. [DOI] [PubMed] [Google Scholar]
- 20.Tagliarino C, Pink JJ, Dubyak GR, Nieminen AL, Boothman DA. Calcium is a key signaling molecule in beta-lapachone-mediated cell death. J Biol Chem. 2001;276:19150–9. doi: 10.1074/jbc.M100730200. [DOI] [PubMed] [Google Scholar]
- 21.Bey EA, Bentle MS, Reinicke KE, Dong Y, Yang CR, Girard L, et al. An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proc Natl Acad Sci U S A. 2007;104:11832–7. doi: 10.1073/pnas.0702176104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pink JJ, Wuerzberger-Davis S, Tagliarino C, Planchon SM, Yang X, Froelich CJ, et al. Activation of a cysteine protease in MCF-7 and T47D breast cancer cells during beta-lapachone-mediated apoptosis. Exp Cell Res. 2000;255:144–55. doi: 10.1006/excr.1999.4790. [DOI] [PubMed] [Google Scholar]
- 23.Savage RE, Tyler AN, Miao XS, Chan TC. Identification of a novel glucosylsulfate conjugate as a metabolite of 3,4-dihydro-2,2-dimethyl-2H-naphtho[1,2-b]pyran-5,6-dione (ARQ 501, beta-lapachone) in mammals. Drug Metab Dispos. 2008;36:753–8. doi: 10.1124/dmd.107.018655. [DOI] [PubMed] [Google Scholar]
- 24.Cullen JJ, Hinkhouse MM, Grady M, Gaut AW, Liu J, Zhang YP, et al. Dicumarol inhibition of NADPH: quinone oxidoreductase induces growth inhibition of pancreatic cancer via a superoxide-mediated mechanism. Cancer Res. 2003;63:5513–20. [PubMed] [Google Scholar]
- 25.Ough M, Lewis A, Bey EA, Gao J, Ritchie JM, Bornmann W, et al. Efficacy of beta-lapachone in pancreatic cancer treatment: exploiting the novel, therapeutic target NQO1. Cancer Biol Ther. 2005;4:95–102. doi: 10.4161/cbt.4.1.1382. [DOI] [PubMed] [Google Scholar]
- 26.Planchon SM, Pink JJ, Tagliarino C, Bornmann WG, Varnes ME, Boothman DA. Beta-lapachone-induced apoptosis in human prostate cancer cells: involvement of NQO1/xip3. Exp Cell Res. 2001;267:95–106. doi: 10.1006/excr.2001.5234. [DOI] [PubMed] [Google Scholar]
- 27.Nasongkla N, Wiedmann AF, Bruening A, Beman M, Ray D, Bornmann WG, et al. Enhancement of solubility and bioavailability of beta-lapachone using cyclodextrin inclusion complexes. Pharm Res. 2003;20:1626–33. doi: 10.1023/a:1026143519395. [DOI] [PubMed] [Google Scholar]
- 28.Li LS, Morales JC, Hwang A, Wagner MW, Boothman DA. DNA mismatch repair-dependent activation of c-Abl/p73alpha/GADD45alpha-mediated apoptosis. J Biol Chem. 2008;283:21394–403. doi: 10.1074/jbc.M709954200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blanco E, Bey EA, Khemtong C, Yang SG, Setti-Guthi J, Chen H, et al. Beta-lapachone micellar nanotherapeutics for non-small cell lung cancer therapy. Cancer Res. 2010;70:3896–904. doi: 10.1158/0008-5472.CAN-09-3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fitzsimmons SA, Workman P, Grever M, Paull K, Camalier R, Lewis AD. Reductase enzyme expression across the National Cancer Institute Tumor Cell Line Panel: correlation with sensitivity to mitomycin C and EO9. J Natl Cancer Inst. 1996;88:259–69. doi: 10.1093/jnci/88.5.259. [DOI] [PubMed] [Google Scholar]
- 31.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 32.Lee YJ, Galoforo SS, Berns CM, Chen JC, Davis BH, Sim JE, et al. Glucose deprivation-induced cytotoxicity and alterations in mitogen-activated protein kinase activation are mediated by oxidative stress in multidrug-resistant human breast carcinoma cells. J Biol Chem. 1998;273:5294–9. doi: 10.1074/jbc.273.9.5294. [DOI] [PubMed] [Google Scholar]
- 33.Sawada F, Inoguchi T, Tsubouchi H, Sasaki S, Fujii M, Maeda Y, et al. Differential effect of sulfonylureas on production of reactive oxygen species and apoptosis in cultured pancreatic beta-cell line, MIN6. Metabolism. 2008;57:1038–45. doi: 10.1016/j.metabol.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 34.Wagner MW, Li LS, Morales JC, Galindo CL, Garner HR, Bornmann WG, et al. Role of c-Abl kinase in DNA mismatch repair-dependent G2 cell cycle checkpoint arrest responses. J Biol Chem. 2008;283:21382–93. doi: 10.1074/jbc.M709953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bey EA, Wuerzberger-Davis SM, Pink JJ, Yang CR, Araki S, Reinicke KE, et al. Mornings with Art, lessons learned: feedback regulation, restriction threshold biology, and redundancy govern molecular stress responses. J Cell Physiol. 2006;209:604–10. doi: 10.1002/jcp.20783. [DOI] [PubMed] [Google Scholar]
- 36.Jimenez RE, Hartwig W, Antoniu BA, Compton CC, Warshaw AL, Fernandez-Del Castillo C. Effect of matrix metalloproteinase inhibition on pancreatic cancer invasion and metastasis: an additive strategy for cancer control. Ann Surg. 2000;231:644–54. doi: 10.1097/00000658-200005000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tagliarino C, Pink JJ, Reinicke KE, Simmers SM, Wuerzberger-Davis SM, Boothman DA. Mu-calpain activation in beta-lapachone-mediated apoptosis. Cancer Biol Ther. 2003;2:141–52. doi: 10.4161/cbt.2.2.237. [DOI] [PubMed] [Google Scholar]
- 38.Reinicke KE, Bey EA, Bentle MS, Pink JJ, Ingalls ST, Hoppel CL, et al. Development of beta-lapachone prodrugs for therapy against human cancer cells with elevated NAD(P)H:quinone oxidoreductase 1 levels. Clin Cancer Res. 2005;11:3055–64. doi: 10.1158/1078-0432.CCR-04-2185. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, Chatterjee K, Alano CC, Kalinowski MA, Honbo N, Karliner JS. Vincristine attenuates N-methyl-N′-nitro-N-nitrosoguanidine-induced poly-(ADP) ribose polymerase activity in cardiomyocytes. J Cardiovasc Pharmacol. 2010;55:219–26. doi: 10.1097/FJC.0b013e3181c87e6c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fagerholm R, Hofstetter B, Tommiska J, Aaltonen K, Vrtel R, Syrja-koski K, et al. NAD(P)H: quinone oxidoreductase 1 NQO1*2 genotype (P187S) is a strong prognostic and predictive factor in breast cancer. Nat Genet. 2008;40:844–53. doi: 10.1038/ng.155. [DOI] [PubMed] [Google Scholar]
- 41.Burris HA, III, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–13. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 42.Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17(Suppl 5):v7–12. doi: 10.1093/annonc/mdj941. [DOI] [PubMed] [Google Scholar]
- 43.Ewald B, Sampath D, Plunkett W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol Cancer Ther. 2007;6:1239–48. doi: 10.1158/1535-7163.MCT-06-0633. [DOI] [PubMed] [Google Scholar]
- 44.Doyle TH, Mornex F, McKenna WG. The clinical implications of gemcitabine radiosensitization. Clin Cancer Res. 2001;7:226–8. [PubMed] [Google Scholar]
- 45.Shewach DS, Hahn TM, Chang E, Hertel LW, Lawrence TS. Metabolism of 2′,2′-difluoro-2′-deoxycytidine and radiation sensitization of human colon carcinoma cells. Cancer Res. 1994;54:3218–23. [PubMed] [Google Scholar]
- 46.Boothman DA, Pardee AB. Inhibition of radiation-induced neoplastic transformation by beta-lapachone. Proc Natl Acad Sci USA. 1989;86:4963–7. doi: 10.1073/pnas.86.13.4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boothman DA, Trask DK, Pardee AB. Inhibition of potentially lethal DNA damage repair in human tumor cells by beta-lapachone, an activator of topoisomerase I. Cancer Res. 1989;49:605–12. [PubMed] [Google Scholar]
- 48.Dong Y, Bey EA, Li LS, Kabbani W, Yan J, Xie XJ, et al. Prostate cancer radiosensitization through poly(ADP-Ribose) polymerase-1 hyperactivation. Cancer Res. 2010;70:8088–96. doi: 10.1158/0008-5472.CAN-10-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dong Y, Chin SF, Blanco E, Bey EA, Kabbani W, Xie XJ, et al. Intratumoral delivery of beta-lapachone via polymer implants for prostate cancer therapy. Clin Cancer Res. 2009;15:131–9. doi: 10.1158/1078-0432.CCR-08-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.