

Abstract
Discussion on improving the power of genome-wide association studies to identify candidate variants and genes is generally centered on issues of maximizing sample size; less attention is given to the role of phenotype definition and ascertainment. The authors used genome-wide data from patients infected with human immunodeficiency virus type 1 (HIV-1) to assess whether differences in type of population (622 seroconverters vs. 636 seroprevalent subjects) or the number of measurements available for defining the phenotype resulted in differences in the effect sizes of associations between single nucleotide polymorphisms and the phenotype, HIV-1 viral load at set point. The effect estimate for the top 100 single nucleotide polymorphisms was 0.092 (95% confidence interval: 0.074, 0.110) log10 viral load (log10 copies of HIV-1 per mL of blood) greater in seroconverters than in seroprevalent subjects. The difference was even larger when the authors focused on chromosome 6 variants (0.153 log10 viral load) or on variants that achieved genome-wide significance (0.232 log10 viral load). The estimates of the genetic effects tended to be slightly larger when more viral load measurements were available, particularly among seroconverters and for variants that achieved genome-wide significance. Differences in phenotype definition and ascertainment may affect the estimated magnitude of genetic effects and should be considered in optimizing power for discovering new associations.
Keywords: genome-wide association study, HIV seropositivity, HIV seroprevalence, phenotype, seroepidemiologic studies
Genome-wide association (GWA) studies have discovered many gene-phenotype associations with strong statistical support (1, 2). Most of them represent small effects (3), and one would like to ensure that statistical power in GWA studies is optimized for the detection of such small effects. In theory, power is better when the sample size is larger and when the genetic effects to be detected are larger. One critical aspect in this regard is the definition and measurement of study phenotype and eligibility criteria (4). Lenient definitions and relaxed eligibility criteria allow for more participants to be eligible but may dilute genetic effects if the phenotype measurements become imprecise (4, 5). More stringent phenotype definitions and demanding eligibility criteria avoid this dilution but may also drastically reduce the available sample size.
This dilemma is crucial in human immunodeficiency virus type 1 (HIV-1) infection, where it is typically easier to accumulate large numbers of samples from patients who are seroprevalent (patients whose time of infection is unknown) than to accumulate data from seroconverters (patients whose date of infection is known). Moreover, for precision of the viral load set point, phenotype may depend on how many measurements are available.
Here, we aimed to explore empirically the estimated changes in the effect sizes of discovered risk alleles when phenotypes of HIV-1 viral load were defined and measured under different criteria in the setting of GWA studies. We used study populations with differences in the stringency of eligibility criteria, and we evaluated their performance in GWA studies aiming to identify genetic determinants of viral load set point in HIV-1 infection. Specifically, we assessed whether the observed genetic effects differed between seroconverters and seroprevalent subjects and whether results were also influenced by the number of available viral load measurements used to define the set point.
MATERIALS AND METHODS
Data
We included participants from the Center for HIV/AIDS Vaccine Immunology (CHAVI) EuroCHAVI Consortium, which was established by the US National Institute of Allergy and Infectious Diseases in 2006 to study the major determinants of host control of HIV-1. The data set comprised 9 cohorts/studies whose investigators agreed to participate in the Host Genetics Core of the EuroCHAVI Consortium. The participating cohorts are: the Swiss HIV Cohort Study; the I.CO.Na Cohort, Italy; the San Raffaele del Monte Tabor Foundation, Italy; Royal Perth Hospital, Australia; IrsiCaixa, Spain; Guy's, King's and St. Thomas’ Hospitals, United Kingdom; the Danish Cohort, Denmark; the Modena Cohort, Italy; and the Hospital Clinic of Barcelona, Spain. All participants included in our study were of European descent and were part of earlier studies of genomic determinants of viral set point (6, 7).
The definitions of seroconverters and seroprevalent subjects were similar to those used by Fellay et al. (7). Seroconverters were defined on the basis of a valid seroconversion date estimation proven by biologic markers: 1) a documented positive test and a documented valid negative testing documentation within 2 years before the first positive test; and 2) 1 or more biologic criteria of primary infection: incomplete Western blot and/or positive p24 antigen and/or high viremia (>1 million copies of HIV-1 per mL of blood) and a consistent dynamic pattern of the biologic parameters (completion of Western blot, negativization of p24 antigen, decrease of peak viremia); a compatible clinical syndrome was considered supporting evidence. Seroprevalent persons, on the other hand, lacked documentation of HIV-negative serology; they were eligible if they had at least 3 viral load results recorded over a period of at least 3 years, diverging by no more than 0.5 log10 copies/mL.
Outcome
Longitudinal viral load data were individually inspected by an experienced blind infectious-disease clinician who eliminated measurements not reflecting the steady state (6). Briefly, viral load results were discarded if they were obtained during the initial peak of viremia observed during primary HIV-1 infection (viral load > 0.25 log10 higher than the average of subsequent viral load) and during the late phase of the disease (part of a significantly ascending viral load slope). In addition, viral load results were discarded when they were obtained during the set point period but were conflicting with other available results and possibly linked to coincident infection, trauma, or vaccination, to laboratory errors, or to data management mistakes (viral load > 0.5 log10 higher or lower than the average of all remaining points). The set point was then calculated as the average of all remaining log10-transformed viral load measurements.
Genotyping
All samples were genotyped on the Illumina HumanHap550 BeadChip (Illumina, Inc., San Diego, California) with 555,352 single nucleotide polymorphisms (SNPs). Quality control steps are described in detail elsewhere (6). We directly genotyped the C-C chemokine receptor type 5 (CCR5) Δ32 gene variant, a validated allele influencing HIV-1 disease (8) that is neither included nor tagged in the genome chip.
Analysis
We considered an additive model wherein we assessed the estimated effects of each SNP on viral load set point (increase or decrease in log scale per allele) within each group (seroconverters and seroprevalents) using a linear regression analysis. For all comparisons, the common allele was considered the reference allele. Gender and age were included as covariates in all analyses. We adjusted for population stratification by using EIGENSTRAT values; 17 principal component axes were found to be significant (P < 0.05) by the Tracy-Widom test and were used in the model (6). The EIGENSTRAT method derives the principal components of the correlations among gene variants and corrects for those correlations in the association tests (9).
In order to evaluate the differences between the results obtained in the seroconverter and seroprevalent groups, we examined a set of SNPs likely to show genuine associations with viral load. Otherwise, for nongenuine associations, an equal number of SNPs would be expected to have a stronger effect versus a weaker effect in either group. We therefore generated the list of the top 100 SNPs with the lowest P values when the results from both groups were combined in a stratified analysis (equivalent to fixed-effects calculations, where each data set is weighted by the inverse of its variance (10)). In a sensitivity analysis, we also examined the list of top 100 SNPs with the lowest P values that were found in the original publication (6) based on 486 seroconverters (the “discovery cohort”) and then compared the results with those for a separate set of seroconverters from a follow-up publication (7). This last analysis compares 2 replication data sets excluding the discovery set (6), and thus it corroborates a scenario wherein one is using different cohorts with different definitions to replicate the original top hits of a GWA study. The effect sizes for a replication effort are expected to be unbiased, while the effect sizes of the top 100 SNPs based on either the original GWA study (6) or the combination of all seroconverter and seroprevalent values are expected to be inflated on average in comparison with the true effects due to the winner's curse (11, 12).
We calculated the difference in effect sizes between seroconverter and seroprevalent groups for each of the 100 SNPs according to each of these approaches. The differences were parameterized so as to be positive if the effect observed in seroconverters was stronger in absolute value—that is, regardless of whether the minor allele was associated with higher or lower viral load. We then combined these differences across all 100 SNPs to estimate an average difference according to fixed- and random-effects calculations. The DerSimonian and Laird random-effects model (13) considers that the differences in the genetic effects in the 2 groups may vary across SNPs and introduces an estimate, τ2, of the between-SNP heterogeneity that makes the confidence intervals larger. Specifically, the standard error (SE) is
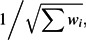 |
where the weights are given by for the fixed-effects models and for the random-effects models. The between-SNP heterogeneity is expressed with the I2 metric and its 95% confidence interval (14, 15).
In further analyses, we limited the calculations to include only SNPs among the top 100 that are located in the major histocompatibility complex region on chromosome 6 or only SNPs that reached genome-wide significance (P < 10−7)—subsets that are likely to include almost entirely genuine associations.
To assess how the number of measurements used for calculation of the viral load set point might affect the results, we modeled the set point as a function of the genotype of each variant (number of risk alleles), the number of viral load measurements used to assess the set point, and their interaction term. Coefficients were parameterized so as to always correspond to a positive effect size (higher viral load set point) for the risk allele. Then the coefficients of the interaction terms for all 100 top SNPs were combined with fixed- and random-effects models. A summary effect greater than 0 means that the increase of the counts of the viral load measurements is associated with a larger effect estimate (a larger impact of the risk allele on the viral load set point).
Stata, version 10 (Stata Corporation, College Station, Texas), and PLINK (16) software were used for the analyses.
RESULTS
Data sets
Clinical and genotyping data were available for 1,686 participants. We excluded 167 African participants and 14 participants with undefined ethnic origin from the study. A total of 715 seroconverters were identified. Of those, 93 were excluded because they had a seroconversion window greater than 2 years. Finally, 622 seroconverters and 636 seroprevalent subjects were included in the study. These persons were a subset of patients included in previous studies (6, 7). Study participants’ characteristics appear in Table 1.
Table 1.
Characteristics of Human Immunodeficiency Virus Type 1 Seroconverters and Seroprevalent Subjects, EuroCHAVI Consortium, 2006a
| Characteristic | All Seroconverters (n = 622) |
Discovery Seroconverters (n = 486) |
Replication Seroconverters (n = 136) |
Seroprevalent Subjects (n = 636) |
||||||||
| % | Mean (SD) | Median (IQRb) | % | Mean (SD) | Median (IQR) | % | Mean (SD) | Median (IQR) | % | Mean (SD) | Median (IQR) | |
| Age, years | 33.9 (9.1) | 34.4 (9.3) | 32.6 (8.9) | 34.6 (10.2) | ||||||||
| Male gender | 74.4 | 75.5 | 70.6 | 75.9 | ||||||||
| Viral load set point, copies of HIV-1 per mL of blood | 3.96 (1.04) | 3.97 (1.08) | 3.88 (0.88) | 3.87 (0.82) | ||||||||
| No. of viral load points available for calculation of the set point | 4 (3–6) | 3 (2–6) | 4 (3–7) | 4 (3–7) | ||||||||
Abbreviations: CHAVI, Center for HIV/AIDS Vaccine Immunology; HIV-1, human immunodeficiency virus type 1; IQR, interquartile range; SD, standard deviation.
The differences in study characteristics between seroconverters and seroprevalent subjects were not statistically significant for any comparisons.
25th–75th percentiles.
Comparison of genetic effect sizes in seroconverters versus seroprevalents
Analysis including all participants.
Web Table 1, which appears on the Journal’s Web site (http://aje.oxfordjournals.org/), presents a list of the top 100 hits resulting from analyses unadjusted and adjusted for the number of viral load measurements. The P values of the associations ranged from 1.32 × 10−4 to 3.21 × 10−19 for the unadjusted analysis and from 1.28 × 10−4 to 2 × 10−18 for the adjusted analysis. Most SNPs mapped to chromosome 6 (n = 57 and n = 56, respectively) (Web Table 1). Five and four SNPs reached genome-wide significance (P < 10−7), respectively. When analysis was limited to seroconverters, 5 and 3 SNPs reached genome-wide significance, respectively, whereas analysis limited to seroprevalent subjects did not reveal any genome-wide significant finding.
Seroconverters showed larger genetic effects than did seroprevalents (Figure 1). For 76 SNPs, the genetic association was stronger in seroconverters, and for 29 of those the difference was nominally significant (P < 0.05), including the top 2 SNPs that were reported as genome-wide significant in the original discovery cohort (6). Conversely, the difference was nominally significant in only 1 of the 24 associations for which seroprevalents showed a stronger association. Most of these 24 SNPs had relatively modest P values, and the best P value was 8.3 × 10−5, thus suggesting limited credibility for a genuine association. Analysis adjusted for viral load counts showed comparable results: 31 differences vs. 1 difference were nominally statistically significant in the seroconverters versus seroprevalents.
Figure 1.

Effect sizes of associations between single nucleotide polymorphisms and human immunodeficiency virus type 1 viral load for seroconverters versus seroprevalent subjects in an analysis that included all participants, EuroCHAVI Consortium, 2006. The effect sizes are parameterized to always reflect a positive difference. The solid line represents the points at which seroconverters and seroprevalent subjects have equal effect sizes. (CHAVI, Center for HIV/AIDS Vaccine Immunology; GWS, genome-wide significance).
The summary difference between the genetic effect estimates in seroconverters and seroprevalents was 0.092 (95% confidence interval (CI): 0.074, 0.110) log10 viral load by fixed-effects calculations, and there was moderate heterogeneity in the differences across the 100 SNPs (I2 = 37%, 95% CI: 19, 50) (Web Figure 1); random-effects summary results were quite similar (Table 2). The summary difference between the effects was comparable when results were adjusted for the number of available viral load measurements (Table 2). The top 2 hits in the combined data—rs2395029, localized in the human leukocyte antigen complex P5 gene (HCP5), and rs9264942, located 35 kilobases upstream of the human leukocyte antigen C gene (HLA-C) on chromosome 6—were the same as previously described (6, 7).
Table 2.
Summary Effect Estimates of the Differences in Genetic Effect Sizes (in Log10 Viral Load Per Copy of the Risk Allele) Between Seroconverters and Seroprevalent Subjects, EuroCHAVI Consortium, 2006a
| Unadjusted for No. of Viral Load Counts |
Adjusted for No. of Viral Load Counts |
|||||||
| Fixed Effects |
Random Effects |
Fixed Effects |
Random Effects |
|||||
| Estimate | 95% CI | Estimate | 95% CI | Estimate | 95% CI | Estimate | 95% CI | |
| Top 100 single nucleotide polymorphisms | ||||||||
| SC vs. SP | 0.092 | 0.074, 0.110 | 0.095 | 0.071, 0.118 | 0.093 | 0.075, 0.111 | 0.095 | 0.071, 0.119 |
| SCr vs. SP | 0.095 | 0.065, 0.125 | 0.095 | 0.065, 0.125 | 0.111 | 0.080, 0.142 | 0.111 | 0.080, 0.142 |
| Chromosome 6 subset | ||||||||
| SC vs. SP | 0.153 | 0.129, 0.177 | 0.153 | 0.129, 0.178 | 0.157 | 0.132, 0.181 | 0.157 | 0.132, 0.183 |
| SCr vs. SP | 0.104 | 0.057, 0.151 | 0.104 | 0.057, 0.151 | 0.111 | 0.063, 0.159 | 0.111 | 0.063, 0.159 |
| Genome-wide significant variants | ||||||||
| SC vs. SP | 0.232 | 0.145, 0.318 | 0.232 | 0.145, 0.322 | 0.247 | 0.148, 0.347 | 0.264 | 0.137, 0.391 |
| SCr vs. SP | 0.111 | −0.096, 0.318 | 0.111 | −0.096, 0.318 | 0.111 | −0.103, 0.325 | 0.111 | −0.103, 0.325 |
Abbreviations: CHAVI, Center for HIV/AIDS Vaccine Immunology; CI, confidence interval; SC, seroconverters; SCr, seroconverters used as replicating datab; SP, seroprevalent subjects.
All analyses were adjusted for age, gender, and population stratification axes.
Excluding the seroconverters of the original discovery data (4).
When the analysis was limited to chromosome 6, the summary difference in the effect sizes between seroconverters and seroprevalents increased to 0.153 (95% CI: 0.129, 0.177) log10 viral load, with consistent results across SNPs (I2 = 0%, 95% CI: 0, 31) (Table 2). The difference was even more prominent when only the 5 genome-wide significant hits were considered: 0.232 (95% CI: 0.145, 0.318) log10 viral load, with limited estimated heterogeneity across SNPs (I2 = 4%, 95% CI: 0, 80). The 5 genome-wide significant hits, which were not in high linkage disequilibrium (r2 < 0.65 for all comparisons), showed comparable estimates of the differences between seroconverters and seroprevalents (Web Figure 1). Random-effects analyses and analyses adjusted for number of viral load measurements yielded similar results (Table 2).
Analysis excluding the discovery cohort.
The associations observed when the discovery cohort was removed from the analysis were more conservative. The P values of the top 100 SNPs ranged from 0.97 to 4.83 × 10−8 for the unadjusted analysis and from 0.97 to 2.59 × 10−7 for the adjusted analysis. Thus, the comparative picture remained similar, with larger effects on average in seroconverters than in seroprevalents. For 75 SNPs, seroconverters showed larger effects, and 4 of the differences were nominally statistically significant. None of the 25 SNPs for which larger effects were observed in seroprevalents were nominally statistically significant. Adjusting for the number of viral load measurements yielded almost identical results: 79 SNPs versus 21 SNPs, with 4 versus 0 differences being nominally statistically significant.
The summary difference between the effect estimates in seroconverters and seroprevalents was 0.095 (95% CI: 0.065, 0.125) for the unadjusted analysis, and the estimated between-SNP heterogeneity was limited (I2 = 0%, 95% CI: 0, 26); results were almost identical for analyses adjusting for the number of viral load measurements (Table 2).
For the 42 SNPs on chromosome 6, the summary effect difference was 0.104 (95% CI: 0.057, 0.151), with no heterogeneity (I2 = 0%, 95% CI: 0, 38) (Table 2).
Impact of the number of viral load measurements
Among seroconverters, the magnitude of the estimated genetic effect for 65 SNPs tended to be larger when a greater number of viral load counts was available, and for 35 SNPs the opposite trend was observed. Only 5 of these interaction terms were nominally significant (2 suggesting a larger estimate of the genetic effect and 3 suggesting a smaller estimate of the genetic effect when a larger number of viral load counts was available). The summary effect of the interaction coefficients suggested that the genetic effect estimate increased by 0.005 (95% CI: 0.002, 0.008) log10 viral load per each additional viral load measurement, with overall consistent results across SNPs (I2 = 0%, 95% CI: 0, 24). For the SNPs that reached genome-wide significance, the summary effect of the interaction coefficients was 0.017 (95% CI: 0.003, 0.032), with overall consistent results across SNPs (I2 = 0%, 95% CI: 0, 79).
Among seroprevalents, the estimated genetic effect for 59 SNPs tended to be larger when a larger number of viral load measurements was available, and for 41 SNPs the opposite trend was observed. Ten interaction coefficients were nominally significant. The summary effect of the interaction coefficient was not nominally significant (0.002 log10 viral load, 95% CI: −0.001, 0.005), with small heterogeneity being observed (I2 = 22%, 95% CI: 0, 39). The summary effect of the interaction coefficients was 0.021 (95% CI: 0.006, 0.036) for the SNPs that reached genome-wide significance, with consistent results across SNPs (I2 = 0%, 95% CI: 0, 79).
CCR5 Δ32 variant
For CCR5 Δ32, the genetic effect estimates showed differences of –0.22 and –0.20 log10 viral load per allele for seroconverters and seroprevalents, respectively (P = 0.9). The number of viral load measurements did not significantly affect the estimate of the genetic effect (interaction coefficient = 0.003, P = 0.24).
DISCUSSION
We have shown that analyses of HIV-1 seroconverters tend to estimate stronger genetic effects of SNPs on the viral load set point than analyses of seroprevalent subjects. The difference was in the range of 0.1 log10 viral load, on average, among the top 100 SNPs emerging from a genome-wide analysis in 1,258 HIV-1-infected patients. The difference was more prominent when we considered only the chromosome 6 SNPs (average = 0.15 log10 viral load), which have higher chances of reflecting true associations, and even more prominent among the SNPs that reached genome-wide significance (average = 0.23 log10 viral load), representing associations with robust credibility. Differences were slightly smaller when we excluded the original discovery data from the comparison. This latter analysis has the benefit of avoiding the winner's curse (11, 12).
Genetic effects may be more accurately estimated in seroconverters because of the more stringent definition, the greater stability of HIV-1 viral load during the first years of infection (17), and the increasing confounding role played by comorbidity during progressive HIV-1 disease. This may explain in part why genetic effect estimates tend to be larger in seroconverters than in seroprevalent subjects. Another possible explanation is that some genetic factors may act mostly early in the course of the disease, a period that is only partly and inconsistently captured for seroprevalent cohorts, in which the date of infection is not known and a variable segment of the early years after seroconversion is missing. Survival bias could also enrich or dilute genetic associations. Some data suggest that the impact of the CCR5 Δ32 and C-C chemokine receptor type 2 (CCR2) 64I variants may indeed differ in early and later stages of HIV-1 disease (18). Different human leukocyte antigen alleles may also alter the rate of progression to acquired immunodeficiency syndrome at distinct intervals after infection (19).
The impact of the number of viral load measurements on the estimate of genetic effects was less prominent, but still discernible. On average, each additional viral load measurement increased the estimate of the genetic effect by 0.005 log10, and the impact was larger when only genome-wide significant SNPs were considered. However, most participants had a relatively limited number of viral load measurements (the interquartile ranges were 3–6 and 3–7 in seroconverters and seroprevalent subjects, respectively). Therefore, although more measurements lead to improved precision for measuring viral load, the benefit is likely to be small. Viral load is currently measured with quite high accuracy (20, 21), and thus even 1 measurement may often give a substantially precise picture of the viral load set point.
Our findings have implications for the choice of populations in studying genetic associations in HIV-1 disease. Restriction of eligibility based on number of viral load measurements is probably unnecessary. Conversely, restriction to seroconverter cohorts may be appropriate for maximizing power of discovery. High-throughput genetic studies in seroprevalent cohorts may produce a much higher proportion of false-negative genetic associations and may suggest nonreplication of otherwise genuine associations that have emerged in seroconverter studies (22). In the typical range of allele frequencies of common variants, when the genetic effect is halved, one needs a sample size approximately 4 times larger to have the same power to detect it (23). The decreases in the effect size estimates seen in seroprevalent groups could well be in the range of halving the genetic effect. In our analysis, the data from seroprevalent subjects did not reveal any variant with genome-wide significance, while seroconverters alone sufficed to reach genome-wide significance for all 5 variants that were genome-wide significant in the combined analysis. Importantly, one should be cautious when claiming that lack of replication of an effect in these 2 types of populations means that an association is not genuine. Our observations and considerations about the influence of phenotype definition and precision are of relevance for other studies dealing with different phenotypes in HIV-1 disease.
Finally, one can also apply a similar approach to GWA studies of other diseases and traits to empirically test the impact of restrictive or lenient inclusion criteria on the estimates of genetic effects. GWA studies have probably had less success for diseases where phenotypes have been more difficult to define and standardize, such as cognitive traits and mental health-related diseases (24), behavioral traits (25), or osteoarthritis (26). Some evidence from other fields, such as obesity, also suggests that the establishment of associations may be dependent on phenotype definition (27), and variability in definitions may cause heterogeneity in effect sizes or even spurious associations (28).
Moreover, the extent of available choices in defining phenotypes differs across fields. Some fields have highly standardized definitions of phenotypes, while others have very disparate practices of defining phenotypes across different teams. In some cases, harmonization of different phenotypes to a common denominator is possible, while in other fields this may not be feasible. When information is available, analyzing multiple phenotypes in the same data set may offer useful biologic insights (29). Moreover, when multiple different definitions and inclusion criteria can be applied to the same data set, empirical analyses may help one identify the most informative definitions that would maximize power for future studies.
Supplementary Material
Acknowledgments
Author affiliations: Institute of Microbiology, University Hospital Center, University of Lausanne, Lausanne, Switzerland (Evangelos Evangelou, Amalio Telenti, Sara Colombo); Department of Hygiene and Epidemiology, School of Medicine, University of Ioannina, Ioannina, Greece (Evangelos Evangelou, John P. A. Ioannidis); Center for Human Genome Variation, Duke Institute for Genome Sciences and Policy, Duke University, Durham, North Carolina (Jacques Fellay, David B. Goldstein); IrsiCaixa Foundation and Hospital Germans Trias i Pujol, Badalona, Spain (Javier Martinez-Picado); Institució Catalana de Recerca i Estudis Avançats, Barcelona, Spain (Javier Martinez-Picado); Department of Infectious Diseases, Copenhagen University Hospital, Rigshospitalet, Copenhagen, Denmark (Niels Obel); School of Medicine and Tufts Medical Center, Tufts University, Boston, Massachusetts (John P. A. Ioannidis); Harvard School of Public Health, Boston, Massachusetts (John P. A. Ioannidis); and Stanford Prevention Research Center, Stanford, California (John P. A. Ioannidis).
Conflict of interest: none declared.
Glossary
Abbreviations
- CCR2
C-C chemokine receptor type 2
- CCR5
C-C chemokine receptor type 5
- CHAVI
Center for HIV/AIDS Vaccine Immunology
- CI
confidence interval
- GWA
genome-wide association
- HCP5
human leukocyte antigen complex P5
- HIV-1
human immunodeficiency virus type 1
- HLA-C
human leukocyte antigen C
- SNP
single nucleotide polymorphism
References
- 1.McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9(5):356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 2.Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118(5):1590–1605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wojczynski MK, Tiwari HK. Definition of phenotype. Adv Genet. 2008;60:75–105. doi: 10.1016/S0065-2660(07)00404-X. [DOI] [PubMed] [Google Scholar]
- 5.Ji F, Yang Y, Haynes C, et al. Computing asymptotic power and sample size for case-control genetic association studies in the presence of phenotype and/or genotype misclassification errors. Stat Appl Genet Mol Biol. 2005;4(1) doi: 10.2202/1544-6115.1184. Article 37. (doi: 10.2202/1544-6115.1184) [DOI] [PubMed] [Google Scholar]
- 6.Fellay J, Shianna KV, Ge D, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317(5840):944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fellay J, Ge D, Shianna KV, et al. Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 2009;5(12):e1000791. doi: 10.1371/journal.pgen.1000791. (doi: 10.1371/journal.pgen.1000791) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ioannidis JP, Rosenberg PS, Goedert JJ, et al. Effects of CCR5-Δ32, CCR2-64I, and SDF-1 3′A alleles on HIV-1 disease progression: an international meta-analysis of individual-patient data. Ann Intern Med. 2001;135(9):782–795. doi: 10.7326/0003-4819-135-9-200111060-00008. [DOI] [PubMed] [Google Scholar]
- 9.Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 10.Lau J, Ioannidis JP, Schmid CH. Quantitative synthesis in systematic reviews. Ann Intern Med. 1997;127(9):820–826. doi: 10.7326/0003-4819-127-9-199711010-00008. [DOI] [PubMed] [Google Scholar]
- 11.Zollner S, Pritchard JK. Overcoming the winner's curse: estimating penetrance parameters from case-control data. Am J Hum Genet. 2007;80(4):605–615. doi: 10.1086/512821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ioannidis JP. Why most discovered true associations are inflated. Epidemiology. 2008;19(5):640–648. doi: 10.1097/EDE.0b013e31818131e7. [DOI] [PubMed] [Google Scholar]
- 13.DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7(3):177–188. doi: 10.1016/0197-2456(86)90046-2. [DOI] [PubMed] [Google Scholar]
- 14.Kavvoura FK, Ioannidis JP. Methods for meta-analysis in genetic association studies: a review of their potential and pitfalls. Hum Genet. 2008;123(1):1–14. doi: 10.1007/s00439-007-0445-9. [DOI] [PubMed] [Google Scholar]
- 15.Ioannidis JP, Patsopoulos NA, Evangelou E. Uncertainty in heterogeneity estimates in meta-analyses. BMJ. 2007;335(7626):914–916. doi: 10.1136/bmj.39343.408449.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Telenti A, Goldstein DB. Genomics meets HIV-1. Nat Rev Microbiol. 2006;4(11):865–873. doi: 10.1038/nrmicro1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mulherin SA, O'Brien TR, Ioannidis JP, et al. Effects of CCR5-Δ32 and CCR2-64I alleles on HIV-1 disease progression: the protection varies with duration of infection. AIDS. 2003;17(3):377–387. doi: 10.1097/01.aids.0000050783.28043.3e. [DOI] [PubMed] [Google Scholar]
- 19.Gao X, Bashirova A, Iversen AK, et al. AIDS restriction HLA allotypes target distinct intervals of HIV-1 pathogenesis. Nat Med. 2005;11(12):1290–1292. doi: 10.1038/nm1333. [DOI] [PubMed] [Google Scholar]
- 20.Schockmel GA, Yerly S, Perrin L. Detection of low HIV-1 RNA levels in plasma. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;14(2):179–183. doi: 10.1097/00042560-199702010-00013. [DOI] [PubMed] [Google Scholar]
- 21.Coste J, Montes B, Reynes J, et al. Comparative evaluation of three assays for the quantitation of human immunodeficiency virus type 1 RNA in plasma. J Med Virol. 1996;50(4):293–302. doi: 10.1002/(SICI)1096-9071(199612)50:4<293::AID-JMV3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 22.Ioannidis JP. Non-replication and inconsistency in the genome-wide association setting. Hum Hered. 2007;64(4):203–213. doi: 10.1159/000103512. [DOI] [PubMed] [Google Scholar]
- 23.Dupont WD, Plummer WD., Jr Power and sample size calculations: a review and computer program. Control Clin Trials. 1990;11(2):116–128. doi: 10.1016/0197-2456(90)90005-m. [DOI] [PubMed] [Google Scholar]
- 24.Sabb FW, Burggren AC, Higier RG, et al. Challenges in phenotype definition in the whole-genome era: multivariate models of memory and intelligence. Neuroscience. 2009;164(1):88–107. doi: 10.1016/j.neuroscience.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tobacco and Genetics Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42(5):441–447. doi: 10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evangelou E, Valdes AM, Kerkhof HJ, et al. Meta-analysis of genome-wide association studies confirms a susceptibility locus for knee osteoarthritis on chromosome 7q22. Ann Rheum Dis. 2011;70(2):349–355. doi: 10.1136/ard.2010.132787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kring SI, Larsen LH, Holst C, et al. Genotype-phenotype associations in obesity dependent on definition of the obesity phenotype. Obes Facts. 2008;1(3):138–145. doi: 10.1159/000137665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heid IM, Huth C, Loos RJ, et al. Meta-analysis of the INSIG2 association with obesity including 74,345 individuals: does heterogeneity of estimates relate to study design? PLoS Genet. 2009;5(10):e1000694. doi: 10.1371/journal.pgen.1000694. (doi: 10.1371/journal.pgen.1000694) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kent JW., Jr Analysis of multiple phenotypes. Genet Epidemiol. 2009;33(suppl 1):S33–S39. doi: 10.1002/gepi.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.